Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.1.1. Mitochondrial DNA Sensitivity Study
2.1.2. Nuclear DNA Sensitivity and Size Selection Study
2.1.3. Mock Degradation Study
2.1.4. Mitochondrial DNA Mixture Study
2.1.5. Telogen Hair Study
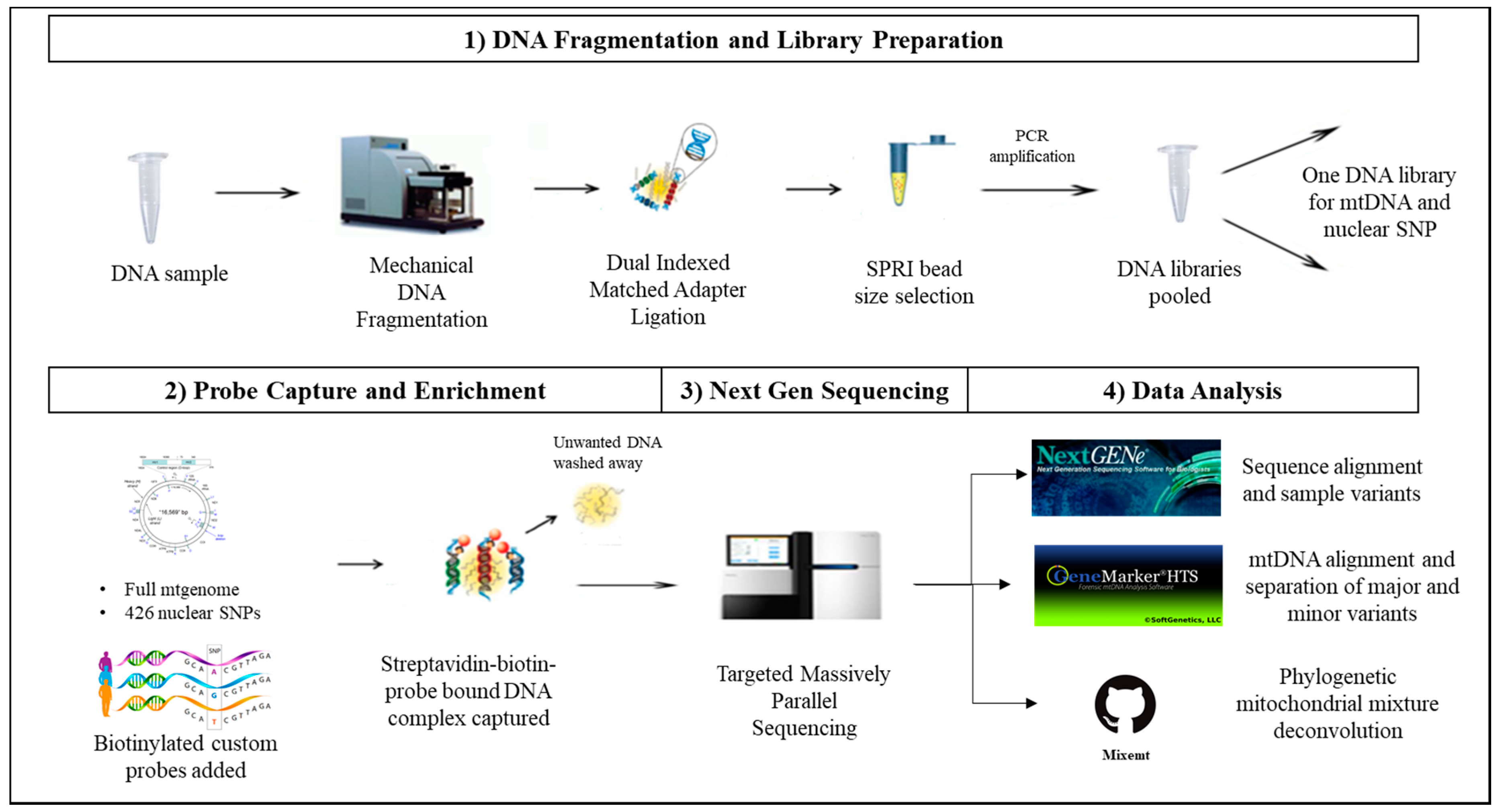
2.2. DNA Fragmentation and Library Preparation
2.3. Probe Capture Enrichment and Next Generation Sequencing
2.4. Data Analysis
3. Results
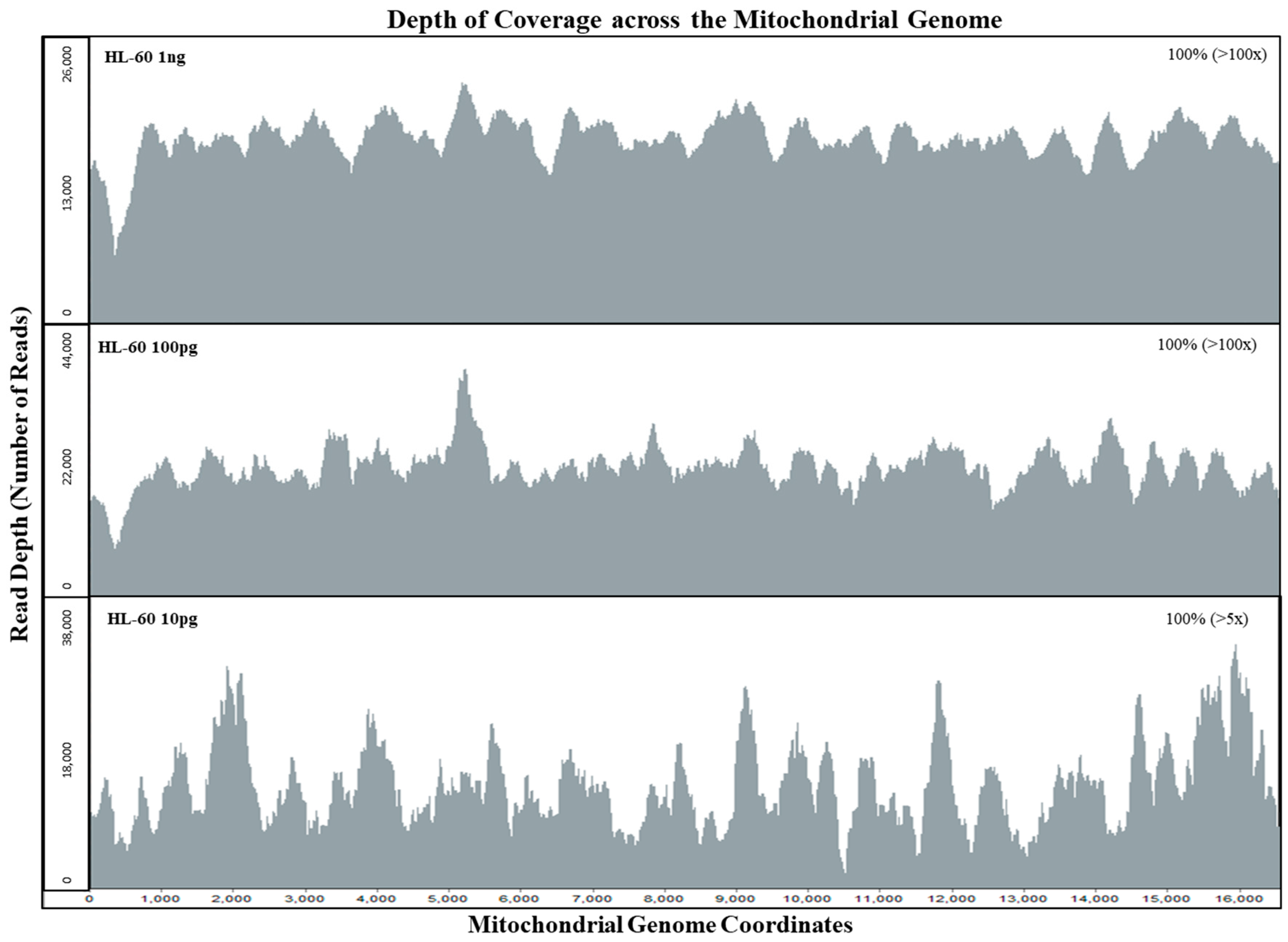
3.1. Mitochondrial DNA Sensitivity Study
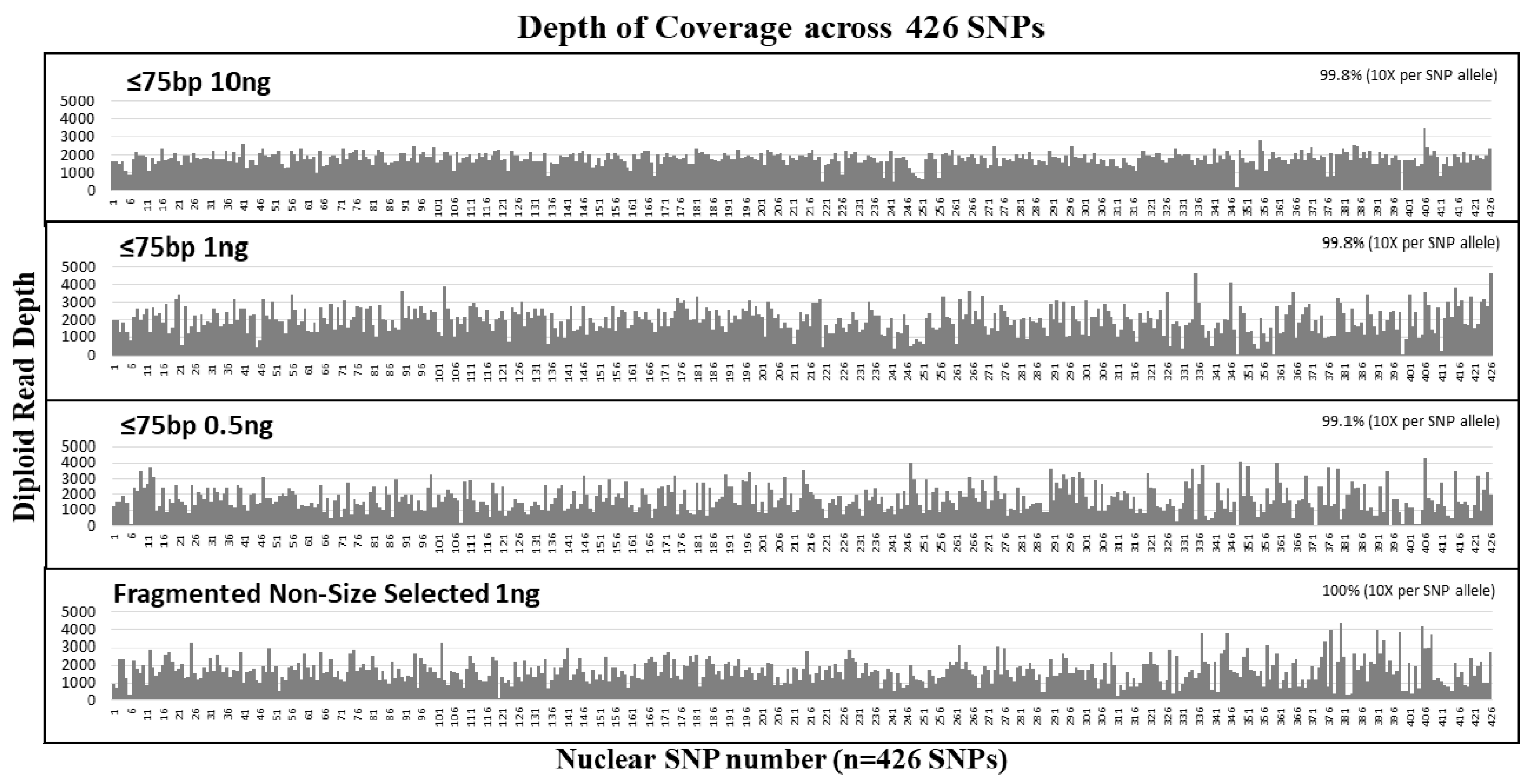
3.2. Nuclear DNA Sensitivity and Size Selection Study
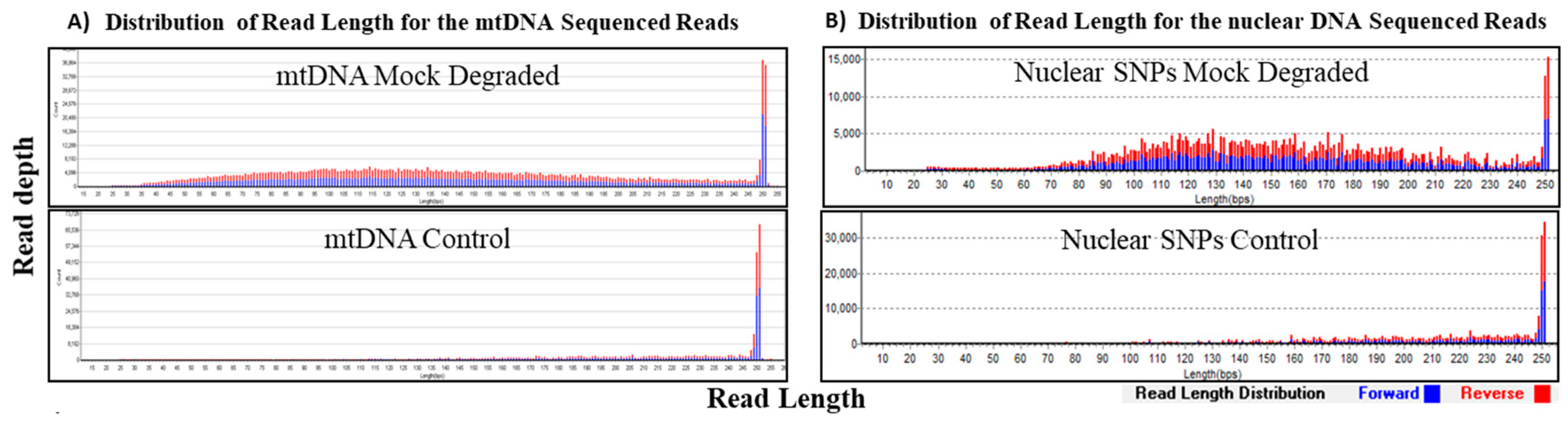
3.3. Mock Degradation Study
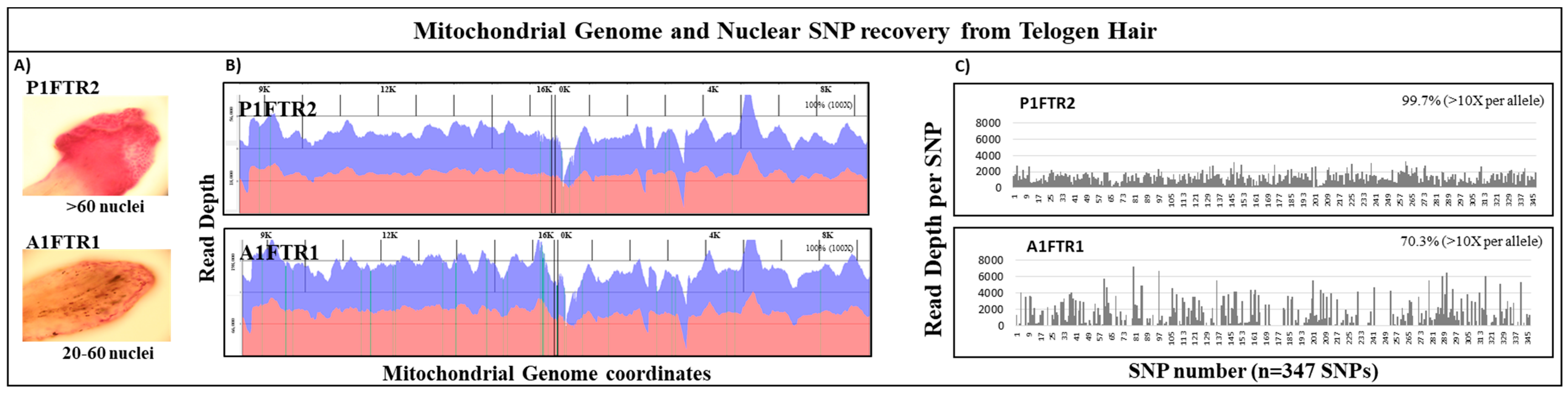
3.4. Mitochondrial DNA and Nuclear SNPs Recovery from Telogen Hairs
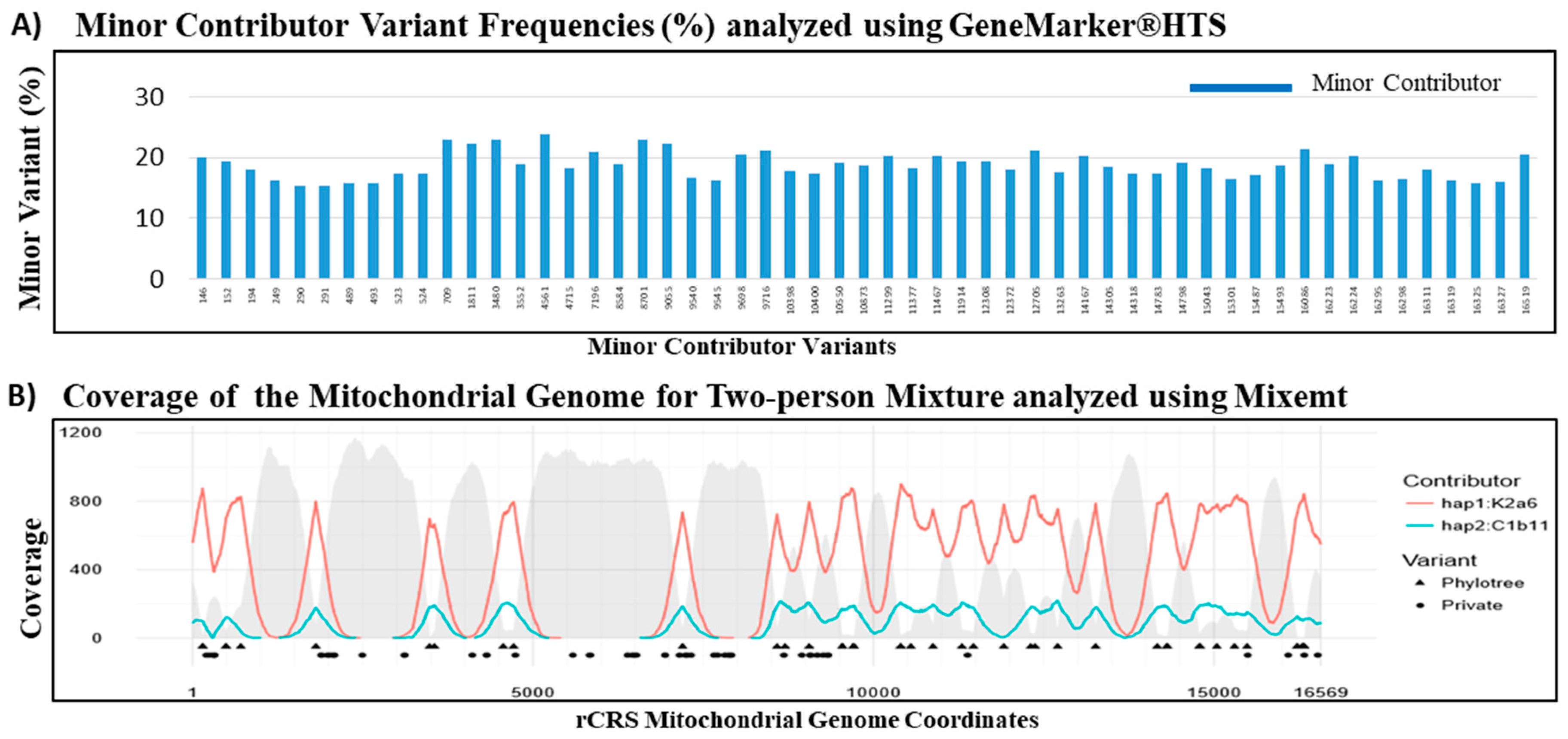
3.5. Mitochondrial DNA Mixture Study
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eduardoff, M.; Xavier, C.; Strobl, C.; Casas-Vargas, A.; Parson, W. Optimized mtDNA control region primer extension capture analysis for forensically relevant samples and highly compromised mtDNA of different age and origin. Genes 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Templeton, J.E.; Brotherton, P.M.; Llamas, B.; Soubrier, J.; Haak, W.; Cooper, A.; Austin, J.J. DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Molto, J.E.; Loreille, O.; Mallott, E.K.; Malhi, R.S.; Fast, S.; Daniels-Higginbotham, J.; Marshall, C.; Parr, R. Complete mitochondrial genome sequencing of a burial from a romano-christian cemetery in the Dakhleh Oasis, Egypt: Preliminary indications. Genes 2017, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Mertes, F.; Elsharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genom. 2011, 10, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Avila-Arcos, M.C.; Cappellini, E.; Romero-Navarro, J.A.; Wales, N.; Moreno-Mayar, J.V.; Rasmussen, M.; Fordyce, S.L.; Montiel, R.; Vielle-Calzada, J.P.; Willerslev, E.; et al. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Sci. Rep. 2011, 1, 74. [Google Scholar] [CrossRef] [PubMed]
- Cummings, N.; King, R.; Rickers, A.; Kaspi, A.; Lunke, S.; Haviv, I.; Jowett, J.B. Combining target enrichment with barcode multiplexing for high throughput SNP discovery. BMC Genom. 2010, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Sio, C.P.; Lu, Y.L.; Chang, H.T.; Hu, C.H.; Pai, T.W. Identification of conserved and polymorphic STRs for personal genomes. BMC Genom. 2014, 15 (Suppl. 10), S3. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Gettings, K.B.; Kiesler, K.M.; Vallone, P.M. Performance of a next generation sequencing SNP assay on degraded DNA. Forensic Sci. Int. Genet. 2015, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, L.; Ralf, A.; van Oven, M.; Kupiec, T.; Chang, J.; Lagace, R.; Kayser, M. Simultaneous whole mitochondrial genome sequencing with short overlapping amplicons suitable for degraded DNA using the ion torrent personal genome machine. Hum. Mutat. 2015, 36, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Just, R.S.; Irwin, J.A.; Parson, W. Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci. Int. Genet. 2015, 18, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Gusmao, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Bar, W.; Brinkmann, B.; Budowle, B.; Carracedo, A.; Gill, P.; Holland, M.; Lincoln, P.J.; Mayr, W.; Morling, N.; Olaisen, B.; et al. Guidelines for mitochondrial DNA typing. DNA Commission of the International Society for Forensic Genetics. Vox Sang. 2000, 79, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Calloway, C.D.; Reynolds, R.L.; Herrin, G.L., Jr.; Anderson, W.W. The frequency of heteroplasmy in the HVII region of mtDNA differs across tissue types and increases with age. Am. J. Hum. Genet. 2000, 66, 1384–1397. [Google Scholar] [CrossRef] [PubMed]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. 2010, 12, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Alaeddini, R.; Walsh, S.J.; Abbas, A. Forensic implications of genetic analyses from degraded DNA—A review. Forensic Sci. Int. Genet. 2010, 4, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Opel, K.L.; Chung, D.T.; Drabek, J.; Butler, J.M.; McCord, B.R. Developmental validation of reduced-size STR Miniplex primer sets. J. Forensic Sci. 2007, 52, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Evett, I.W.; Buffery, C.; Willott, G.; Stoney, D. A guide to interpreting single locus profiles of DNA mixtures in forensic cases. J. Forensic Sci. Soc. 1991, 31, 41–47. [Google Scholar] [CrossRef]
- Butler, J.M.; Shen, Y.; McCord, B.R. The development of reduced size STR amplicons as tools for analysis of degraded DNA. J. Forensic Sci. 2003, 48, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.A.; Dobbins, A.E.; Pulker, H.K.; Butler, J.M.; Vallone, P.M.; Coble, M.D.; Parson, W.; Berger, B.; Grubwieser, P.; Mogensen, H.S.; et al. Analysis of artificially degraded DNA using STRs and SNPs--results of a collaborative European (EDNAP) exercise. Forensic Sci. Int. 2006, 164, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B. SNP typing strategies. Forensic Sci. Int. 2004, 146, S139–S142. [Google Scholar] [PubMed]
- Pakstis, A.J.; Speed, W.C.; Fang, R.; Hyland, F.C.; Furtado, M.R.; Kidd, J.R.; Kidd, K.K. SNPs for a universal individual identification panel. Hum. Genet. 2010, 127, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Wilson, M.R.; DiZinno, J.A.; Stauffer, C.; Fasano, M.A.; Holland, M.M.; Monson, K.L. Mitochondrial DNA regions HVI and HVII population data. Forensic Sci. Int. 1999, 103, 23–35. [Google Scholar] [CrossRef]
- Melton, T.; Dimick, G.; Higgins, B.; Lindstrom, L.; Nelson, K. Forensic mitochondrial DNA analysis of 691 casework hairs. J. Forensic Sci. 2005, 50, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Calloway, C.D.; Kim, H.; Erlich, H.A. Resolution of DNA mixtures and analysis of degraded DNA using the 454 DNA sequencing techonology. Available online: https://www.ncjrs.gov/App/Publications/abstract.aspx?ID=271240 (accessed on 7 December 2017).
- Winters, M.; Monroe, C.; Barta, J.L.; Kemp, B.M. Are we fishing or catching? Evaluating the efficiency of bait capture of CODIS fragments. Forensic Sci. Int. Genet. 2017, 29, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Maricic, T.; Whitten, M.; Paabo, S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE 2010, 5, e14004. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Feitsma, H.; Nijman, I.J.; de Bruijn, E.; van der Zaag, P.J.; Guryev, V.; Cuppen, E. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010, 38, e116. [Google Scholar] [CrossRef] [PubMed]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Chilamakuri, C.S.; Lorenz, S.; Madoui, M.A.; Vodak, D.; Sun, J.; Hovig, E.; Myklebost, O.; Meza-Zepeda, L.A. Performance comparison of four exome capture systems for deep sequencing. BMC Genom. 2014, 15, 449. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.J.; Chen, R.; Lam, H.Y.; Karczewski, K.J.; Chen, R.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Sulonen, A.M.; Ellonen, P.; Almusa, H.; Lepisto, M.; Eldfors, S.; Hannula, S.; Miettinen, T.; Tyynismaa, H.; Salo, P.; Heckman, C.; et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011, 12, R94. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.M.; Devault, A.M.; Kuch, M.; Murgha, Y.E.; Rouillard, J.M.; Poinar, H.N. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 2014, 31, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Davalos, D.I.; Llamas, B.; Gaunitz, C.; Fages, A.; Gamba, C.; Soubrier, J.; Librado, P.; Seguin-Orlando, A.; Pruvost, M.; Alfarhan, A.H.; et al. Experimental conditions improving in-solution target enrichment for ancient DNA. Mol. Ecol. Resour. 2017, 17, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillen, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Briggs, A.W.; Krause, J.; Prufer, K.; Burbano, H.A.; Siebauer, M.; Lachmann, M.; Paabo, S. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009, 28, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Ptak, S.E.; Briggs, A.W.; Ronan, M.T.; Simons, J.F.; Du, L.; Egholm, M.; Rothberg, J.M.; Paunovic, M.; et al. Analysis of one million base pairs of Neanderthal DNA. Nature 2006, 444, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Malaspinas, A.S.; Krause, J.; Briggs, A.W.; Johnson, P.L.; Uhler, C.; Meyer, M.; Good, J.M.; Maricic, T.; Stenzel, U.; et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 2008, 134, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Briggs, A.W.; Good, J.M.; Green, R.E.; Krause, J.; Maricic, T.; Stenzel, U.; Lalueza-Fox, C.; Rudan, P.; Brajkovic, D.; Kucan, Z.; et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 2009, 325, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.; Green, R.E.; Kircher, M.; Krause, J.; Patterson, N.; Durand, E.Y.; Viola, B.; Briggs, A.W.; Stenzel, U.; Johnson, P.L.; et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 2010, 468, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, D. Optimization and Validation of a Probe Capture NGS Assay for Sequencing the Whole Mitochondrial Genome on Forensically Relevant Samples. Master’s Thesis, Forensic Science, University of California, Davis, CA, USA, 2013. [Google Scholar]
- Bose, N. Development of a Nuclear SNP Probe Capture Assay for Massively Parallel Sequencing of Degraded and Mixed DNA Samples. Master’s Thesis, Forensic Science, University of California, Davis, CA, USA, 2016. [Google Scholar]
- Timken, M.D.; Swango, K.L.; Orrego, C.; Buoncristiani, M.R. A duplex real-time qPCR assay for the quantification of human nuclear and mitochondrial DNA in forensic samples: Implications for quantifying DNA in degraded samples. J. Forensic Sci. 2005, 50, 1044–1060. [Google Scholar] [CrossRef] [PubMed]
- Aceves, M. Development of a Real-Time qPCR Assay for the Evaluation of Human Nuclear and Mitochondrial DNA for Use in Missing Persons Casework. Master’s Thesis, Forensic Science, University of California, Davis, CA, USA, 2012. [Google Scholar]
- Almada, G. Improving Telogen Hair Analysis by Predicting Nuclear and Mitochondrial DNA Success for Massively Parallel Sequencing Using Microscopic and qpCR Methods. Master’s Thesis, Forencis Science, University of California, Davis, CA, USA, 2017. [Google Scholar]
- Mixemt. Available online: https://github.com/svohr/mixemt (accessed on 13 November 2017).
- Shih, S. Characterization of Germline Heteroplasmy in Mother-Offspring Pairs Using Next Generation Sequencing. Master’s Thesis, Forensic Science, Universiy of California, Davis, CA, USA, 2017. [Google Scholar]
- Anderon, S.; Bankier, A.T.; Barrel, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- GeneMakerHTS Software Quick Start Guide. Available online: http://www.softgenetics.com/GeneMarkerHTS.php (accessed on 19 January 2017).
- Vohr, S.H.; Gordon, R.; Eizenga, J.M.; Erlich, H.A.; Calloway, C.D.; Green, R.E. A phylogenetic approach for haplotype analysis of sequence data from complex mitochondrial mixtures. Forensic Sci. Int. Genet. 2017, 30, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Minaschek, G.; Bereiter-Hahn, J.; Bertholdt, G. Quantitation of the volume of liquid injected into cells by means of pressure. Exp. Cell Res. 1989, 183, 434–442. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Chou, L.S.; Liu, C.S.; Boese, B.; Zhang, X.; Mao, R. DNA sequence capture and enrichment by microarray followed by next-generation sequencing for targeted resequencing: Neurofibromatosis type 1 gene as a model. Clin. Chem. 2010, 56, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; van Daal, A. Forensically relevant SNP classes. Biotechniques 2008, 44, 603–608, 610. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Stivers, D.N.; Su, B.; Zhong, Y.; Budowle, B. The utility of short tandem repeat loci beyond human identification: Implications for development of new DNA typing systems. Electrophoresis 1999, 20, 1682–1696. [Google Scholar] [CrossRef]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Grigorenko, E.L.; Kajuna, S.L.; Karoma, N.J.; Kungulilo, S.; Kim, J.J.; Lu, R.B.; Odunsi, A.; et al. Developing a SNP panel for forensic identification of individuals. Forensic Sci. Int. 2006, 164, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Zhang, J.; Hui, A.B.; Wong, N.; Lau, T.K.; Leung, T.N.; Lo, K.W.; Huang, D.W.; Lo, Y.M. Size distributions of maternal and fetal DNA in maternal plasma. Clin. Chem. 2004, 50, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M.; Pack, E.D.; McElhoe, J.A. Evaluation of GeneMarker((R)) HTS for improved alignment of mtDNA MPS data, haplotype determination, and heteroplasmy assessment. Forensic Sci. Int. Genet. 2017, 28, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Porreca, G.J.; Zhang, K.; Li, J.B.; Xie, B.; Austin, D.; Vassallo, S.L.; LeProust, E.M.; Peck, B.J.; Emig, C.J.; Dahl, F.; et al. Multiplex amplification of large sets of human exons. Nat. Methods 2007, 4, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Erlich, H.A.; Calloway, C.D. Analysis of mixtures using next generation sequencing of mitochondrial DNA hypervariable regions. Croat. Med. J. 2015, 56, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.M.; McQuillan, M.R.; O’Hanlon, K.A. Second generation sequencing allows for mtDNA mixture deconvolution and high resolution detection of heteroplasmy. Croat. Med. J. 2011, 52, 299–313. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total DNA Amount | PCR Cycle Number |
|---|---|
| ≤10 ng–1 ng | 13 |
| ≤1 ng–500 pg | 17 |
| ≤500 pg–50 pg | 20 |
| ≤50 pg–10 pg | 24 |
| PCR Duplicates Included | PCR Duplicates Removed | ||||||
|---|---|---|---|---|---|---|---|
| HL 60 Samples | Input mtDNA Copies | Total Reads | Alignment (%) | Avg. Read Length (bp) | Avg. Coverage Per Base | Avg. Read Length (bp) | Avg. Coverage Per Base |
| 1 ng | 200,000 | 604,052 | 89.66% | 190 | 6474 | 181 | 1226 |
| 100 pg | 20,000 | 631,716 | 89.17% | 200 | 7043 | 178 | 183 |
| 10 pg | 2000 | 656,556 | 88.54% | 195 | 7197 | 157 | 22 |
| PCR Duplicates Included | PCR Duplicates Removed | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Input Amount (ng) | Total Reads | Avg. Read Length (bp) | Avg. Diploid Read Depth per SNP | SNP Coverage (n = 426 SNPs) | Locus/Allele dropout | Total Reads | Avg. Read Length (bp) | Avg. Diploid Read Depth per SNP | SNP Coverage (n = 426 SNPs) | Locus/Allele dropout |
| ≤75 bp | 10 | 4,606,916 | 78 | 1811 ± 377 | 425 (99.8%) | 1/0 | 2,476,408 | 78 | 517 ± 93 | 424 (99.5%) | 2/0 |
| 1 | 4,431,052 | 79 | 1978 ± 746 | 425 (99.8%) | 1/2 | 2,638,898 | 78 | 217 ± 97 | 423 (99.3%) | 3/1 | |
| 0.5 | 3,679,914 | 78 | 1721 ± 820 | 422 (99.1%) | 4/6 | 2,199,585 | 78 | 190 ± 115 | 421 (98.8%) | 5/10 | |
| Control | 1 | 3,648,297 | 130* | 1690 ± 697 | 426 (100%) | 0/3 | 2,259,909 | 127 | 336 ± 154 | 425 (99.8%) | 1/3 |
| Mock Degradation Study for mtDNA and nuclear SNPs (Input Amount 1 ng) | ||||
|---|---|---|---|---|
| Sample Type | Average Read Length (bp) | Total Reads | Average Depth of Coverage | Coverage (%) |
| mtDNA Mock Degraded | 146 | 990,269 | 7148 | 100 (100×) |
| mtDNA Control | 211 | 328,256 | 4130 | 100 (100×) |
| Nuclear DNA Mock Degraded | 147 | 2,614,853 | 968 * | 409 (96.0%) ** |
| Nuclear DNA Control | 194 | 1,988,568 | 498 * | 360 (84.5%) ** |
| Nuclei Count | mtDNA Recovery | Nuclear SNPs Recovery | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Number Nuclei | mtDNA Copies | Avg. Read Length (bp) | Total Reads | Alignment (%) | Avg. Coverage | Coverage ** (%) | nuDNA amt. (ng) | Total Reads | Avg. Read Length (bp) | Avg. Coverage | SNP coverage *** |
| P1FTR2 | >60 | 270,000 | 185 | 3,926,604 | 92.92% | 46,624 | 100 | 1 | 3,741,800 | 204 | 1035 | 346 (99.7%) * |
| A1FTR1 | 20–60 | 540,000 | 193 | 14,015,748 | 96.14% | 184,491 | 100 | 1 | 4,083,881 | 217 | 1068 | 244 (70.3%) * |
| Mixture | Haplogroups | Contributor Proportions (%) | ||
|---|---|---|---|---|
| GeneMarker®HTS | Mixemt | GeneMarker®HTS | Mixemt | |
| C163 (Major) | K2a6 | K2a6 | 79.58 | 82.2 |
| H104 (Minor) | C1b11 | C1b11 | 18.68 | 17.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, S.Y.; Bose, N.; Gonçalves, A.B.R.; Erlich, H.A.; Calloway, C.D. Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples. Genes 2018, 9, 49. https://doi.org/10.3390/genes9010049
Shih SY, Bose N, Gonçalves ABR, Erlich HA, Calloway CD. Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples. Genes. 2018; 9(1):49. https://doi.org/10.3390/genes9010049
Chicago/Turabian StyleShih, Shelly Y., Nikhil Bose, Anna Beatriz R. Gonçalves, Henry A. Erlich, and Cassandra D. Calloway. 2018. "Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples" Genes 9, no. 1: 49. https://doi.org/10.3390/genes9010049
APA StyleShih, S. Y., Bose, N., Gonçalves, A. B. R., Erlich, H. A., & Calloway, C. D. (2018). Applications of Probe Capture Enrichment Next Generation Sequencing for Whole Mitochondrial Genome and 426 Nuclear SNPs for Forensically Challenging Samples. Genes, 9(1), 49. https://doi.org/10.3390/genes9010049