
Genome-Wide Identification and Characterization of Salinity Stress-Responsive miRNAs in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides)


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Salt Treatment
2.2. Small RNA Library Construction and Sequencing
2.3. miRNA Identification
2.4. Detection of Differentially Expressed miRNAs
2.5. Target Gene Prediction
2.6. Validation of the Expression of miRNAs by qRT-PCR Analysis
3. Results
3.1. Small RNA Sequencing
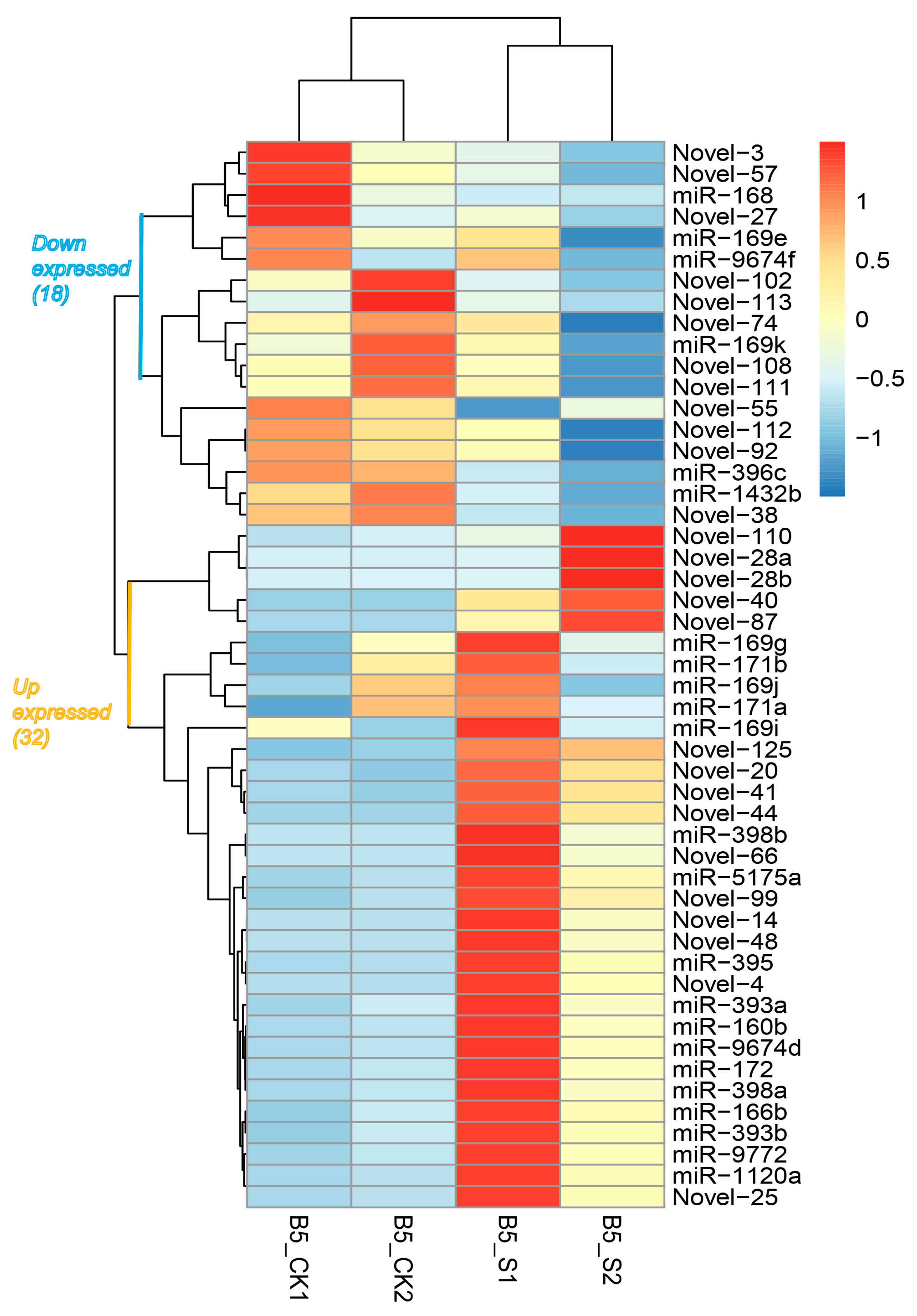
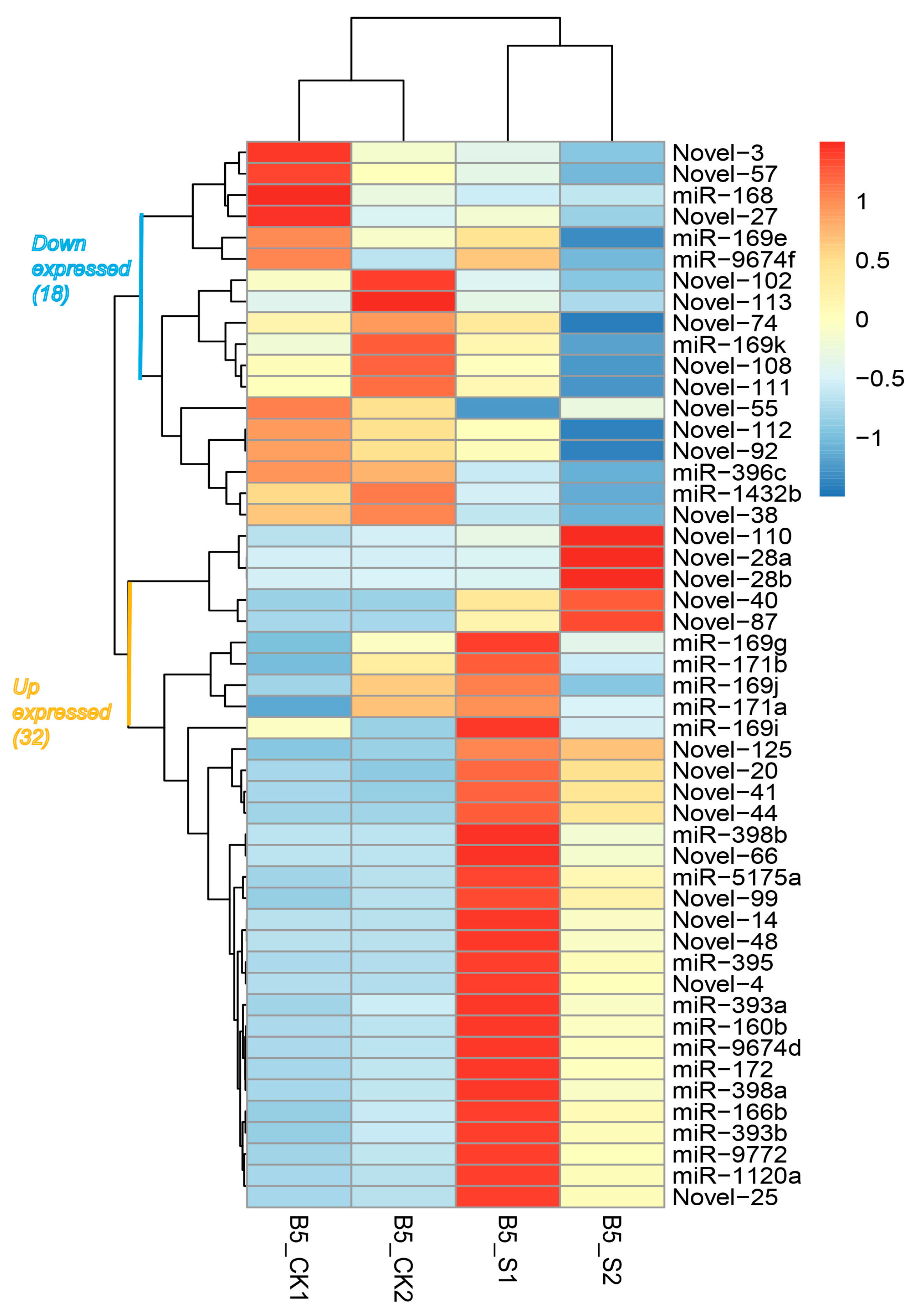
3.2. Identification and Characterization of Salinity-Responsive miRNAs
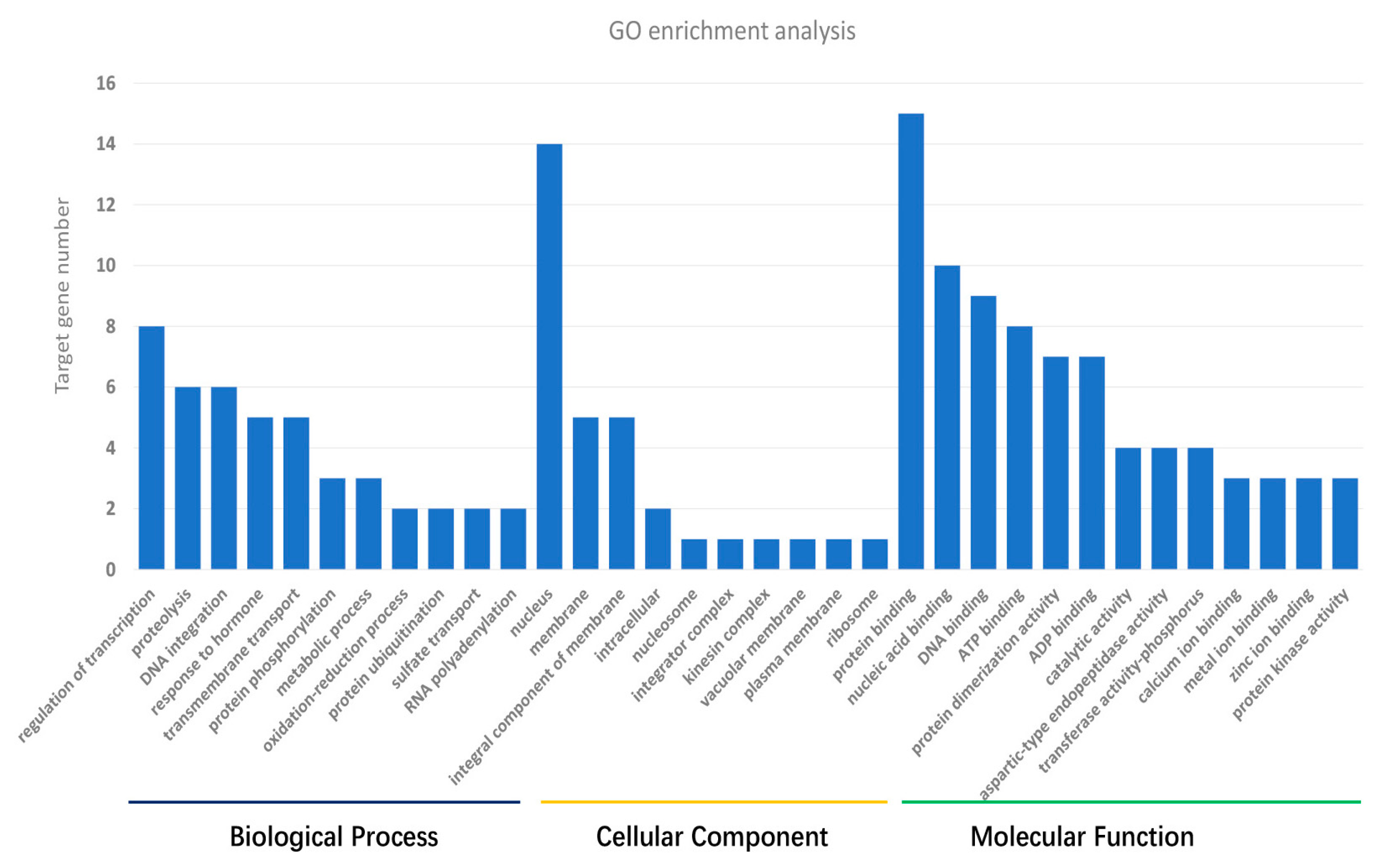
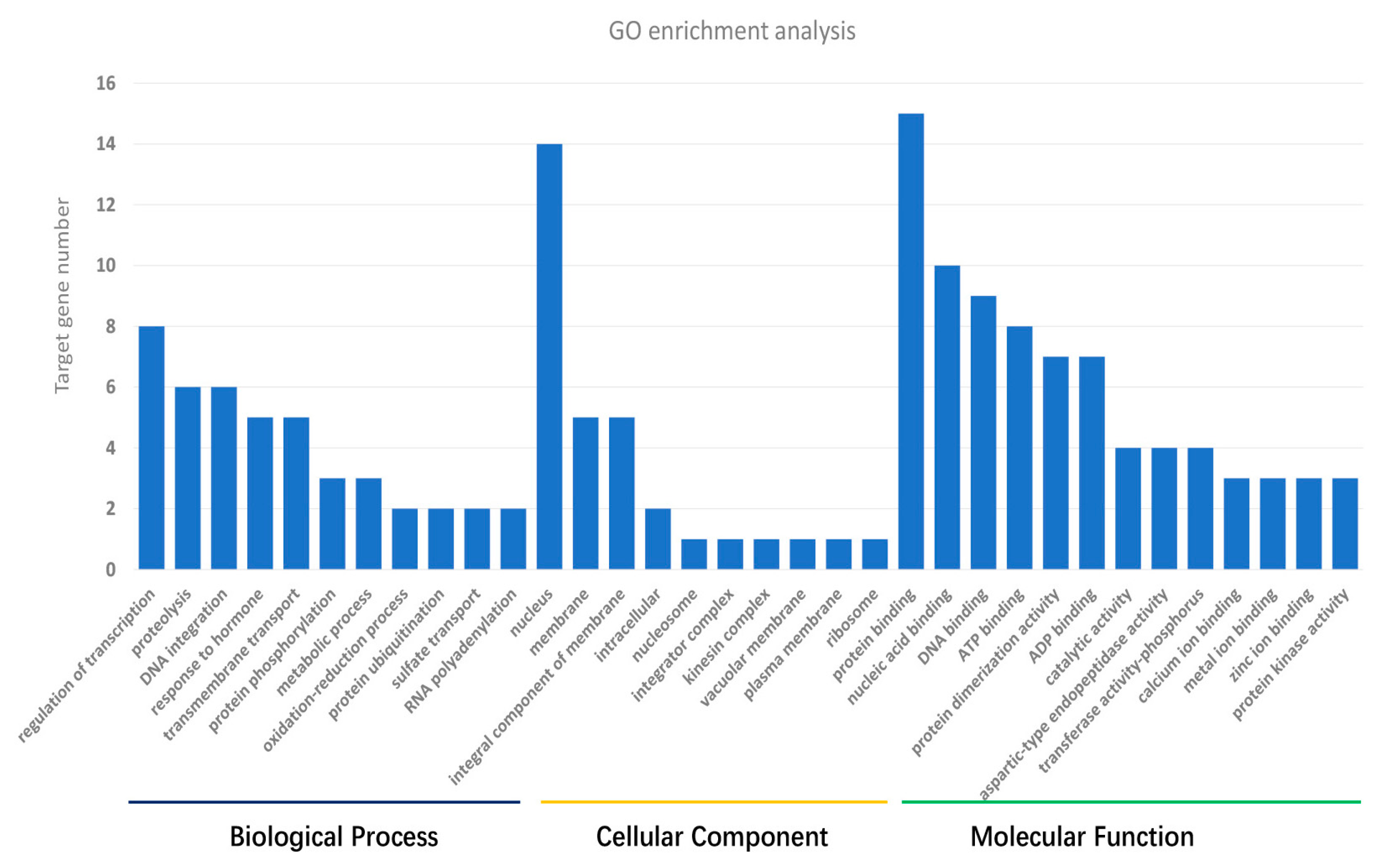
3.3. Target Gene Prediction and Annotation
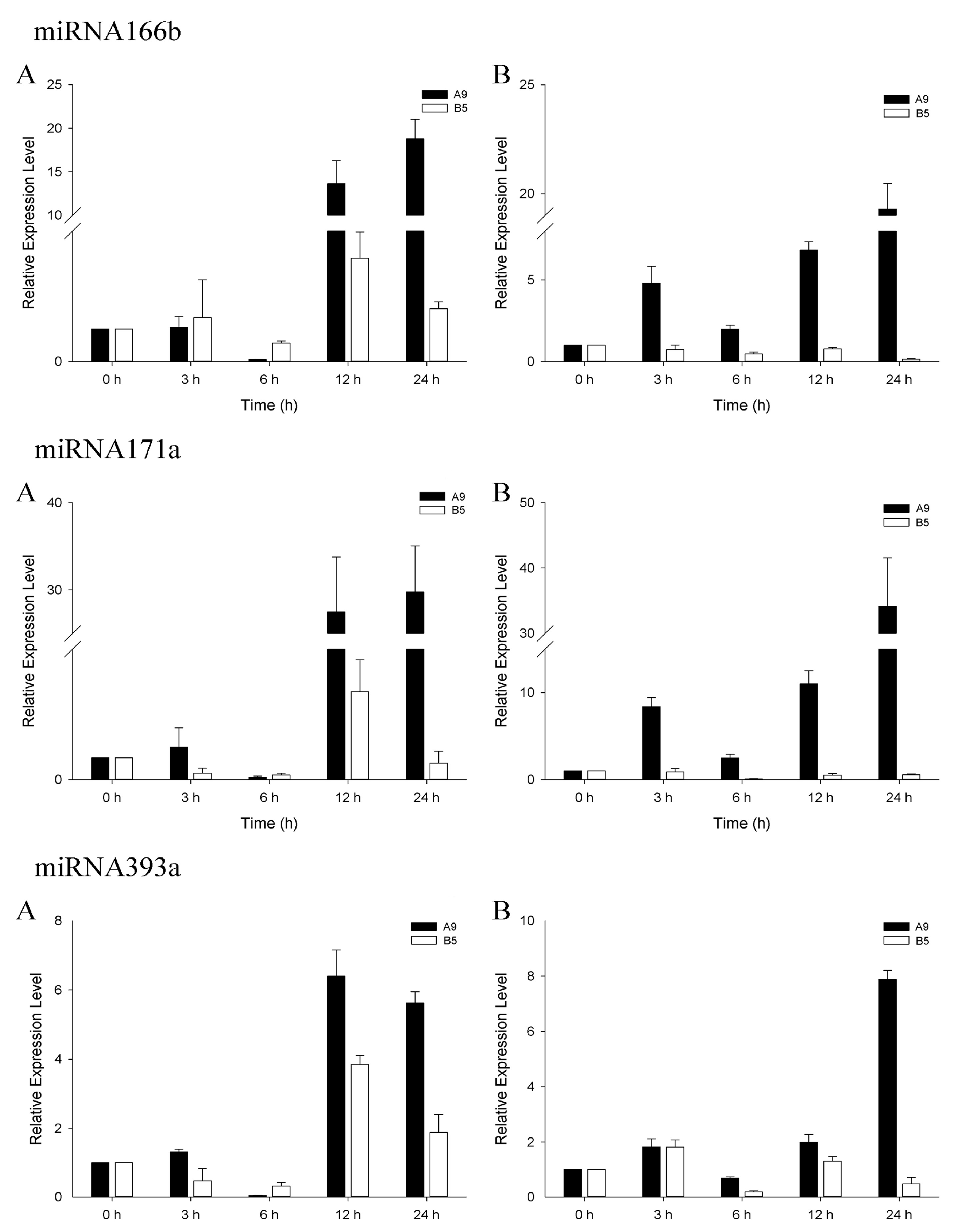
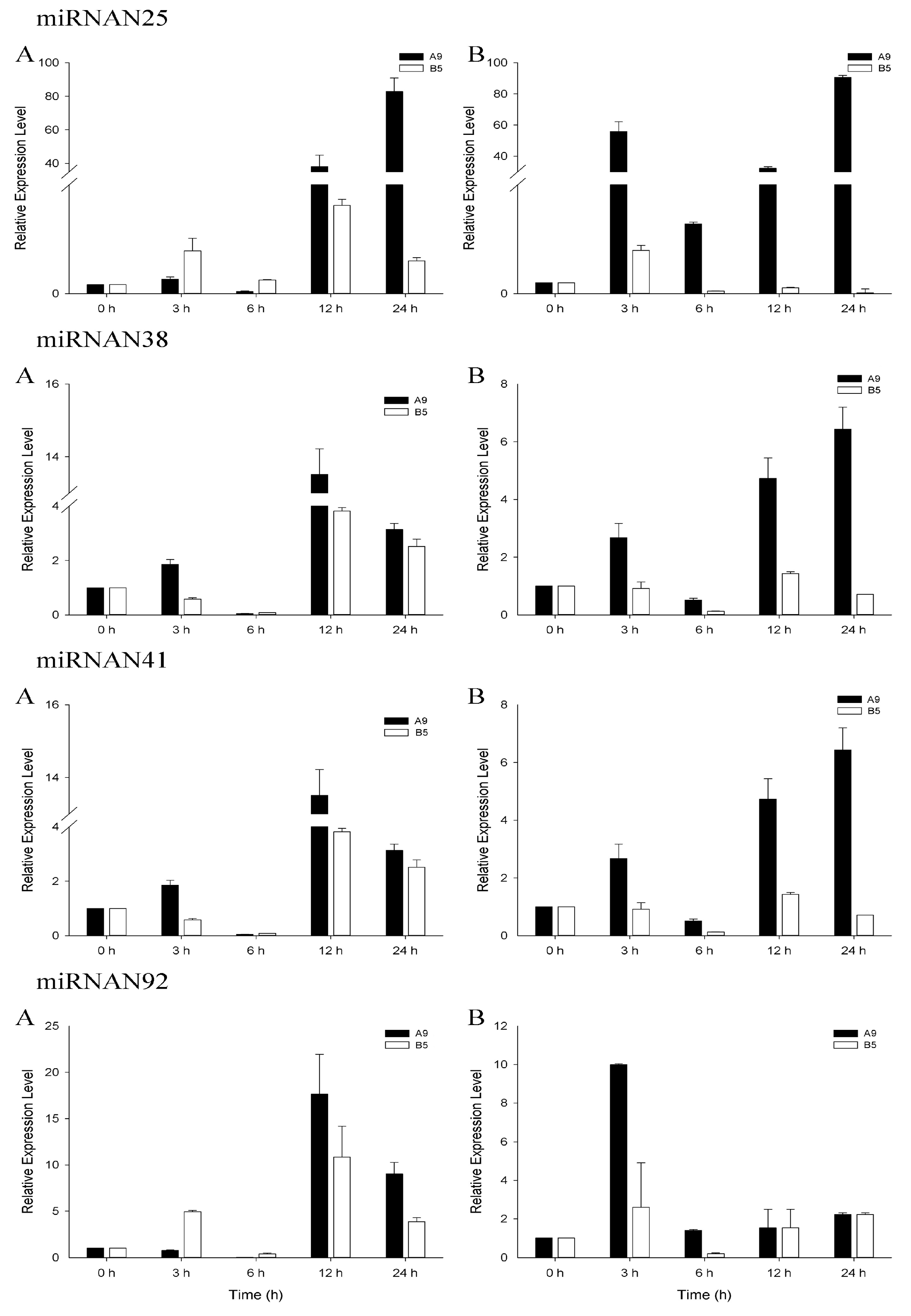
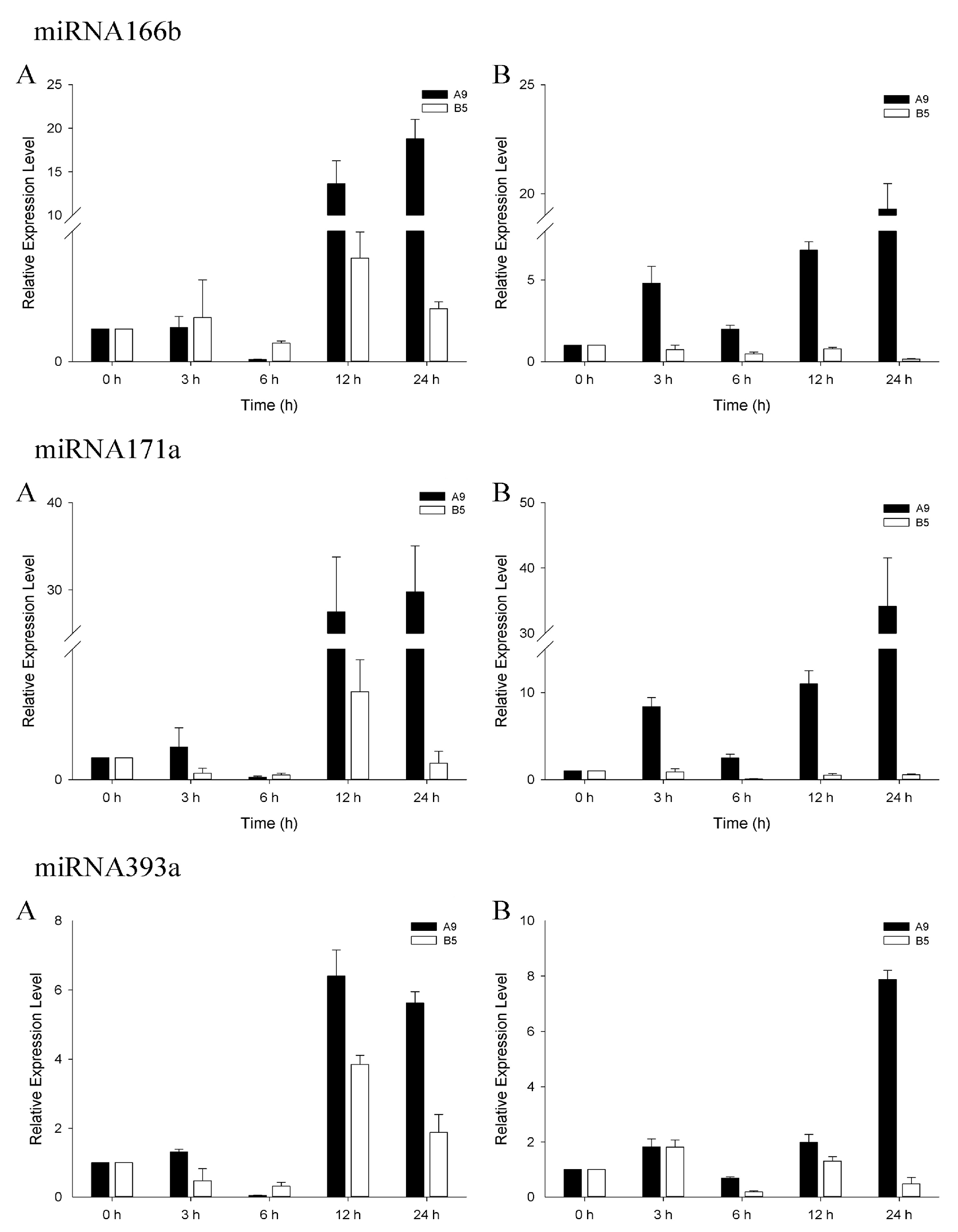
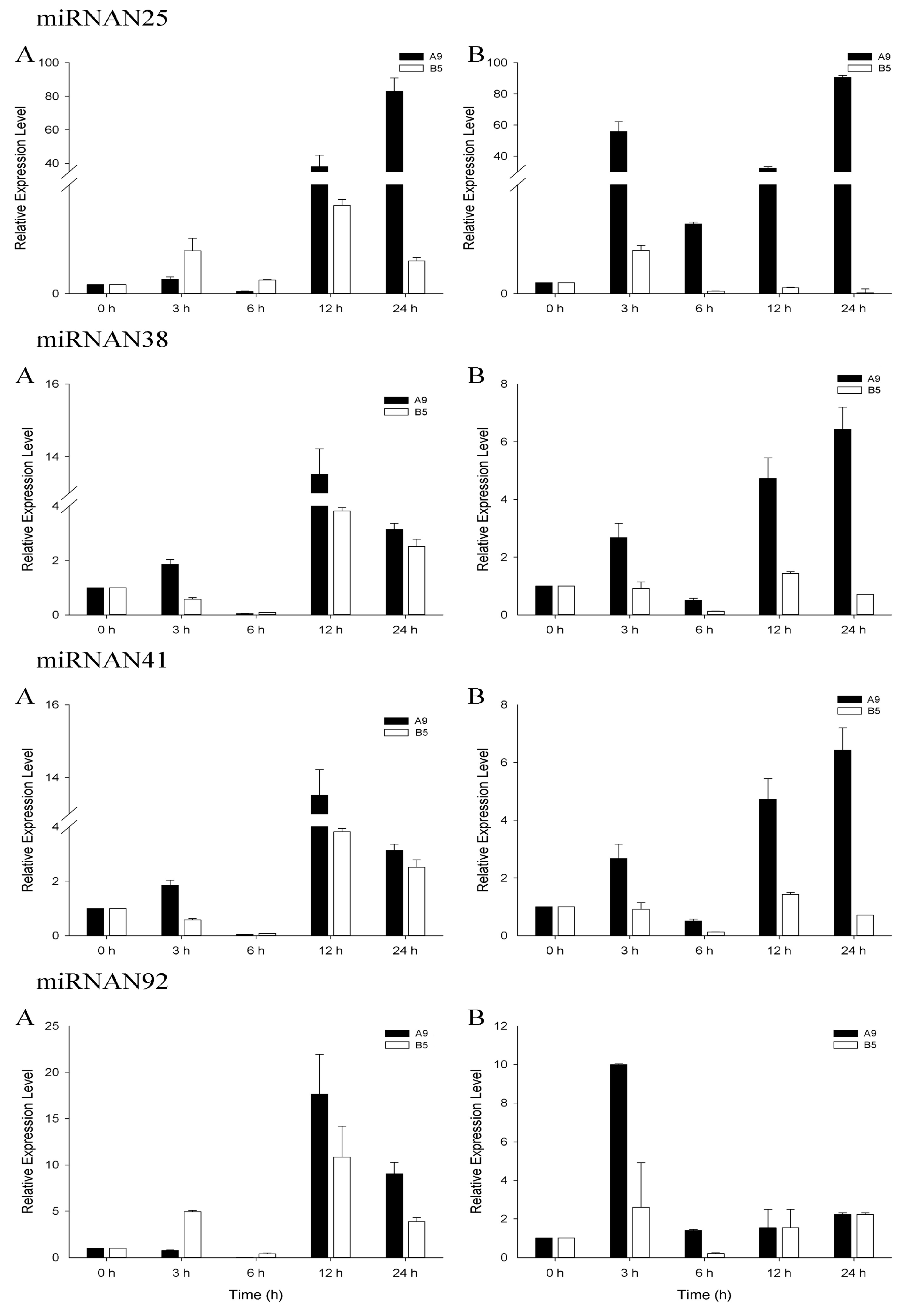
3.4. qRT-PCR Analysis of Salinity-Related miRNAs
4. Discussion
4.1. Genome Wide Identification of miRNAs in Wild Emmer Using High-Throughput Sequencing
4.2. Salinity Stress-Responsive miRNAs and Their Targets in Wild Emmer
4.3. miRNAs Play Critical Roles in Salinity Tolerance of Wild Emmer
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peleg, Z.; Fahima, T.; Korol, A.B.; Abbo, S.; Saranga, Y. Genetic analysis of wheat domestication and evolution under domestication. J. Exp. Bot. 2011, 62, 5051–5061. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, B.; Langridge, P.; Budak, H. Abiotic stress mirnomes in the triticeae. Funct. Integr. Genom. 2017, 17, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Nevo, E.; Chen, G.X. Drought and salt tolerances in wild relatives for wheat and barley improvement. Plant Cell Environ. 2010, 33, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Shavrukov, Y.; Langridge, P.; Tester, M.; Nevo, E. Wide genetic diversity of salinity tolerance, sodium exclusion and growth in wild emmer wheat, Triticum dicoccoides. Breed. Sci. 2010, 60, 426–435. [Google Scholar] [CrossRef]
- Nevo, E.; Krugman, T.; Beiles, A. Genetic-resources for salt tolerance in the wild progenitors of wheat (Triticum dicoccoides) and barley (Hordeum spontaneum) in Israel. Plant Breed. 1993, 110, 338–341. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Shukla, P.S.; Gupta, K.; Jha, B. Bioengineering for salinity tolerance in plants: State of the art. Mol. Biotechnol. 2013, 54, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.C.; Wang, L.; Cui, L.C.; Feng, K.W.; Liu, F.Y.; Du, X.H.; Tong, W.; Nie, X.J.; Ji, W.Q.; Weining, S. Global identification of microRNAs and their targets in barley under salinity stress. PLoS ONE 2015, 10, e0137990. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, S.; Horie, T.; Hauser, F.; Deinlein, U.; Schroeder, J.I.; Uozumi, N. HKT transporters mediate salt stress resistance in plants: From structure and function to the field. Curr. Opin. Biotechnol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Akpinar, B.A. Plant miRNAs: Biogenesis, organization and origins. Funct. Integr. Genom. 2015, 15, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Pathak, H.; Sharma, S.K.; Kala, Y.K.; Nirjal, M.K.; Singh, G.P.; Goswami, S.; Rai, R.D. Novel and conserved heat-responsive microRNAs in wheat (Triticum aestivum L.). Funct. Integr. Genom. 2015, 15, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.; Wu, J.; Zhou, Y.; Xiao, J.; Feng, J.; Zhao, W.; Xiang, S.; Jiang, G.; Chen, J.Y. Computational analysis of drought stress-associated miRNAs and miRNA co-regulation network in Physcomitrella patens. Genom. Proteom. Bioinform. 2011, 9, 37–44. [Google Scholar] [CrossRef]
- Mendoza-Soto, A.B.; Sanchez, F.; Hernandez, G. MicroRNAs as regulators in plant metal toxicity response. Front. Plant Sci. 2012, 3, 105. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, R.; Rodriguez, R.E.; Debernardi, J.M.; Palatnik, J.F.; Casati, P. Repression of growth regulating factors by the microRNA396 inhibits cell proliferation by UV-b radiation in Arabidopsis leaves. Plant Cell 2013, 25, 3570–3583. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.L.; Stewart, C.N.; Taki, F.A.; He, Q.L.; Liu, H.W.; Zhang, B.H. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol. J. 2014, 12, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; He, S.P.; Gong, W.F.; Sun, J.L.; Pan, Z.E.; Xu, F.F.; Lu, Y.L.; Du, X.M. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genom. 2014, 15, 700. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Bai, X.; Yang, L.A.; Lv, D.K.; Pan, X.; Li, Y.; Cai, H.; Ji, W.; Chen, Q.; Zhu, Y.M. Osa-mir393: A salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 2011, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, L.F.; Wang, H.; Liu, Z.J.; Zhang, Z.X.; Zheng, Y.L. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Q.L.; Zhang, B.H. Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 2013, 530, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Meena, N.L.; Sharma, I.; Sharma, P. Differential regulation of microRNAs in response to osmotic, salt and cold stresses in wheat. Mol. Biol. Rep. 2014, 41, 4623–4629. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sun, Y.F.; Song, N.; Wei, J.P.; Wang, X.J.; Feng, H.; Yin, Z.Y.; Kang, Z.S. MicroRNAs involving in cold, wounding and salt stresses in Triticum aestivum L. Plant Physiol. Biochem. 2014, 80, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kantar, M.; Lucas, S.J.; Budak, H. MiRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 2011, 233, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Kantar, M.; Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genom. 2015, 15, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. Mirbase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Dsouza, M.; Larsen, N.; Overbeek, R. Searching for patterns in genomic data. Trends Genet. 1997, 13, 497–498. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Wu, P.; Han, S.H.; Chen, T.; Qin, G.X.; Li, L.; Guo, X.J. Involvement of microRNAs in infection of silkworm with Bombyx mori cytoplasmic polyhedrosis virus (BMCPV). PLoS ONE 2013, 8, e68209. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Wu, R.M.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta D(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, F.; Zhu, J.; Zhang, S.; Yang, Y.; Chen, T.; Tang, B.; Dong, L.; Ding, N.; Zhang, Q.; et al. GSA: Genome sequence archive. Genom. Proteom. Bioinform. 2017, 15, 14–18. [Google Scholar] [CrossRef] [PubMed]
- BIG Data Center Members. The big data center: From deposition to integration to translation. Nucleic Acids Res. 2017, 45, D18–D24. [Google Scholar]
- Wu, Y.; Guo, J.; Cai, Y.M.; Gong, X.L.; Xiong, X.M.; Qi, W.W.; Pang, Q.Y.; Wang, X.M.; Wang, Y. Genome-wide identification and characterization of Eutrema salsugineum microRNAs for salt tolerance. Physiol. Plant. 2016, 157, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Fahad, S.; Aqeel, M.; Ali, U.; Amanullah; Anwar, S.; Baloch, S.K.; Zainab, M. MiRNAs: Major modulators for crop growth and development under abiotic stresses. Biotechnol. Lett. 2017, 39, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Eren, H.; Pekmezci, M.Y.; Okay, S.; Turktas, M.; Inal, B.; Ilhan, E.; Atak, M.; Erayman, M.; Unver, T. Hexaploid wheat (Triticum aestivum) root mirnome analysis in response to salt stress. Ann. Appl. Biol. 2015, 167, 208–216. [Google Scholar] [CrossRef]
- Carnavale Bottino, M.; Rosario, S.; Grativol, C.; Thiebaut, F.; Rojas, C.A.; Farrineli, L.; Hemerly, A.S.; Ferreira, P.C. High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 2013, 8, e59423. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.R.; Zeng, Y.L.; Yi, X.Y.; Zhao, L.J.; Zhang, Y.F. Small RNA deep sequencing reveals the important role of microRNAs in the halophyte Halostachys caspica. Plant Biotechnol. J. 2015, 13, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Zandkarimi, H.; Bedre, R.; Solis, J.; Mangu, V.; Baisakh, N. Sequencing and expression analysis of salt-responsive miRNAs and target genes in the halophyte smooth cordgrass (Spartina alternifolia loisel). Mol. Biol. Rep. 2015, 42, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, B.; Budak, H. Wheat miRNA ancestors: Evident by transcriptome analysis of A, B, and D genome donors. Funct. Integr. Genom. 2017, 17, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. MiRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Shriram, V.; Kumar, V.; Devarumath, R.M.; Khare, T.S.; Wani, S.H. MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar] [CrossRef] [PubMed]
- Ozhuner, E.; Eldem, V.; Ipek, A.; Okay, S.; Sakcali, S.; Zhang, B.H.; Boke, H.; Unver, T. Boron stress responsive microRNAs and their targets in barley. PLoS ONE 2013, 8, e59543. [Google Scholar] [CrossRef] [PubMed]
- Inal, B.; Turktas, M.; Eren, H.; Ilhan, E.; Okay, S.; Atak, M.; Erayman, M.; Unver, T. Genome-wide fungal stress responsive miRNA expression in wheat. Planta 2014, 240, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Kantar, M.; Bulut, R.; Akpinar, B.A. Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 2015, 235, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.F.; Wang, R.; Ou, X.J.; Fang, Z.M.; Tian, C.G.; Duan, J.; Wang, Y.Q.; Zhang, M.Y. OsTIR1 and osAFB2 downregulation via osmir393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS ONE 2012, 7, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, M.J.; Terrile, M.C.; Windels, D.; Lombardo, M.C.; Bartoli, C.G.; Vazquez, F.; Estelle, M.; Casalongue, C.A. Mir393 regulation of auxin signaling and redox-related components during acclimation to salinity in Arabidopsis. PLoS ONE 2014, 9, e107678. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, B.A.; Budak, H. Dissecting miRNAs in wheat d genome progenitor, Aegilops tauschii. Front. Plant Sci. 2016, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Budak, H.; Khan, Z.; Kantar, M. History and current status of wheat miRNAs using next-generation sequencing and their roles in development and stress. Brief. Funct. Genom. 2015, 14, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.Z.; Nie, X.J.; Wang, L.; Du, X.H.; Biradar, S.S.; Jia, X.O.; Weining, S. Identification and characterization of microRNAs from barley (Hordeum vulgare L.) by high-throughput sequencing. Int. J. Mol. Sci. 2012, 13, 2973–2984. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, E.; Verelst, W.; Horner, D.S.; Gianfranceschi, L.; Piccolo, V.; Inze, D.; Pe, M.E.; Mica, E. Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol. Plant 2013, 6, 423–443. [Google Scholar] [CrossRef] [PubMed]
- Ganie, S.A.; Dey, N.; Mondal, T.K. Promoter methylation regulates the abundance of osa-miR393a in contrasting rice genotypes under salinity stress. Funct. Integr. Genom. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S1 | S2 | S_Total | Ck1 | Ck2 | Ck_Total | |||
|---|---|---|---|---|---|---|---|---|
| Reads | Reads | Reads | % | Reads | Reads | Reads | % | |
| Total reads | 16,267,700 | 18,193,591 | 34,461,291 | 100 | 17,162,819 | 15,876,321 | 33,039,140 | 100 |
| High quality (size ≥ 18 nt) | 11,906,016 | 16,651,061 | 28,557,077 | 82.87 | 14,783,969 | 13,438,116 | 28,222,085 | 85.42 |
| Redundancy (%) | 80.65 | 83.73 | ||||||
| Total of perfectly matched | 7,594,637 | 9,486,577 | 17,081,214 | 59.81 | 5,470,601 | 5,799,131 | 11,269,732 | 39.93 |
| Unique reads | 2,487,193 | 3,093,686 | 5,580,879 | 19.54 | 2,117,167 | 2,448,167 | 4,565,334 | 16.18 |
| Unique reads matched | 1,035,449 | 1,066,097 | 2,101,546 | 37.66 | 369,710 | 558,073 | 927,783 | 20.32 |
| Class | Unique Reads | % | Total Reads | % |
|---|---|---|---|---|
| Total reads | 2,243,768 | 100 | 28,349,876 | 100 |
| rRNAa | 135,117 | 6.02 | 5,574,737 | 19.66 |
| tRNA | 27,037 | 1.20 | 1,392,454 | 4.91 |
| snoRNA | 4094 | 0.18 | 52,837 | 0.19 |
| Other | 14,361 | 0.64 | 276,293 | 0.97 |
| High repeats (matched genome times >20) | 95,407 | 4.25 | 3,189,419 | 11.25 |
| Low expression (sequence times = 1) | 1,550,607 | 69.11 | 1,550,607 | 5.47 |
| Total Potential miRNA | 417,145 | 18.59 | 16,313,529 | 57.54 |
| Name | Sequence | Length | p-Value | Ratio | Mark |
|---|---|---|---|---|---|
| miR-1120a | TTCCGTCTCATAATATAAGAA | 21 | 0.000630604 | 13.3564 | UP |
| miR-1432b | ATCAGGAGAGATGACACCGAC | 21 | 0.001282166 | 0.89426 | DOWN |
| miR-160b | TGCCTGGCTCCCTGAATGCCA | 21 | 0.000299524 | 13.2333 | UP |
| miR-166b | TCGGACCAGGCTTCAATCCCT | 21 | 0.016126572 | 9.08411 | UP |
| miR-168 | CCCGCCTTGCACCAAGTGA | 19 | 7.46 × 10−7 | 0.12191 | DOWN |
| miR-169e | GGCAGTCTCCTTGGCTAGC | 19 | 0.000734617 | 0.74931 | DOWN |
| miR-169g | GGCAAGTCCGTCCTTGGCTAC | 21 | 0.006909309 | 1.13138 | UP |
| miR-169i | CAGCCAAGGATGACTTGCCGA | 21 | 0.036371524 | 1.19995 | UP |
| miR-169j | CAAGTTGTTCTTGGCTAGC | 19 | 0.015899833 | 1.0635 | UP |
| miR-169k | TGGGCAAGTCACCCTGGCTACC | 22 | 0.001990317 | 0.90879 | DOWN |
| miR-171a | TGAGCCGAACCAATATCACTC | 21 | 0.00400738 | 1.04654 | UP |
| miR-171b | TGATTGAGCCGTGCCAATATC | 21 | 0.008975612 | 1.13702 | UP |
| miR-172 | AGAATCTTGATGATGCTGCAT | 21 | 0.001014298 | 14.092 | UP |
| miR-393a | TTCCAAAGGGATCGCATTGAT | 21 | 0.013664601 | 11.4734 | UP |
| miR-393b | TCCAAAGGGATCGCATTGATC | 21 | 0.016875444 | 9.49652 | UP |
| miR-395 | TGAAGTGTTTGGGGGAACTC | 20 | 0.005497432 | 8.12411 | UP |
| miR-396c | TTCCACAGCTTTCTTGAACTG | 21 | 1.35 × 10−7 | 0.29308 | DOWN |
| miR-398a | TGTTTTCCCAGGTCACCCCTT | 21 | 0.03744688 | 4.247 | UP |
| miR-398b | TGTGTTCTCAGGTCACCCCTT | 21 | 0.010706403 | 7.30687 | UP |
| miR-5175a | TTCCAAATTACTCGTCGTGGT | 21 | 0.020159266 | 4.5196 | UP |
| miR-9674d | ATAGCATCATCCATCCTGCCA | 21 | 0.005919157 | 10.5192 | UP |
| miR-9674f | GTAGGATGGCTGGTGCTATGG | 21 | 0.003070376 | 0.83586 | DOWN |
| miR-9772 | TGAGATGAGATTACCCCATAC | 21 | 0.008646577 | 9.88001 | UP |
| Novel-102 | CGCTGTCTCCGACATGGACC | 20 | 0.000307346 | 0.27861 | DOWN |
| Novel-108 | TCAGGAAGGACCGCATCATC | 20 | 0.043754142 | 0.84898 | DOWN |
| Novel-110 | AGTTTTTTCTACGACACTTTAGATTCT | 27 | 0.04597715 | 7.5706 | UP |
| Novel-111 | GTCTCTGCCAATTCTTCGTGT | 21 | 0.02294524 | 0.68635 | DOWN |
| Novel-112 | TCAGGAAGGACTGCATTATC | 20 | 0.010667151 | 0.60942 | DOWN |
| Novel-113 | CCCGCTGTCTCCGACATG | 18 | 0.00241642 | 0.37701 | DOWN |
| Novel-125 | TGCCTTTAAGGCACCTGCCTTT | 22 | 0.011360968 | 7.19148 | UP |
| Novel-14 | ACAACGTTACAAAGAACT | 18 | 0.011088055 | 6.70541 | UP |
| Novel-20 | CTCTCCTGTAGAAATAGGCACCGA | 24 | 0.044368782 | 5.26639 | UP |
| Novel-25 | TTCCGTCTCATAATATAAGAA | 21 | 0.014815363 | 13.3564 | UP |
| Novel-27 | GTGTTCTCAGGTCGCCCCCGC | 21 | 0.006439364 | 0.5309 | DOWN |
| Novel-28a | ACAAGATATTGGGTATTTCTGTCTTTATT | 29 | 0.012777427 | 6.47692 | UP |
| Novel-28b | AACAAGATATTGGGTATTTCTGTCTTTATT | 30 | 0.000878252 | 33.9326 | UP |
| Novel-3 | TACGCAGAGTGAATCGGTC | 19 | 0.000906629 | 0.27046 | DOWN |
| Novel-38 | TAGCGAAATTCCTTGTCGGGT | 21 | 0.004373032 | 0.45084 | DOWN |
| Novel-4 | CGAGGACCTTGGTTGAGCCTG | 21 | 0.002502204 | 7.85936 | UP |
| Novel-40 | TAGTAGCACCTTAGGATGGCATA | 23 | 0.014233197 | 5.35626 | UP |
| Novel-41 | TATCCTCGTCGTATTCTTTATA | 22 | 0.010294018 | 21.6221 | UP |
| Novel-44 | AGTAATTTTGGACGGAGGGAG | 21 | 0.005345682 | 4.99639 | UP |
| Novel-48 | TATTTGTTTGCAGAGGGAGTA | 21 | 0.019775829 | 4.616 | UP |
| Novel-55 | ATAAACCGGGTTTTCTGAAGCACC | 24 | 0.035691617 | 0.64187 | DOWN |
| Novel-57 | TCATTTGGAACTCGCCGGTGC | 21 | 0.002525248 | 0.43535 | DOWN |
| Novel-66 | CCCATGGATTGGCTAGTTCCT | 21 | 0.04578047 | 4.32044 | UP |
| Novel-74 | TGCATCATTTGGAACTCGCC | 20 | 0.015178025 | 0.72326 | DOWN |
| Novel-87 | AAATGGAATTTAACTCTTTCATGCT | 25 | 0.039866375 | 4.0404 | UP |
| Novel-92 | AGCCAACAACCTCCTAGTTCC | 21 | 0.018601675 | 0.75506 | DOWN |
| Novel-99 | ATGCCGTGTTGTTCTGAAAGAA | 22 | 0.024139855 | 9.38665 | UP |
| Triticum dicoccoides | Aegilops tauschii | Triticum aestivum | Hordeum Vulgare | Brachypodium distachyon | Putative Target | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D | S | D | S | D | S | D | S | D | S | ||
| miR1074 | UP | Malate dehydrogenase | |||||||||
| miR1120 | UP | ||||||||||
| miR1134 | UP | ||||||||||
| miR1432 | UP | DOWN | Calcium binding or Calcium ATPases | ||||||||
| miR1435 | UP | ||||||||||
| miR1450 | UP | UP | Mn superoxide dismutase | ||||||||
| miR156 | UP | UP | UP | UP | UP | UP | SPL | ||||
| miR159 | UP | UP | MYB65/MYB33 | ||||||||
| miR160 | UP | UP | DOWN | Metal transporter NRAMP2-like | |||||||
| miR164 | UP/DOWN | UP | NAC TF, Got1/Sft2-like, NAM | ||||||||
| miR166 | DOWN | UP | UP | UP/DOWN | HD-Zip TF | ||||||
| miR167 | UP | UP/DOWN | DOWN | UP | Auxin response factor | ||||||
| miR168 | DOWN | DOWN | CCAAT-binding TF | ||||||||
| miR169 | UP | DOWN | UP | Polyol transporter 5-like | |||||||
| miR171 | DOWN | UP | UP | UP/DOWN | UP/DOWN | UP | Auxin response factor | ||||
| miR172 | UP | DOWN | DOWN | Argonaute/AP2/EREBP TF | |||||||
| miR1867 | UP | DUF1242 superfamily | |||||||||
| miR1881 | UP | UP | |||||||||
| miR319 | DOWN | DOWN | TCP TF21 | ||||||||
| miR3440 | DOWN | ||||||||||
| miR3710 | UP | VAMP protein SEC22 | |||||||||
| miR393 | UP | UP | auxin F-box | ||||||||
| miR395 | UP | UP | DOWN | UP | ATP sulfurylase, APS4/F-box | ||||||
| miR396 | DOWN | DOWN | UP | DOWN | UP | GRF1/GRF2, GRF2 & SYP131 | |||||
| miR398 | Up | UP | programmed cell death protein 4 | ||||||||
| miR408 | UP | Laccase | |||||||||
| miR444 | UP | DOWN | DOWN | MADS-box TF 27-like | |||||||
| miR474 | UP | Kinesin, PPR family | |||||||||
| miR482 | UP | UP | Rab15B protein | ||||||||
| miR5021 | UP | ||||||||||
| miR5024 | UP | ||||||||||
| miR5049 | D-E | UP | 60S ribosomal protein l36 | ||||||||
| miR5083 | DOWN | ||||||||||
| miR5175 | UP | DOWN | UP | ACC-like oxidase | |||||||
| miR5205 | DOWN | D-E | DOWN | ||||||||
| miR5227 | UP | 6-phosphofructokinase 2-like | |||||||||
| miR5266 | DOWN | ||||||||||
| miR528 | DOWN | DOWN | UP/DOWN | XBAT32 | |||||||
| miR529 | UP | E3 ubiquitin ligase RNF14-like/bHLH TF | |||||||||
| miR535 | UP/DOWN | 5.8S ribosomal RNA gene | |||||||||
| miR5523 | DOWN | ||||||||||
| miR5568 | D-E | ||||||||||
| miR5655 | UP | TF bHLH135-like | |||||||||
| miR6108 | UP | Alpha tubulin | |||||||||
| miR6220 | D-E | ||||||||||
| miR6222 | DOWN | ||||||||||
| miR6248 | UP | ||||||||||
| miR7714 | UP | ||||||||||
| miR894 | UP | Zinc finger and C2 domain protein-like | |||||||||
| miR896 | DOWN | ||||||||||
| miR903 | DOWN | 26S ribosomal RNA gene | |||||||||
| miR9674 | UP/DOWN | ||||||||||
| miR9772 | UP | ||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, K.; Nie, X.; Cui, L.; Deng, P.; Wang, M.; Song, W. Genome-Wide Identification and Characterization of Salinity Stress-Responsive miRNAs in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides). Genes 2017, 8, 156. https://doi.org/10.3390/genes8060156
Feng K, Nie X, Cui L, Deng P, Wang M, Song W. Genome-Wide Identification and Characterization of Salinity Stress-Responsive miRNAs in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides). Genes. 2017; 8(6):156. https://doi.org/10.3390/genes8060156
Chicago/Turabian StyleFeng, Kewei, Xiaojun Nie, Licao Cui, Pingchuan Deng, Mengxing Wang, and Weining Song. 2017. "Genome-Wide Identification and Characterization of Salinity Stress-Responsive miRNAs in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides)" Genes 8, no. 6: 156. https://doi.org/10.3390/genes8060156