A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network

Abstract
1. Introduction
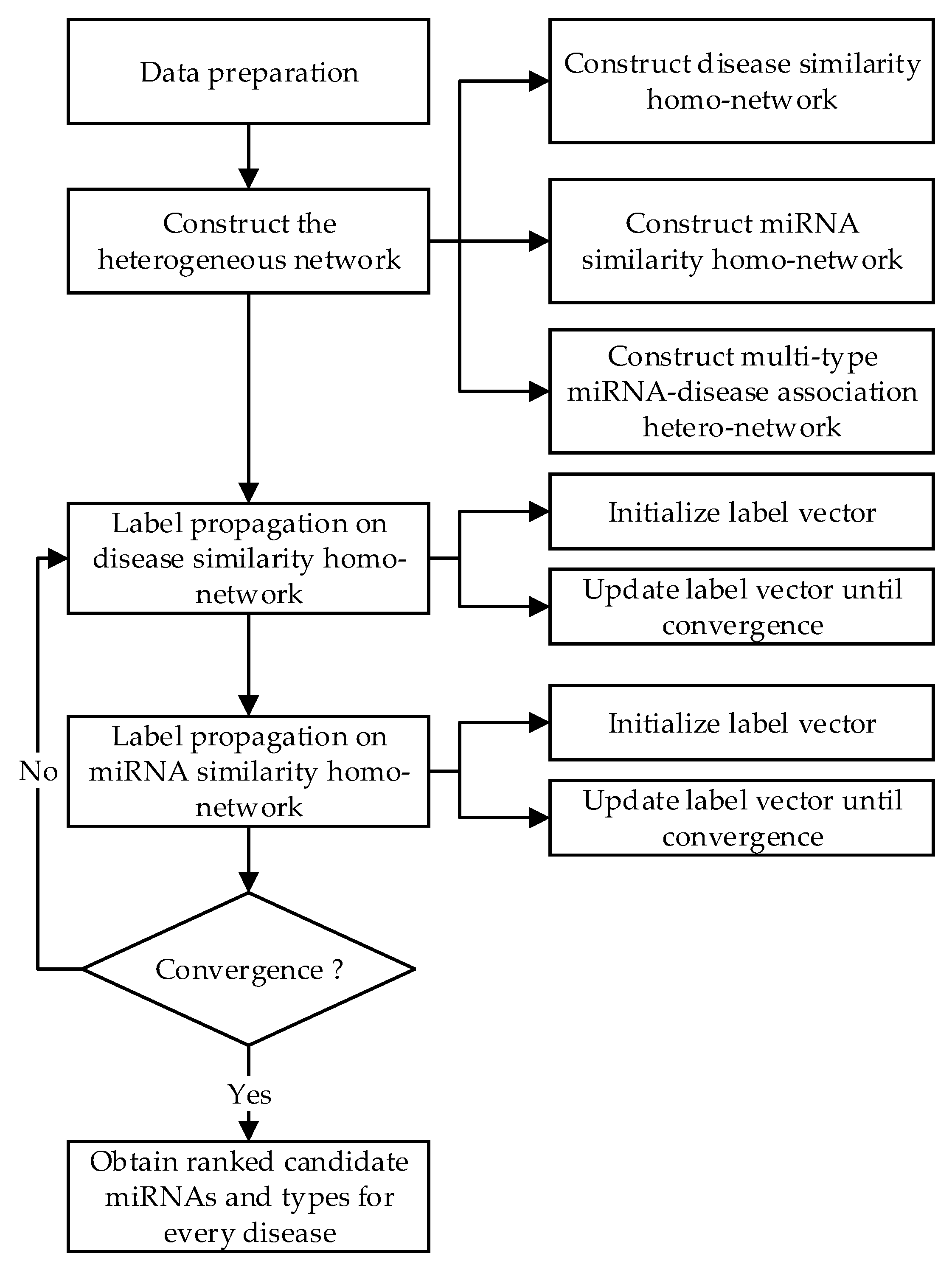
2. Materials and Methods
2.1. Data Preparation
2.2. Construct Disease Similarity Homo-Network
2.3. Construction of the miRNA Similarity Homo-Network
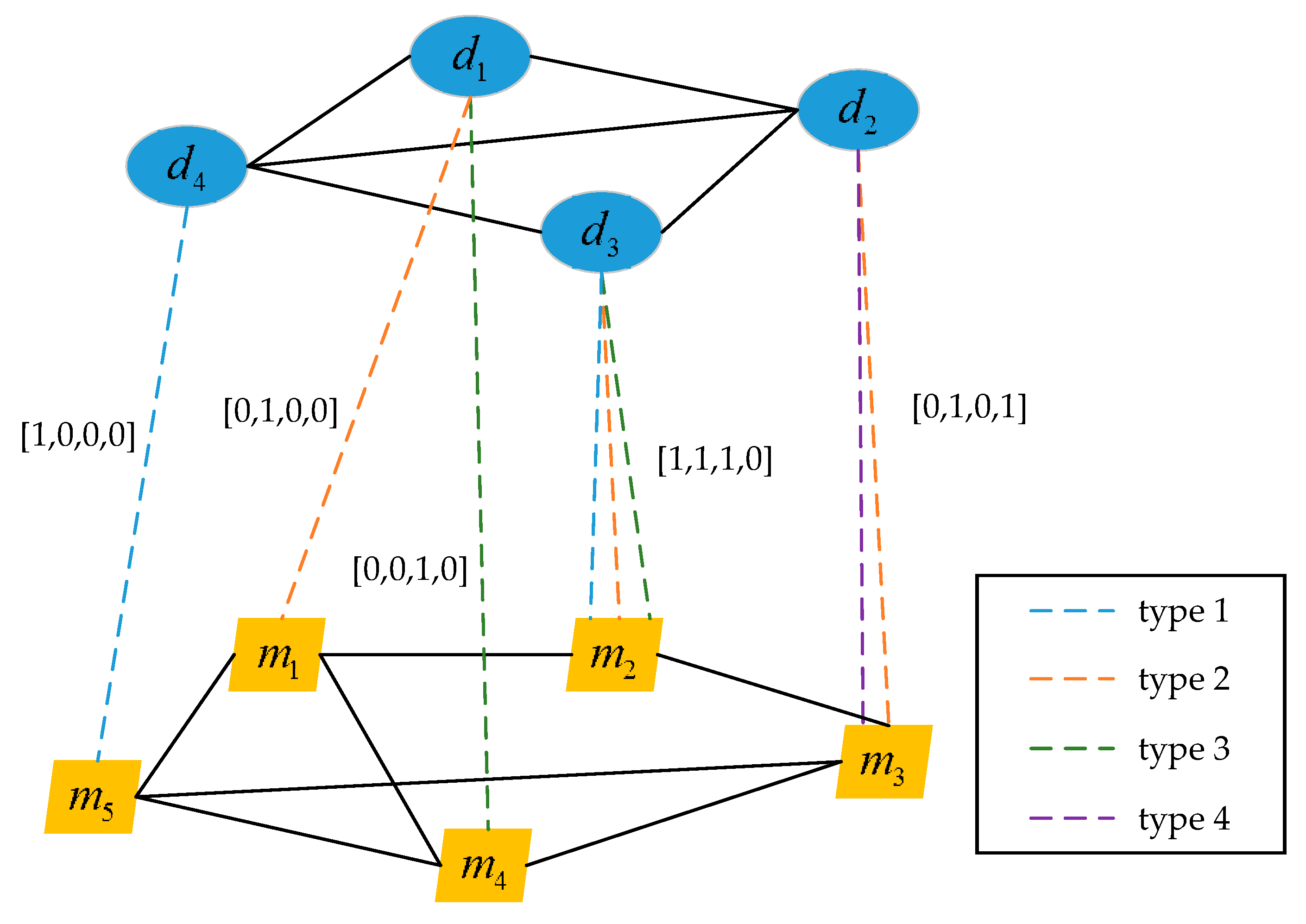
2.4. Construction of the Multi-Type miRNA-Disease Association Hetero-Network
2.5. Network-Based Label Propagation Algorithm for Predicting Multiple miRNA-Disease Associations
3. Results
3.1. Performance Evaluation
3.2. Comparison with the Restricted Boltzmann Machine Model for Predicting Multiple Types of miRNA-Disease Associations Method
3.3. Effect of the Parameters
3.4. Case Studies of Lung Cancer and Breast Cancer
3.5. Web Server for Network-Based Label Propagation Algorithm to Predicting Multiple miRNA-Disease Association Method
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of miRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis Elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.W.; Ferland-McCollough, D.; Jackson, T.J.; Bushell, M. MicroRNAs in cancer management. Lancet Oncol. 2012, 13, e249–e258. [Google Scholar] [CrossRef]
- Hatziapostolou, M.; Polytarchou, C.; Aggelidou, E.; Drakaki, A.; Poultsides, G.A.; Jaeger, S.A.; Ogata, H.; Karin, M.; Struhl, K.; Hadzopoulou-Cladaras, M. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011, 147, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhang, Y.; Wei, J.; Jia, W.; Ge, Z.; Zhang, Z.; Liu, X. MicroRNA-21 promotes hepatocellular carcinoma HepG2 cell proliferation through repression of mitogen-activated protein kinase-kinase 3. BMC Cancer 2013, 13, 469. [Google Scholar] [CrossRef] [PubMed]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2012, 481, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Zhang, T.; Teng, M.; Liu, Y.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010, 4, S2. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z. Correction: Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Z. Similarity-based methods for potential human microRNA-disease association prediction. BMC Med. Genom. 2013, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Bo, L.; Li, X.; Li, K. Network consistency projection for human miRNA-disease associations inference. Sci. Rep. 2016, 6, 36054. [Google Scholar] [CrossRef] [PubMed]
- Mørk, S.; Pletscher-Frankild, S.; Caro, A.P.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA–disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, G.; Jin, S.; Li, Y.; Wang, Y. Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.-X.; Lv, J.-Y.; Li, Y.-S.; Xiao, Y.; Shao, T.-T.; Huo, X.; Li, X.; Zou, Y.; Han, Q.-L. Prioritizing candidate disease miRNAs by topological features in the miRNA target–dysregulated network: Case study of prostate cancer. Mol. Cancer Ther. 2011, 10, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, C.; Gardès, J. Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 2016, 6, 27036. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.B.; Chiu, C.M.; Hsu, S.D.; Huang, W.Y.; Chien, C.H.; Lee, T.Y.; Huang, H.D. miRTar: An integrated system for identifying miRNA-target interactions in human. BMC Bioinform. 2011, 12, 300. [Google Scholar] [CrossRef] [PubMed]
- Pio, G.; Ceci, M.; Malerba, D.; D’Elia, D. ComiRNet: A web-based system for the analysis of miRNA-gene regulatory networks. BMC Bioinform. 2015, 16 (Suppl. S9), S7. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; Li, Z.; Deng, L.; Zhang, Y.; Dai, Q. RBMMMDA: Predicting multiple types of disease-microRNA associations. Sci. Rep. 2015, 5, 13877. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Allison, K.R.; Bonneau, R. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Ceci, M.; Pio, G.; Kuzmanovski, V.; Džeroski, S. Semi-supervised multi-view learning for gene network reconstruction. PLoS ONE 2015, 10, e0144031. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-Y.; Zhang, S.-W.; Zhang, S.-Y. Prediction of drug–target interaction by label propagation with mutual interaction information derived from heterogeneous network. Mol. BioSyst. 2016, 12, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; Yan, S.; Kassim, A. Learning a propagable graph for semisupervised learning: Classification and regression. IEEE Trans. Knowl. Data Eng. 2012, 24, 114–126. [Google Scholar]
- Huang, Y.-F.; Yeh, H.-Y.; Soo, V.-W. Inferring drug-disease associations from integration of chemical, genomic and phenotype data using network propagation. BMC Med. Genom. 2013, 6, S4. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Bousquet, O.; Lal, T.N.; Weston, J.; Olkopf, B.S. Learning with local and global consistency. Adv. Neural Inf. Process. Syst. 2004, 16, 321–328. [Google Scholar]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.H.; Kuang, R. A heterogeneous label propagation algorithm for disease gene discovery. In Proceedings of the 10th SIAM International Conference on Data Mining, Columbus, OH, USA, 29 April–1 May 2010; pp. 583–594. [Google Scholar]
- Castro, D.; Moreira, M.; Gouveia, A.M.; Pozza, D.H.; Mello, R.A.D. MicroRNAs in lung cancer. Oncotarget 2017, 8, 81679–81685. [Google Scholar] [CrossRef] [PubMed]
- Sittka, A.; Schmeck, B. MicroRNAs in the lung. Adv. Exp. Med. Biol. 2013, 774, 121–134. [Google Scholar] [PubMed]
- Tominaga, E.; Yuasa, K.; Shimazaki, S.; Hijikata, T. MicroRNA-1 targets Slug and endows lung cancer A549 cells with epithelial and anti-tumorigenic properties. Exp. Cell Res. 2013, 319, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Y.; Shen, H.; Li, H.; Long, L.; Hui, L.; Xu, W. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS Lett. 2013, 587, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Haddadin, S.; Wang, Y.; Gu, L.Q.; Perry, M.C.; Freter, C.E.; Wang, M.X. Plasma microRNAs as novel biomarkers for early detection of lung cancer. Int. J. Clin. Exp. Pathol. 2011, 4, 575–586. [Google Scholar] [PubMed]
- Han, H.S.; Yun, J.; Lim, S.N.; Han, J.H.; Lee, K.H.; Kim, S.T.; Kang, M.H.; Son, S.M.; Lee, Y.M.; Choi, S.Y. Downregulation of cell-free miR-198 as a diagnostic biomarker for lung adenocarcinoma-associated malignant pleural effusion. Int. J. Cancer 2013, 133, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, N.; Delpu, Y.; Suriawinata, A.A.; Bournet, B.; Bureau, C.; Selves, J.; Tsongalis, G.J.; Dufresne, M.; Buscail, L.; Cordelier, P. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin. Chem. 2010, 56, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Chari, R.; Lockwood, W.W.; Lam, S.; Lam, W.L. miR-101 DNA copy loss is a prominent subtype specific event in lung cancer. J. Thorac. Oncol. 2011, 6, 1594–1598. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Hsiao, T.H.; Hong, T.M.; Chen, H.Y.; Kao, S.H.; Wang, W.L.; Yu, S.L.; Lin, C.W.; Yang, P.C. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PLoS ONE 2014, 9, e96765. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ma, Z.; Li, Y.; Zhao, B.; Wang, D.; Jin, Y.; Jin, Y. miR-143 inhibits cell proliferation by targeting autophagy-related 2B in non-small cell lung cancer H1299 cells. Mol. Med. Rep. 2015, 11, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.L.; Hou, P.P.; Li, Y.L.; Wang, D.T.; Yuan, T.W.; Wei, J.L.; Zhao, B.T.; Lou, J.T.; Zhao, X.T.; Jin, Y. MicroRNA-34a inhibits the proliferation and promotes the apoptosis of non-small cell lung cancer H1299 cell line by targeting TGFβR2. Tumour Biol. 2015, 36, 2481–2490. [Google Scholar] [CrossRef] [PubMed]
- Shang, A.Q.; Xie, Y.N.; Wang, J.; Sun, L.; Wei, J.; Lu, W.Y.; Lan, J.Y.; Wang, W.W.; Wang, L.; Wang, L.L. Predicative values of serum microRNA-22 and microRNA-126 levels for non-small cell lung cancer development and metastasis: A case-control study. Neoplasma 2017, 64, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Li, B.J.; Lu, H.W.; Chen, Y.; Lu, C.; Zhu, R.X.; Liu, S.H.; Yi, Q.T.; Li, J.; Song, C.H. Serum miR-152, miR-148a, miR-148b, and miR-21 as novel biomarkers in non-small cell lung cancer screening. Tumour Biol. 2015, 36, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Joerger, M.; Baty, F.; Früh, M.; Droege, C.; Stahel, R.A.; Betticher, D.C.; Moos, R.V.; Ochsenbein, A.; Pless, M.; Gautschi, O. Circulating microRNA profiling in patients with advanced non-squamous NSCLC receiving bevacizumab/erlotinib followed by platinum-based chemotherapy at progression (SAKK 19/05). Lung Cancer 2014, 85, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zang, A.; Shang, Y.; Yang, H.; Song, Z.; Wang, Z.; Ren, L.; Wei, Y.; Hu, L.; Shi, H. MicroRNA-146a rs2910164 polymorphism is associated with susceptibility to non-small cell lung cancer in the chinese population. Med. Oncol. 2014, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Druesne-Pecollo, N.; Touvier, M.; Barrandon, E.; Chan, D.S.; Norat, T.; Zelek, L.; Hercberg, S.; Latino-Martel, P. Excess body weight and second primary cancer risk after breast cancer: A systematic review and meta-analysis of prospective studies. Breast Cancer Res. Treat. 2012, 135, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast cancer: Epidemiology and etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, Q.; Liu, Y.; Hao, X.; Zhang, S.; Zhang, J. Analysis of miR-205 and miR-155 expression in the blood of breast cancer patients. Chin. J. Cancer Res. 2013, 25, 46–54. [Google Scholar] [PubMed]
- Sun, Y.; Wang, M.; Lin, G.; Sun, S.; Li, X.; Qi, J.; Li, J. Serum microRNA-155 as a potential biomarker to track disease in breast cancer. PLoS ONE 2012, 7, e47003. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.; Lai, Y.; Liao, Y.; Liu, R.; Qiu, W. Hsa-miR-1 suppresses breast cancer development by down-regulating K-ras and long non-coding RNA MALAT1. Int. J. Biol. Macromol. 2015, 81, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Ouchida, M.; Kanzaki, H.; Ito, S.; Hanafusa, H.; Jitsumori, Y.; Tamaru, S.; Shimizu, K. Novel direct targets of miR-19a identified in breast cancer cells by a quantitative proteomic approach. PLoS ONE 2012, 7, e44095. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, Y.; He, Y.; Mao, Y. miR-19b promotes breast cancer metastasis through targeting mylip and its related cell adhesion molecules. Oncotarget 2017, 8, 64330–64343. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Zhang, S.; Shan, C.; Zhou, L.; Zhou, Z. MicroRNA-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the EGFR/Akt signaling pathway. FEBS J. 2013, 280, 3962–3974. [Google Scholar] [CrossRef] [PubMed]
- Stückrath, I.; Rack, B.; Janni, W.; Jäger, B.; Pantel, K.; Schwarzenbach, H. Aberrant plasma levels of circulating miR-16, miR-107, miR-130a and miR-146a are associated with lymph node metastasis and receptor status of breast cancer patients. Oncotarget 2015, 6, 13387–13401. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Müller, M.; Römermann, D.; Länger, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J. Pathol. 2008, 214, 17–24. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithms | RBMMMDA | NLPMMDA |
|---|---|---|
| AUC | 0.8606 | 0.9739 |
| Data | Known four types of miRNA-disease associations | Disease semantic similarity, miRNA functional similarity, Gaussian interaction profile kernel similarity and known four types of miRNA-disease associations |
| Application | Cannot be applied to isolated diseases | Cannot be applied to isolated diseases |
| Parameters | Use the previous value | Select by the performance of experiments |
| model | Supervised learning | Semi-supervised learning |
| Case study | Lung cancer: 33 of top 50 | Lung cancer: 44 of top 50 |
| Breast cancer: 17 of top 50 | Breast cancer: 37 of top 50 |
| AUC | AUPR | ||
|---|---|---|---|
| 0.1 | 0.1 | 0.9738 | 0.9320 |
| 0.2 | 0.2 | 0.9739 | 0.9323 |
| 0.3 | 0.3 | 0.9738 | 0.9309 |
| 0.4 | 0.4 | 0.9720 | 0.9302 |
| 0.5 | 0.5 | 0.5 | 0.5 |
| 0.6 | 0.6 | 0.8173 | 0.6490 |
| 0.7 | 0.7 | 0.8076 | 0.6409 |
| 0.8 | 0.8 | 0.7900 | 0.6251 |
| 0.9 | 0.9 | 0.7559 | 0.5962 |
| miRNAs | Types | PMID | miRNAs | Types | PMID |
|---|---|---|---|---|---|
| hsa-mir-499a | genetics | unconfirmed | hsa-mir-19a | target | 27588137;25604748;28592790 |
| hsa-mir-146a | genetics | 25154761;24144839;29127520 | hsa-let-7f | target | 29017393 |
| hsa-mir-133a | target | 24816813;22089643;25518741 | hsa-mir-15a | target | 25442346;24500260;25874488 |
| hsa-mir-126 | circulation | 28253725;27093275;29266846 | hsa-mir-206 | target | 26919096;26075299;25522678 |
| hsa-mir-17 | genetics | 17384677 | hsa-mir-16 | genetics | unconfirmed |
| hsa-mir-21 | circulation | 25501703;25421010;29163821 | hsa-mir-126 | target | 18602365;22510476;29277611 |
| hsa-mir-143 | target | 25322940;25003638;24070896 | hsa-mir-125b | target | 28713974 |
| hsa-mir-34a | target | 25501507;25038915;24983493 | hsa-mir-218 | target | 21159652;24247270;24705471 |
| hsa-mir-20a | genetics | 17384677 | hsa-mir-17 | circulation | 23263848 |
| hsa-mir-29a | circulation | 24928469 | hsa-let-7e | target | unconfirmed |
| hsa-mir-200c | target | 24997798;24205206;23708087 | hsa-mir-20a | target | 24722426 |
| hsa-mir-17 | target | 24755562;24722426;29289833 | hsa-mir-219 | target | 28714014 |
| hsa-mir-92a | genetics | unconfirmed | hsa-mir-222 | target | 21042732 |
| hsa-mir-20a | circulation | 25421010 | hsa-mir-19b | target | 28364280 |
| hsa-mir-34a | epigenetics | 18719384 | hsa-mir-429 | target | 24866238;27602157 |
| hsa-mir-34b | epigenetics | 24130071;22047961;21383543 | hsa-mir-223 | circulation | 28356944;25421010;29212284 |
| hsa-mir-18a | genetics | unconfirmed | hsa-mir-18a | target | 28471447 |
| hsa-mir-200b | target | 22139708 ;28731781;28615992 | hsa-mir-122 | circulation | 24282590;25926378 |
| hsa-mir-155 | target | 22027557 ;29260515;28939896 | hsa-let-7a | target | 21097396 |
| hsa-mir-16 | target | 25435430;23954293;29138833 | hsa-mir-15a | genetics | unconfirmed |
| hsa-mir-34c | epigenetics | 24130071;22047961;21383543 | hsa-mir-124 | epigenetics | 17308079 |
| hsa-mir-221 | target | 18246122;21042732;19962668 | hsa-mir-92a | target | 23820254 |
| hsa-mir-183 | target | 18840437;26951513;27593936 | hsa-mir-133b | target | 22883469;19654003;29328427 |
| hsa-mir-214 | target | 28396596;26462018;28396596 | hsa-mir-155 | genetics | 28225782 |
| hsa-mir-146a | circulation | 28678319;25755772;24531034 | hsa-mir-203 | target | 25140799;24040137;28921827 |
| miRNAs | Types | PMID | miRNAs | Types | PMID |
|---|---|---|---|---|---|
| hsa-mir-16 | genetics | 16754881;17012848 | hsa-mir-127 | target | 24282530;24155205;25477702 |
| hsa-mir-1 | target | 26275461;26926567;26497855 | hsa-let-7i | target | 24662829;21826373; |
| hsa-mir-126 | circulation | 28683441 | hsa-let-7a | genetics | 26681038 |
| hsa-mir-19a | target | 22952885;23831570;27596294 | hsa-mir-106b | target | 27519168;27325313;28518139 |
| hsa-let-7a | target | 24172884 | hsa-mir-219 | target | Unconfirmed |
| hsa-mir-19b | target | 28969074;28731027;27602768 | hsa-let-7f | genetics | 23042301 |
| hsa-mir-92a | genetics | Unconfirmed | hsa-mir-127 | epigenetics | 27998789 |
| hsa-mir-223 | circulation | Unconfirmed | hsa-mir-15b | target | 25783158 |
| hsa-mir-18a | target | 19684618;25069832;21755340 | hsa-mir-143 | target | 28746466;28559978;28588724;27121210 |
| hsa-mir-29a | circulation | Unconfirmed | hsa-mir-19b | circulation | Unconfirmed |
| hsa-let-7c | target | 25388283 | hsa-mir-199a | circulation | 26476723;25906045 |
| hsa-mir-125b | genetics | 19738052 | hsa-let-7e | genetics | Unconfirmed |
| hsa-mir-133a | target | 23786162;29207145;26107945 | hsa-mir-145 | circulation | 23334650 |
| hsa-mir-15a | target | 27596816;27713175;28655885 | hsa-mir-155 | genetics | 26095675 |
| hsa-let-7d | target | 22081076 | hsa-let-7d | genetics | Unconfirmed |
| hsa-let-7f | target | 22407818;25552929 | hsa-mir-218 | circulation | Unconfirmed |
| hsa-mir-29b | epigenetics | 24297604 | hsa-mir-221 | circulation | 25009660;22156446 |
| hsa-mir-214 | target | 24577056;25738546;28071724 | hsa-mir-146a | target | 27175941;25596948;25712342 |
| hsa-mir-9 | epigenetics | 26519551;17948228 | hsa-mir-124 | epigenetics | Unconfirmed |
| hsa-mir-146a | circulation | 27197674;26033453;23898484 | hsa-mir-19a | circulation | 24938880;24416156 |
| hsa-let-7e | target | Unconfirmed | hsa-let-7g | target | 21868760 |
| hsa-mir-18a | circulation | 24694649;23705859;28109133 | hsa-mir-106a | target | 27325313 |
| hsa-mir-25 | target | 25026296;29310680;28188287 | hsa-mir-9 | circulation | Unconfirmed |
| hsa-let-7b | target | 21826373;24264599;23339187;22761738 | hsa-mir-145 | genetics | Unconfirmed |
| hsa-mir-92a | target | 28881597;29162724;28881597 | hsa-mir-19b | epigenetics | Unconfirmed |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Yin, J.; Zhang, X. A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network. Genes 2018, 9, 139. https://doi.org/10.3390/genes9030139
Zhang X, Yin J, Zhang X. A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network. Genes. 2018; 9(3):139. https://doi.org/10.3390/genes9030139
Chicago/Turabian StyleZhang, Xiaotian, Jian Yin, and Xu Zhang. 2018. "A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network" Genes 9, no. 3: 139. https://doi.org/10.3390/genes9030139
APA StyleZhang, X., Yin, J., & Zhang, X. (2018). A Semi-Supervised Learning Algorithm for Predicting Four Types MiRNA-Disease Associations by Mutual Information in a Heterogeneous Network. Genes, 9(3), 139. https://doi.org/10.3390/genes9030139