The Genome of the Beluga Whale (Delphinapterus leucas)

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Methods, Results and Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stewart, B.E.; Stewart, R.E.A. Mammalian Species Delphinapterus leucas. J. Mammal. 1989, 1–8. [Google Scholar] [CrossRef]
- O’Corry-Crowe, G.M. Beluga whale Delphinapterus leucas. In Encyclopedia of Marine Mammals, 1st ed.; Perrin, W.F., Würsig, B.G., Thewissen, J.G.M., Eds.; Academic Press: San Diego, CA, USA, 2002; pp. 94–99. [Google Scholar]
- Vandervalk, B.P.; Yang, C.; Xue, Z.; Raghavan, K.; Chu, J.; Mohamadi, H.; Jackman, S.D.; Chiu, R.; Warren, R.L.; Birol, I. Konnector v2.0: Pseudo-long reads from paired-end sequencing data. BMC Med. Genom. 2015, 8 (Suppl. 3), S1. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a bloom filter. Genom. Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.A.; Warren, R.L.; Vandervalk, B.P.; Kucuk, E.; Khan, H.; Gibb, E.A.; Pandoh, P.; Kirk, H.; Zhao, Y.; Jones, M.; et al. The North American bullfrog draft genome provides insight into hormonal regulation of long noncoding RNA. Nat. Commun. 2017, 8, 1433. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L. RAILS and Cobbler: Scaffolding and automated finishing of draft genomes using long DNA sequences. J. Open Source Softw. 2016, 1, 116. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:1303.3997v1301. [Google Scholar]
- Warren, R.L.; Chen, Y.; Vandervalk, B.P.; Behsaz, B.; Lagman, A.; Jones, S.J.M.; Birol, I. LINKS: Scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 2015, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Paulino, D.; Warren, R.L.; Vandervalk, B.P.; Raymond, A.; Jackman, S.D.; Birol, I. Sealer: A scalable gap-closing application for finishing draft genomes. BMC Bioinform. 2015, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4.11.1–4.11.39. [Google Scholar] [CrossRef]
- RepeatMasker; Open-4.0; Smit, A.F.A.; Hubley, R.; Green, P. (Eds.) Institute for Systems Biolog: Seattle, WA, USA, 2013–2015; Available online: http://www.repeatmasker.org (accessed on 12 September 2017).
- Stanke, M.; Tzvetkova, A.; Morgenstern, B. AUGUSTUS at EGASP: Using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006, 7, S11. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Liu, Y.; Thomas, G.W.; Vinar, T.; Alfoldi, J.; Deng, J.; Dugan, S.; van Elk, C.E.; Hunter, M.E.; Joshi, V.; et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015, 47, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. Cell Mol. Biol. 2015, 83, 189–212. [Google Scholar] [CrossRef] [PubMed]
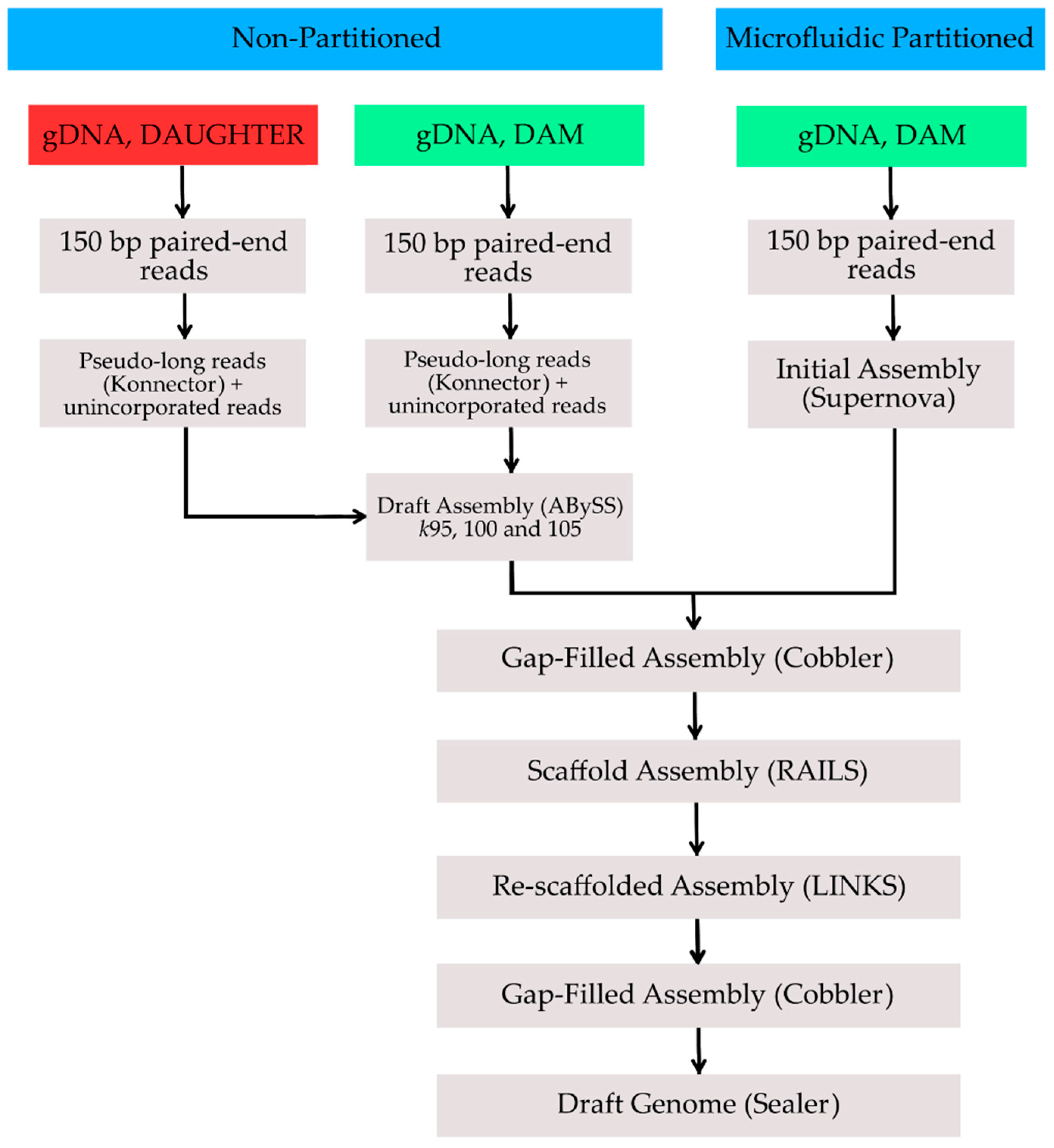


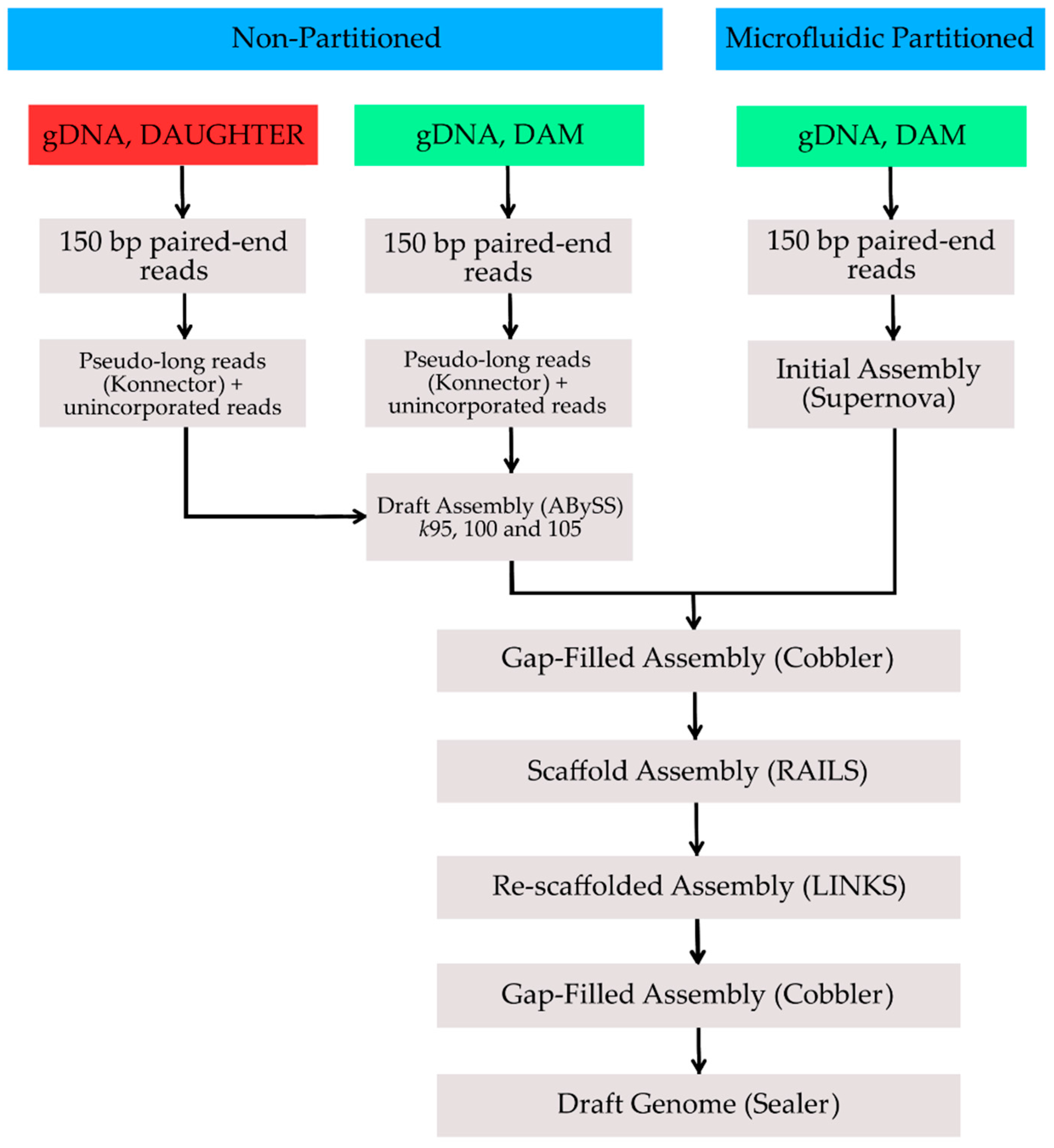{kind=link}
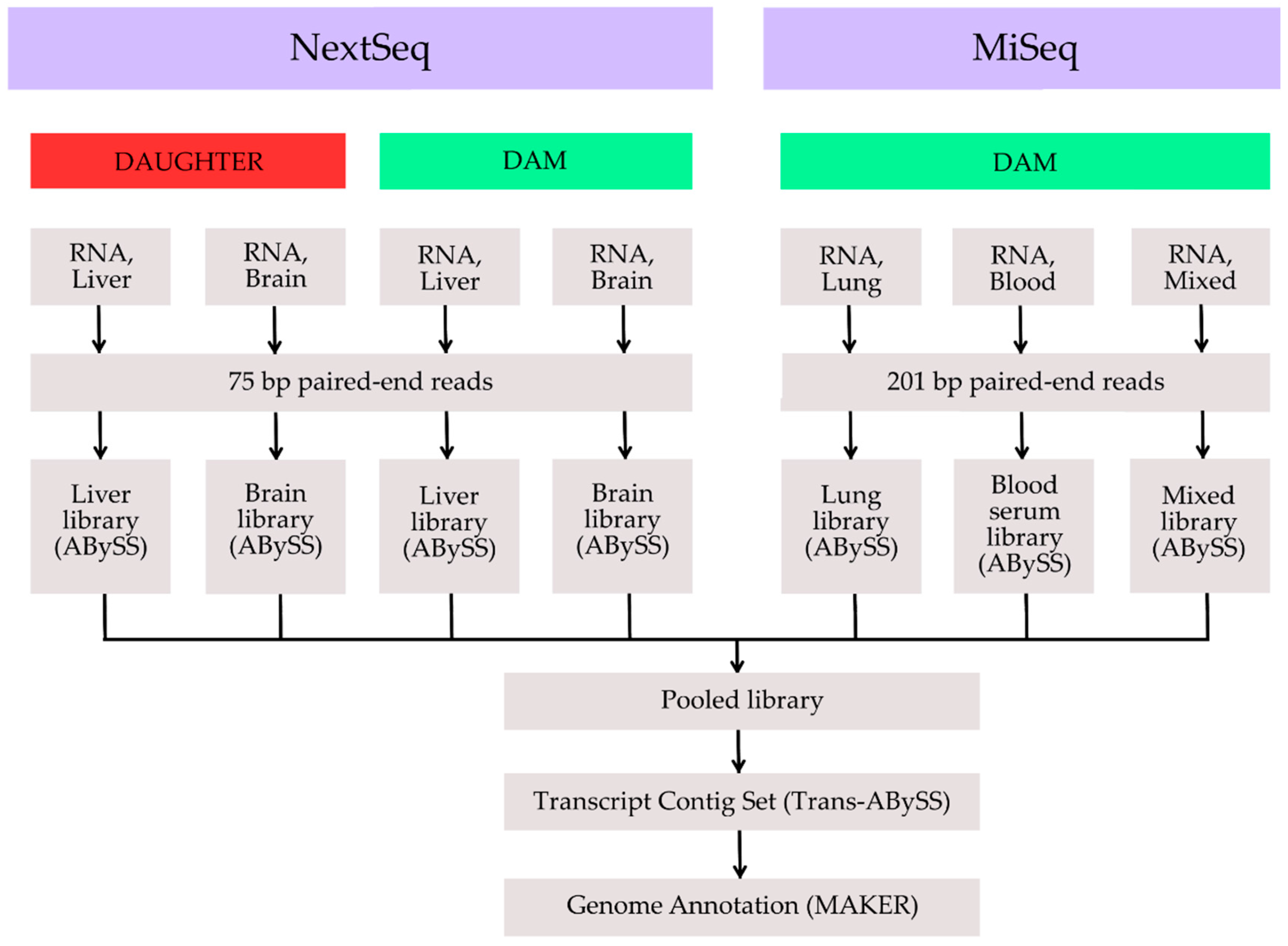{kind=link}
| Assembly | Total Size (Gbp) | No. of Gaps | No. of Scaffolds | Scaffold N50 (bp) | Longest Scaffold (bp) | BUSCO Complete Genes | BUSCO Complete + Fragmented Genes |
|---|---|---|---|---|---|---|---|
| ABySS-pe | 2.325 + 0.216% in gaps | 210,782 | 102,940 | 58,545 | 997,316 | 4153 (66.42%) | 4689 (74.99%) |
| Supernova | 2.314 + 1.40% in gaps | 30,858 | 8930 | 16.79 × 106 | 78 × 106 | 5667 (90.63%) | 5911 (94.53%) |
| Rails/Cobbler | 2.327 + 1.37% in gaps | 26,898 | 6971 | 19.59 × 106 | 95 × 106 | 5669 (90.66%) | 5915 (94.59%) |
| Sealer | 2.327 + 1.36% in gaps | 25,839 | 6971 | 19.59 × 106 | 95 × 106 | 5669 (90.66%) | 5915 (94.59%) |
| Tissue | n | n:N50 | Min | N80 | N50 | N20 | Max | Sum | Read Count |
|---|---|---|---|---|---|---|---|---|---|
| Liver, dam | 960,722 | 117,511 | 74 | 144 | 420 | 1542 | 47,312 | 246.3 × 106 | 239.4 × 106 |
| Brain, dam | 2,019,281 | 247,296 | 74 | 193 | 587 | 2013 | 18,656 | 691 × 106 | 247.0 × 106 |
| Liver, daughter | 854,394 | 99,263 | 74 | 145 | 555 | 1538 | 47,494 | 235.4 × 106 | 241.2 × 106 |
| Brain daughter | 2,258,624 | 260,327 | 74 | 198 | 653 | 2219 | 19,796 | 806.2 × 106 | 270.4 × 106 |
| Lung, dam | 1,170,674 | 374,225 | 162 | 201 | 282 | 504 | 5091 | 339.2 × 106 | 25.6 × 106 |
| Mixed, dam | 860,603 | 305,220 | 149 | 186 | 220 | 352 | 4751 | 208.2 × 106 | 26.6 × 106 |
| Serum, dam | 1,244,441 | 516,834 | 135 | 195 | 203 | 287 | 8034 | 281.5 × 106 | 23.8 × 106 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, S.J.M.; Taylor, G.A.; Chan, S.; Warren, R.L.; Hammond, S.A.; Bilobram, S.; Mordecai, G.; Suttle, C.A.; Miller, K.M.; Schulze, A.; et al. The Genome of the Beluga Whale (Delphinapterus leucas). Genes 2017, 8, 378. https://doi.org/10.3390/genes8120378
Jones SJM, Taylor GA, Chan S, Warren RL, Hammond SA, Bilobram S, Mordecai G, Suttle CA, Miller KM, Schulze A, et al. The Genome of the Beluga Whale (Delphinapterus leucas). Genes. 2017; 8(12):378. https://doi.org/10.3390/genes8120378
Chicago/Turabian StyleJones, Steven J. M., Gregory A. Taylor, Simon Chan, René L. Warren, S. Austin Hammond, Steven Bilobram, Gideon Mordecai, Curtis A. Suttle, Kristina M. Miller, Angela Schulze, and et al. 2017. "The Genome of the Beluga Whale (Delphinapterus leucas)" Genes 8, no. 12: 378. https://doi.org/10.3390/genes8120378
APA StyleJones, S. J. M., Taylor, G. A., Chan, S., Warren, R. L., Hammond, S. A., Bilobram, S., Mordecai, G., Suttle, C. A., Miller, K. M., Schulze, A., Chan, A. M., Jones, S. J., Tse, K., Li, I., Cheung, D., Mungall, K. L., Choo, C., Ally, A., Dhalla, N., ... Haulena, M. (2017). The Genome of the Beluga Whale (Delphinapterus leucas). Genes, 8(12), 378. https://doi.org/10.3390/genes8120378