Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression



Abstract
:1. Introduction
2. Gene Expression Studies in Immune Cells
3. Candidate Gene Identification
3.1. Cis-Regulated Genes
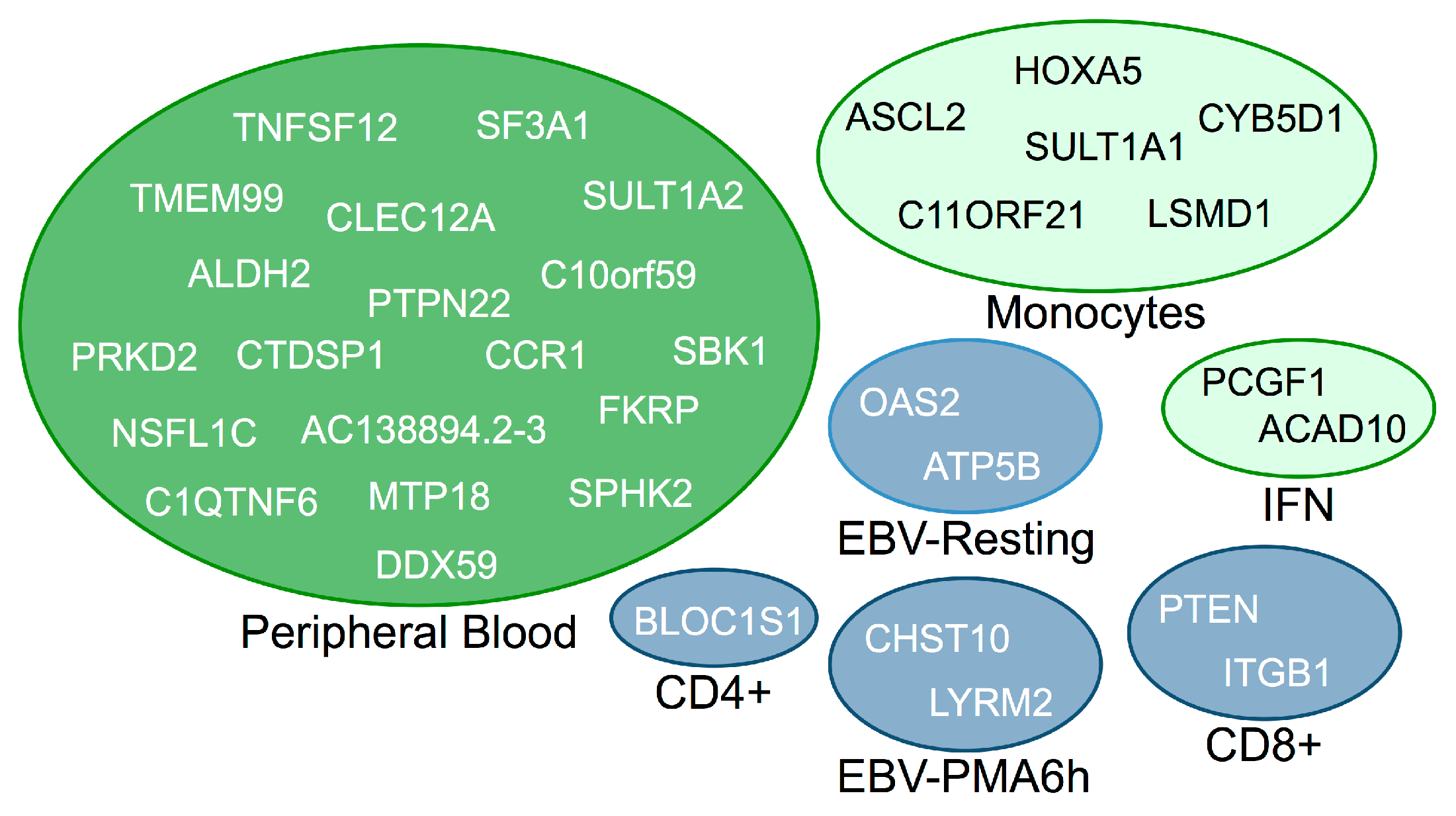
3.2. Trans-Regulated Genes
4. Alternative Methods for Studying eQTLs Associated with Disease SNPs
4.1. Prediction of Functionality of Disease Associated Variants
4.2. Gene Expression Quantification
4.3. Batch Effect Correction and Removing Unwanted Variations
4.4. Identify and Remove Known and Hidden Confounding Factors in the Normalized Expression Data
4.5. Expression Quantitative Trait Locus (eQTL) Analysis
4.6. Statistical Significance and Permutation Analysis
4.7. Colocalization: Overlap between eQTL for a Gene and GWAS SNPs for Disease
4.8. Joint eQTL Analysis for Multiple Cell Types/Tissues
4.9. Imputation of Gene Expression Profiles
4.10. Chromatin Conformation Capture and Linking GWAS SNPs to Target Genes
4.11. Pathway, Network, and Enrichment Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Onengut-Gumuscu, S.; Chen, W.M.; Burren, O.; Cooper, N.J.; Quinlan, A.R.; Mychaleckyj, J.C.; Farber, E.; Bonnie, J.K.; Szpak, M.; Schofield, E.; et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015, 47, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Bradfield, J.P.; Qu, H.Q.; Wang, K.; Zhang, H.; Sleiman, P.M.; Kim, C.E.; Mentch, F.D.; Qiu, H.; Glessner, J.T.; Thomas, K.A.; et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011, 7, e1002293. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Morahan, G.; Mehta, M.; James, I.; Chen, W.M.; Akolkar, B.; Erlich, H.A.; Hilner, J.E.; Julier, C.; Nerup, J.; Nierras, C.; et al. Tests for Genetic Interactions in Type 1 Diabetes: Linkage and Stratification Analyses of 4422 Affected Sib-Pairs. Diabetes 2011, 60, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.D.; Smyth, D.J.; Smiles, A.M.; Plagnol, V.; Walker, N.M.; Allen, J.E.; Downes, K.; Barrett, J.C.; Healy, B.C.; Mychaleckyj, J.C.; et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 2008, 40, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007, 39, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.J.; Plagnol, V.; Walker, N.M.; Cooper, J.D.; Downes, K.; Yang, J.H.; Howson, J.M.; Stevens, H.; McManus, R.; Wijmenga, C.; et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Engl. J. Med. 2008, 359, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Downes, K.; Howson, J.M.; Nutland, S.; Stevens, H.E.; Walker, N.M.; Todd, J.A. Evidence of association with type 1 diabetes in the SLC11A1 gene region. BMC Med. Genet. 2011, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Swafford, A.D.; Howson, J.M.; Davison, L.J.; Wallace, C.; Smyth, D.J.; Schuilenburg, H.; Maisuria-Armer, M.; Mistry, T.; Lenardo, M.J.; Todd, J.A. An allele of IKZF1 (Ikaros) conferring susceptibility to childhood acute lymphoblastic leukemia protects against type 1 diabetes. Diabetes 2011, 60, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Singal, D.P.; Blajchman, M.A. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973, 22, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Miletic, A.V.; Bottini, N.; Mustelin, T. Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity 2007, 40, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, A.; Zeller, M.; Fernandez, A., Jr.; Zalcberg, L.J.; Bartlett, R.J.; Ricordi, C.; Pietropaolo, M.; Eisenbarth, G.S.; Bennett, S.T.; Patel, D.D. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 1997, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Nistico, L.; Buzzetti, R.; Pritchard, L.E.; Van der Auwera, B.; Giovannini, C.; Bosi, E.; Larrad, M.T.; Rios, M.S.; Chow, C.C.; Cockram, C.S.; et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Hum. Mol. Genet. 1996, 5, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.E.; Cooper, J.D.; Brusko, T.; Walker, N.M.; Smyth, D.J.; Bailey, R.; Bourget, K.; Plagnol, V.; Field, S.; Atkinson, M.; et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat. Genet. 2007, 39, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Mehta, M.; Nguyen, Q.T.; Larma, I.; Boehm, B.O.; Pociot, F.; Concannon, P.; Morahan, G. Systematic Evaluation of Genes and Genetic Variants Associated with Type 1 Diabetes Susceptibility. J. Immunol. 2016, 196, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 2001, 29, 1165–1188. [Google Scholar]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Makino, S.; Radhakrishnan, J.; Plant, K.; Leslie, S.; Dilthey, A.; Ellis, P.; Langford, C.; Vannberg, F.O.; Knight, J.C. Genetics of gene expression in primary immune cells identifies cell type–specific master regulators and roles of HLA alleles. Nat. Genet. 2012, 44, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Ye, C.; Villani, A.C.; Raj, T.; Li, W.; Eisenhaure, T.M.; Imboywa, S.H.; Chipendo, P.I.; Ran, F.A.; Slowikowski, K.; et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 2014, 343, 1246980. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.E.; Lyons, P.A.; Lee, J.C.; Richard, A.C.; Fortune, M.D.; Newcombe, P.J.; Richardson, S.; Smith, K.G. Insight into genotype-phenotype associations through eQTL mapping in multiple cell types in health and immune-mediated disease. PLoS Genet. 2016, 12, e1005908. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Arends, D.; Esko, T.; Peters, M.J.; Schurmann, C.; Schramm, K.; Kettunen, J.; Yaghootkar, H.; Fairfax, B.P.; Tserel, L.; et al. Cell specific eQTL analysis without sorting cells. PLoS Genet. 2014, 11, e1005223. [Google Scholar]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorow, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Corradin, O.; Saiakhova, A.; Akhtar-Zaidi, B.; Myeroff, L.; Willis, J.; Cowper-Sal, L.R.; Lupien, M.; Markowitz, S.; Scacheri, P.C. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beki, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Whitaker, J.W.; Huang, C.C.; Cherkas, Y.; Lamberth, S.L.; Brodmerkel, C.; Curran, M.E.; Dobrin, R. Integrative genomic deconvolution of rheumatoid arthritis GWAS loci into gene and cell type associations. Genome Biol. 2016, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Hakonarson, H.; Qu, H.Q.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes 2008, 57, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Ogden, L.; Wadwa, R.P.; Hamman, R.F.; Mayer-Davis, E.J.; Liese, A.D.; D’Agostino, R., Jr.; Marcovina, S.; Dabelea, D. Lipid and lipoprotein profiles in youth with and without type 1 diabetes: The SEARCH for Diabetes in Youth case-control study. Diabetes Care 2009, 32, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Pastor, C.; Rotellar, F.; Silva, C.; Rodríguez, A.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Vendrell, J.; et al. Influence of morbid obesity and insulin resistance on gene expression levels of AQP7 in visceral adipose tissue and AQP9 in liver. Obes. Surg. 2008, 18, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S. The UPS in diabetes and obesity. BMC Biochem. 2008, 9, S6. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Morahan, G. Using Systems Genetics to Understanding the Etiology of Complex Disease. Methods Mol. Biol. 2017, 1488, 597–606. [Google Scholar] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- FANTOM Consortium; the RIKEN PMI; CLST (DGT). A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep-learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Gulko, B.; Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017, 49, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Musil, M.; Štourač, J.; Zendulka, J.; Damborský, J.; Brezovský, J. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput. Biol. 2016, 12, e1004962. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Arloth, J.; Bader, D.M.; Röh, S.; Altmann, A. Re-Annotator: Annotation pipeline for microarray probe sequences. PLoS ONE 2015, 10, e0139516. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Scherer, A. Batch Effects and Noise in Microarray Experiments: Sources and Solutions. In Wiley Series Probability Statistics; Wiley-Blackwell: Oxford, UK, 2009; p. 272. [Google Scholar]
- Wu, Z.; Aryee, M.J. Subset quantile normalization using negative control features. J. Comput. Biol. 2010, 17, 1385–1395. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Storey, J.D. sva: Surrogate Variable Analysis. In R Package; Version 3.25.0; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Westra, H.J.; Jansen, R.C.; Fehrmann, R.S.; te Meerman, G.J.; van Heel, D.; Wijmenga, C.; Franke, L. MixupMapper: Correcting sample mix-ups in genome-wide datasets increases power to detect small genetic effects. Bioinformatics 2011, 27, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.I.; Bates, D.M.; Rosa, G.J.M.; Gianola, D.; Weigel, K.A. Technical note: An R package for fitting generalized linear mixed models in animal breeding. J. Anim. Sci. 2010, 88, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Stegle, O.; Parts, L.; Piipari, M.; Winn, J.; Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012, 7, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.E.; Gupta, S.; Ashar, F.N.; Bader, J.S.; West, A.B. Arking DE RNA-Seq optimization with eQTL gold standards. BMC Genom. 2013, 14, 892. [Google Scholar] [CrossRef] [PubMed]
- Tracy, C.A.; Widom, H. Level-Spacing Distributions and the Airy Kernel. Commun. Math. Phys. 1994, 159, 151–174. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Dabney, A.; Storey, J.D.; Warnes, G.R. qvalue: Q-value estimation for false discovery rate control. In R Package; Version 1.20.0; R Foundation for Statistical Computing: Vienna, Austria, 2009. [Google Scholar]
- He, X.; Fuller, C.K.; Song, Y.; Meng, Q.; Zhang, B.; Yang, X.; Li, H. Sherlock: Detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am. J. Hum. Genet. 2013, 92, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Berisa, T.; Liu, J.Z.; Ségurel, L.; Tung, J.Y.; Hinds, D.A. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 2016, 48, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Petretto, E.; Bottolo, L.; Langley, S.R.; Heinig, M.; McDermott-Roe, C. New insights into the genetic control of gene expression using a Bayesian multi-tissue approach. PLoS Comput. Biol. 2010, 6, e1000737. [Google Scholar] [CrossRef] [PubMed]
- Flutre, T.; Wen, X.; Pritchard, J.; Stephens, M. A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet. 2013, 9, e1003486. [Google Scholar] [CrossRef] [PubMed]
- The GTEx Consortium; Ardlie, K.G.; Deluca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648660. [Google Scholar]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; GTEx Consortium; Nicolae, D.L.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; AC’t Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, G.P.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Battle, A.; Mostafavi, S.; Zhu, X.; Potash, J.B.; Weissman, M.M.; McCormick, C.; Haudenschild, C.D.; Beckman, K.B.; Shi, J.; Mei, R.; et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014, 24, 14–24. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Schofield, E.C.; Carver, T.; Achuthan, P.; Freire-Pritchett, P.; Spivakov, M.; Todd, J.A.; Burren, O.S. CHiCP: A web-based tool for the integrative and interactive visualization of promoter capture Hi-C datasets. Bioinformatics 2016, 32, 2511–2513. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Iotchkova, V.; Ritchie, G.R.S.; Geihs, M.; Morganella, S.; Min, J.L.; Walter, K.; Timpson, N.J.; Dunham, I.; Birney, E.; Soranzo, N.; et al. GARFIELD—GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction. Biorxiv 2016. [Google Scholar] [CrossRef]
- Schmidt, E.M.; Zhang, J.; Zhou, W.; Chen, J.; Mohlke, K.L.; Chen, Y.E. Gregor: Evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 2015, 31, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
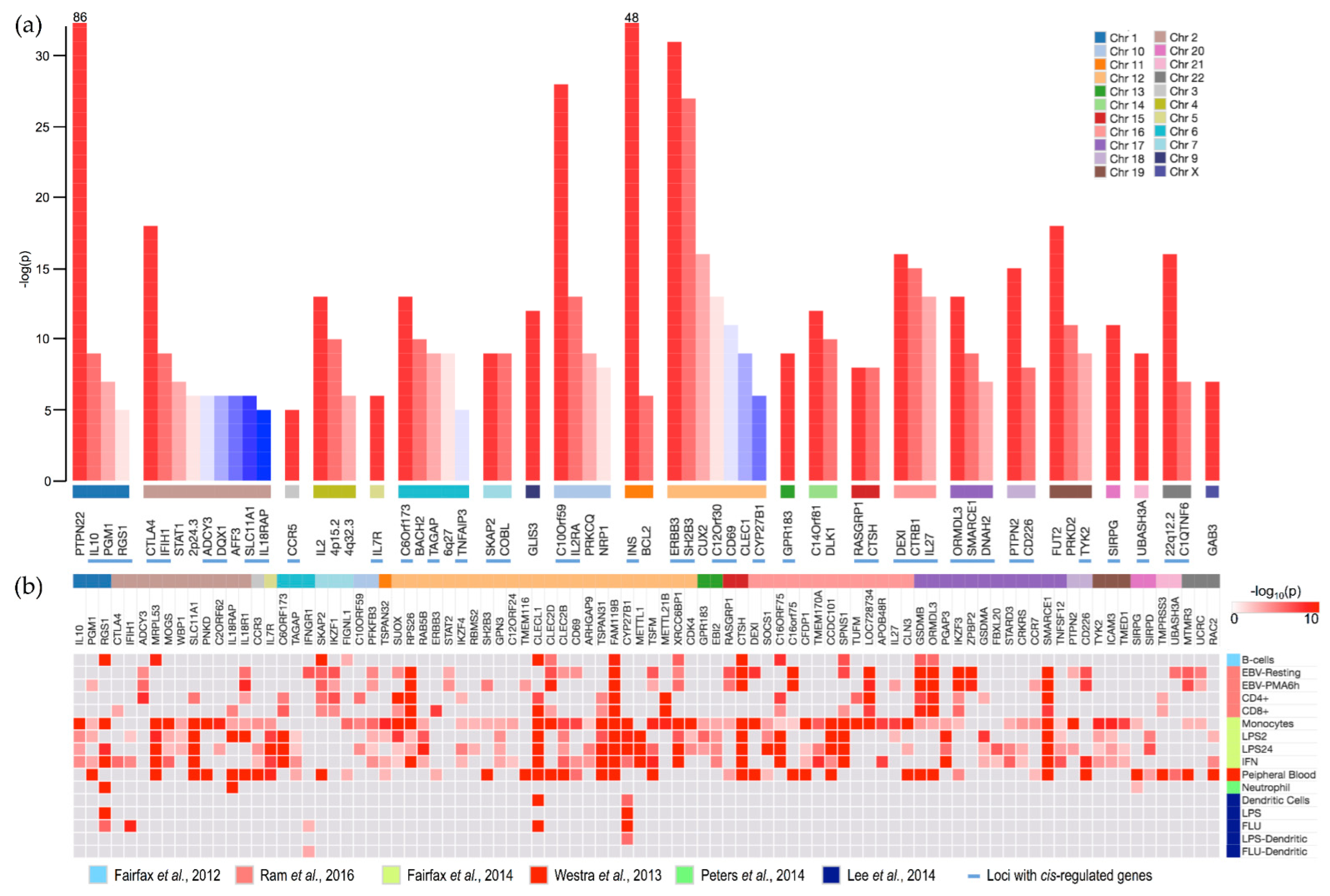

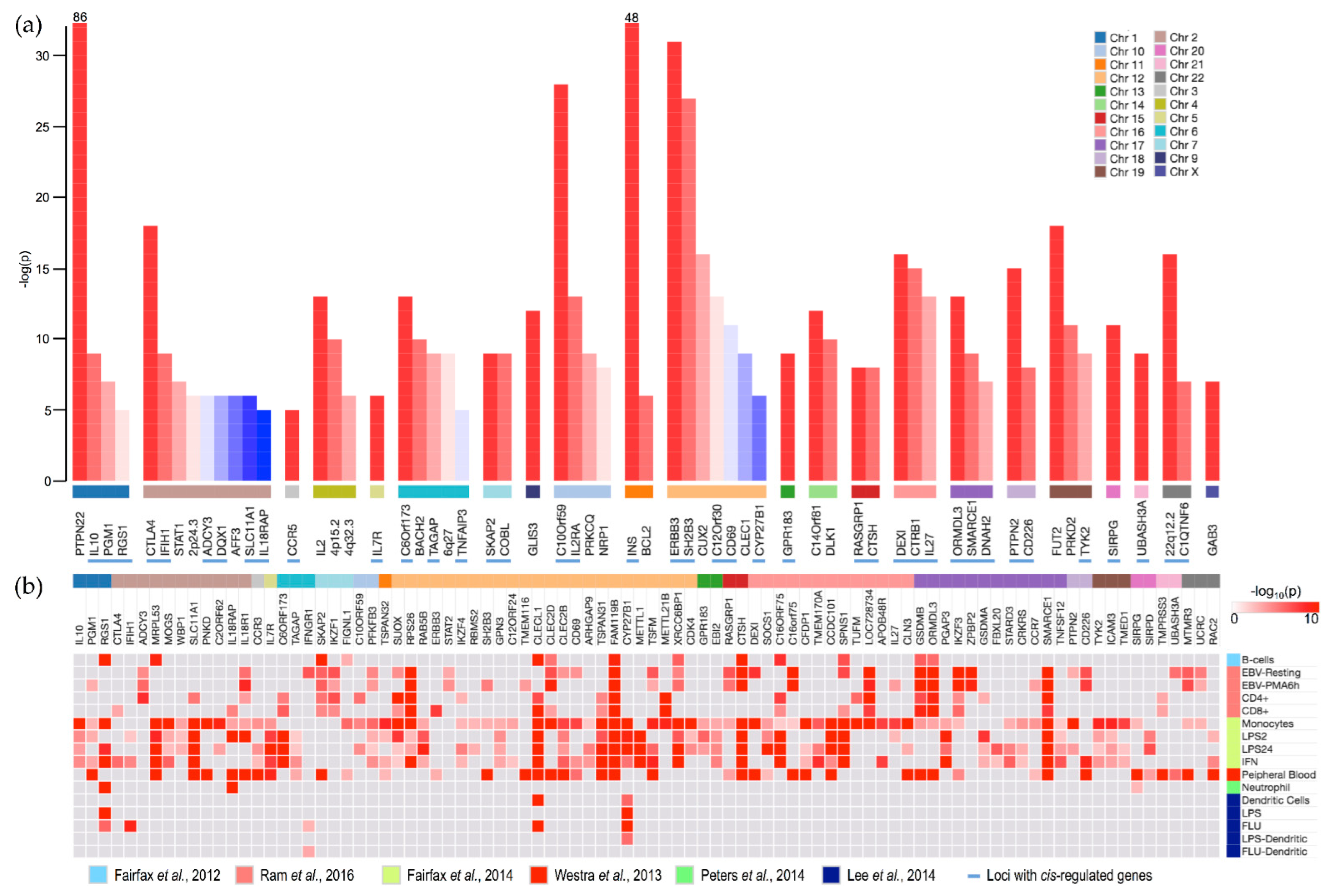{kind=link}
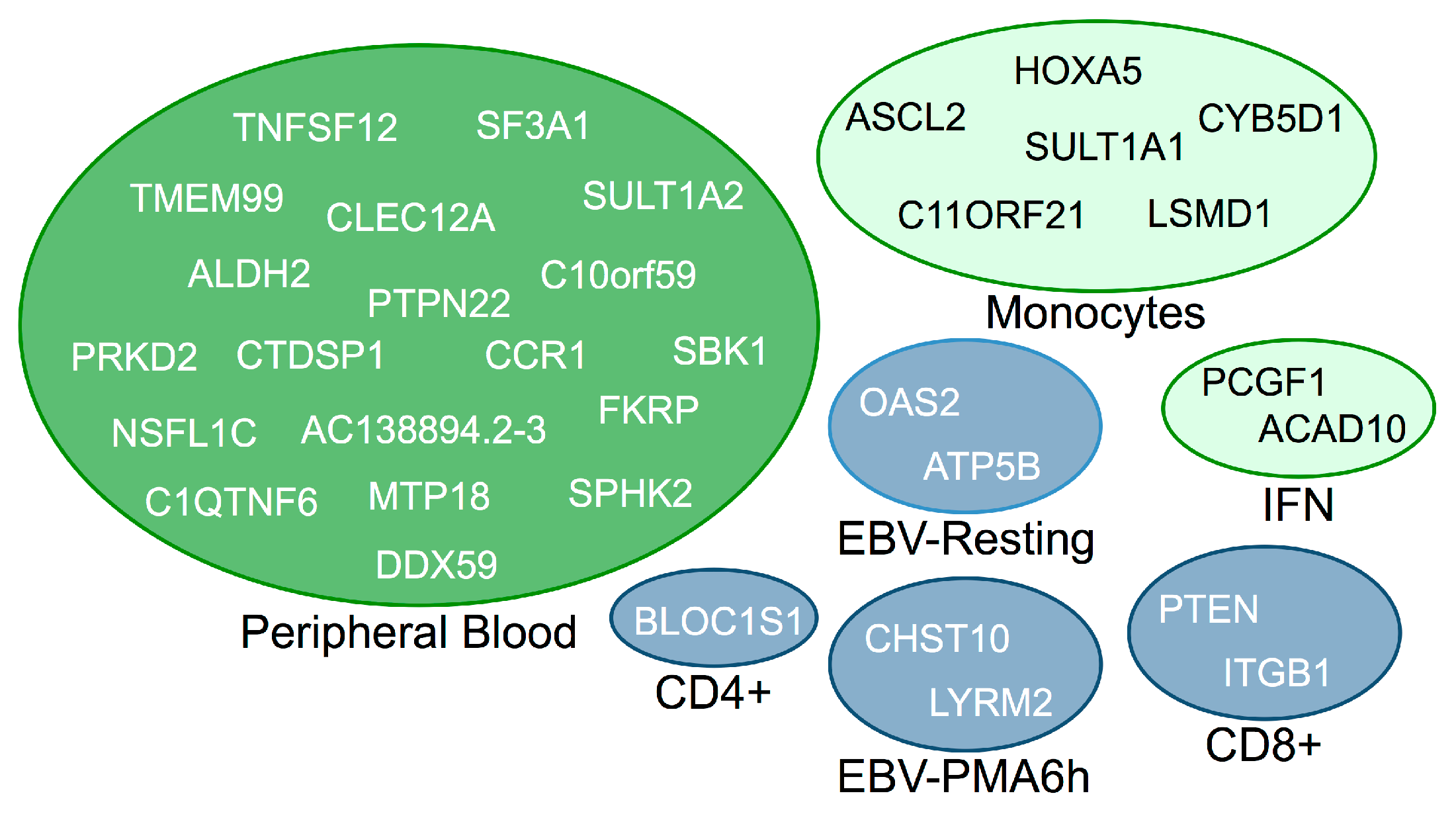{kind=link}
| T1D SNP | Locus | Chr | Gene | B | EBV | PMA | CD4 | CD8 | Mono | LPS2 | LPS24 | IFN | Blood |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs7111341 | INS | 11p15.5 | AQP9 | 6.0 | 5.6 | ||||||||
| IP6K2 | 14.2 | 18.2 | 11.9 | 17.8 | 20.7 | 16.9 | 7.7 | 13.3 | 10.0 | ||||
| LAP3P2 | 51.7 | 14.6 | 14.4 | 13.9 | |||||||||
| rs11171739 | ERBB3 | 12q13.2 | MAFG-AS1 | 6.9 | 5.6 | 17.0 | 19.9 | ||||||
| MIR130A | 36.2 | 27.8 | 25.7 | 21.8 | |||||||||
| MIR1471 | 6.9 | 5.6 | 8.5 | 13.0 | |||||||||
| DPF2 | 6.8 | 12.0 | |||||||||||
| rs3184504 | SH2B3 | 12q24.12 | UBE2L6 | 7.9 | 4.7 | 7.9 | |||||||
| STAT1 | 5.5 | 7.5 | |||||||||||
| rs17696736 | C12Orf30 | UBE2L6 | 5.7 | 5.3 |
| EBV Cells | Peripheral Blood | ||
|---|---|---|---|
| SNP | Trans-Gene | SNP | Trans-Gene |
| rs1990760 | LOC643997 | rs1701704 | CCL5 |
| rs10499194 | TUBB6 | rs1701704 | CRLF3 |
| rs7804356 | SLC39A8 | rs2058660 | CYP2C19 |
| rs12251307 | DERA | rs3184504 | FOS |
| rs947474 | MEIS2 | rs3184504 | GBP4 |
| rs3842727 | ID2 | rs11171739 | MIF |
| rs1738074 | IRF8 | rs3184504 | NALP12 |
| rs1265565 | NCOA7 | CD4+ | |
| rs12908309 | FAHD1 | rs1265565 | ZMYM5 |
| rs2290400 | TEX9 | rs11711054 | GRAMD1B |
| rs763361 | P2RY11 | Monocytes | |
| rs7221109 | EIF5A | rs11171739 | KCTD11 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ram, R.; Morahan, G. Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression. Genes 2017, 8, 167. https://doi.org/10.3390/genes8060167
Ram R, Morahan G. Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression. Genes. 2017; 8(6):167. https://doi.org/10.3390/genes8060167
Chicago/Turabian StyleRam, Ramesh, and Grant Morahan. 2017. "Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression" Genes 8, no. 6: 167. https://doi.org/10.3390/genes8060167
APA StyleRam, R., & Morahan, G. (2017). Effects of Type 1 Diabetes Risk Alleles on Immune Cell Gene Expression. Genes, 8(6), 167. https://doi.org/10.3390/genes8060167