1. Introduction
Many species produce RNAs whose sequences differ from the DNA from which they are transcribed. The processes that result in these site-specific alterations are collectively referred to as RNA editing [
1]. Editing mechanisms vary widely, resulting in changes at either the base or nucleotide level [
2]. Mitochondrial RNAs are extensively edited in a number of organisms. Maturation of higher plant mitochondrial RNAs involves hundreds of C to U changes resulting from targeted post-transcriptional deamination reactions, while some lower plant mitochondrial RNAs contain thousands of C to U changes and hundreds of U to C changes [
3]. Base changes of multiple types are also prevalent in mitochondrial RNAs in certain dinoflagellates [
4,
5], although the underlying mechanism remains unclear. Widespread changes at the nucleotide level occur in the mitochondria of kinetoplastid protozoa and myxomycetes. Kinetoplast RNAs are processed by a series of guide RNA-directed cleavage and ligation reactions, resulting in the insertion of thousands of added uridines and removal of hundreds of encoded Us [
6], while over 1300 non-encoded nucleotides are added to mitochondrial RNAs in
Physarum polycephalum [
7].
At least three distinct RNA editing mechanisms are used during mitochondrial gene expression in
P. polycephalum [
8]. The majority of editing events in
P. polycephalum mitochondria involve the precise insertion of non-encoded nucleotides (nt), ~95% (1255 of 1324 nt) of which are single C insertions. Other ribonucleotides are also added at a limited number of defined sites, either singly or in pairs. Rarer forms of editing include a deletion of three encoded A residues [
9], the replacement of the 5′ nucleotide of two mitochondrial tRNAs [
10], and four C to U changes [
11]. In total, site-specific changes are present in 37 of the 39 mRNAs, all three rRNAs, and the five tRNAs encoded in the
P. polycephalum mitochondrial genome. Editing is extremely accurate and highly efficient based on both characterization of transcripts from individual genes [
9,
10,
11,
12,
13,
14,
15,
16] and sequencing of the entire mitochondrial transcriptome [
7].
The mechanism of nucleotide insertion in
P. polycephalum mitochondria is distinct from the guide RNA-directed U insertion/deletion form of editing described in trypanosomes and the polymerase stuttering utilized in viral systems [
17]. We are interested in elucidating the
cis-acting elements and
trans-acting factors involved in RNA sequence alterations within
P. polycephalum mitochondria, and have established two in vitro systems in which to study editing, both of which rely on transcription/editing complexes formed in vivo [
18,
19]. Using these systems, we have demonstrated that insertion editing occurs co-transcriptionally, i.e., the extra nucleotides are added at the 3′ end of the nascent RNA during synthesis [
17], and that the template elements required for the insertion of non-encoded nucleotides are limited to ~18 base pairs (bp) centered around the editing site [
20]. However, in order to definitively identify the
cis-elements that direct editing of
P. polycephalum mitochondrial transcripts, it will be necessary to achieve transcription and editing from a template that can be easily manipulated in vitro.
Electroporation has been used previously to introduce DNA into isolated mitochondria. An early study using mammalian mitochondria demonstrated DNA uptake and the effects of field strength on mitochondrial function, but did not examine transcriptional activity [
21]. This methodology was subsequently adapted to study transcription, splicing, and RNA editing in mitochondria from wheat [
22,
23,
24,
25,
26,
27], maize and sorghum [
28,
29], and potato [
30,
31]. Accurate C to U editing of transcripts derived from chimeric genes was observed in each of these systems, allowing localization of the
cis-acting signals essential for editing [
22,
25].
Here we describe the development of methods for the introduction of DNA into P. polycephalum mitochondria and provide evidence that exogenous DNA is expressed, albeit at low levels. In these initial studies, no evidence of either nucleotide insertion or C to U changes was observed in transcripts from the introduced templates. However, transcription from endogenous genes is not significantly affected by electroporation at moderate field strengths, and the transcripts synthesized from the mitochondrial genome after electroporation are still largely edited. Thus, the editing apparatus is not inactivated under these conditions, providing a starting point for future investigations of editing signals.
2. Materials and Methods
2.1. DNAs Used for Electroporation
All plasmids used in this study contained the promoter region that drives transcription of the mitochondrial ribosomal RNA genes (large and small subunit rRNAs and 5S rRNA). The plasmid (#809) used in the experiments in
Figure 1,
Figure 2B, and
Figure S1 contains the entire 125 bp intergenic region upstream of the
P. polycephalum mitochondrial large subunit rRNA (
LSU) gene and 97 bp of the transcribed portion of the
LSU gene fused via a linker to 445 bp derived from the
P. polycephalum mitochondrial
cox1 gene and inserted into the HindIII and EcoRI sites of vector pBSM13+ (Agilent Technologies, Santa Clara, CA, USA). The linker sequence provides a unique primer binding site which, along with the 5 bp block mutation introduced near the 3′ end of the
cox1 portion (shown in
Figure S1), ensures that reverse transcription PCR (RT-PCR) products derived from fusion gene transcripts can be distinguished from those arising from the endogenous
cox1 gene. The 847 bp PCR product used in the experiments shown in
Figure 1 and
Figure 2A contained the entire insert plus flanking sequences derived from the vector. The PCR product was purified using a QIAquick PCR purification kit (Qiagen, Germantown, MD, USA) followed by passage through a Micro Bio-Spin P30 column (Bio Rad, Hercules, CA, USA), concentrated via ethanol precipitation, and resuspended in PCR-grade water at a final concentration of 0.5 μg/μL prior to electroporation. The plasmid used in the experiment shown in
Figure 3 (#819) contains the entire 125 bp intergenic region upstream of the
P. polycephalum mitochondrial
LSU gene and 15 bp of the transcribed portion of the
LSU gene inserted into the
HindIII and
SalI sites of pBSM13+. The
LSU gene was omitted from the dot blot to avoid potential hybridization to
LSU-vector fusion transcripts. The plasmid used in the experiment shown in
Figure 4 (#802) contains 70 bp of the intergenic region immediately upstream of the
P. polycephalum mitochondrial
LSU gene and 97 bp of the transcribed portion of the
LSU gene inserted into the
PstI and
XbaI sites of pBSM13+.
2.2. Mitochondrial Isolation
Although
P. polycephalum mitochondria isolated via differential centrifugation synthesize run-on transcripts that are fully edited, when further purified on Percoll (GE Healthcare Life Sciences, Pittsburgh, PA, USA) gradients, few sediment at the appropriate density, resulting in very low yields. To enhance mitochondrial yields, we explored a broad range of conditions, ultimately arriving at the conditions described below. The mitochondria in these preparations are uniform in size and essentially free of nuclei, pigment granules, and other contaminants (
Figure 1, left). They are also transcriptionally active and synthesize edited RNAs (see Results).
Microplasmodia were cultured at 26 °C in 300 mL SDM medium [
32] in a 2-L baffled flask to mid-log phase, then poured into a large beaker on ice and allowed to settle by gravity. The medium was decanted and the cells washed by swirling gently in 300 mL ice-cold sclerotia salts (19 mM citric acid, 0.3 mM FeCl
2, 2.4 mM MgSO
4, 8.1 mM CaCl
2, 0.4 mM MnCl
2, 0.1 mM ZnSO
4, 2.9 mM KH
2PO
4, pH 4.8) [
33]. The liquid was again decanted and the cells were resuspended in 75 mL ice-cold sclerotia salts and split evenly into two 50 mL conical tubes. Cells were pelleted by centrifugation for 2 min at ~800×
g and the liquid was again decanted. Each pellet was washed twice with sterile ice cold distilled water, vortexing gently and pelleting as above. After removing as much liquid as possible, cells were transferred to a 55 mL Wheaton homogenizer (Millville, NJ, USA) on ice, rinsing the tubes with 15 mL of ice cold buffered sucrose (BSS, 10 mM Tris (pH 7.5)/0.25 M sucrose). Cells were broken open by douncing for 5 strokes and α-amylase was added to the lysate at a final concentration of 40–80 μg/mL. After 10 min on ice, cells were dounced another 5 strokes and the homogenate was examined using phase contrast microscopy to determine if additional strokes were needed. The lysate was then filtered to remove residual clumps and slime prior to layering over Percoll gradients consisting of steps of 1.082, 1.062, and 1.044 g/mL in 2 mM Tris (pH 7.5)/0.25 M sucrose. Gradients were centrifuged for 1 min at 25,000×
g and the mitochondrial bands were transferred to a fresh tube. Mitochondria were then diluted by slowly layering 2.5 volumes of 2 mM Tris (pH 7.5)/0.25 M sucrose with gentle mixing prior to centrifugation at 7600×
g for 10 min at 4 °C. Mitochondria were resuspended in 0.33 M sucrose and the protein content was determined (Bio-Rad Protein Assay) prior to pelleting at 5100×
g for 5 min at 4 °C. The final mitochondrial pellets were resuspended in 0.33 M sucrose and kept on ice until use.
2.3. Electroporation
Mitochondria at protein concentrations of 5–10 μg/μL were split into 50 μL aliquots and mixed with plasmid DNA (0.5–1 μg) or purified PCR fragment (30–500 ng) as appropriate. Mitochondria were then transferred to a cold 0.1 cm gap electroporation cuvette (Bio Rad) and electroporated using an Eppendorf Electroporator 2510 (Hauppauge, NY, USA) at 10 μF, 600 Ω, and field strengths between 5 kV/cm and 20 kV/cm. The samples were immediately transferred to a fresh tube and the cuvette was rinsed with 50 μL 0.33 M sucrose, which was added to the sample. Samples were then used for microscopy and/or transcription and editing assays as noted.
2.4. Microscopy
Mitochondria were concentrated by pelleting and resuspended in 0.33 M sucrose prior to microscopy. Images were collected with a Retiga EXI camera (Q-imaging Vancouver, BC, Canada) mounted on a Leica DMI 6000 B inverted microscope (Leica Biosystems, Wetzlar, Germany) using a 63× objective (1.4 NA Plan Apo) with a 1.6× mag changer for a resulting magnification of 1008×. Mitochondria were visualized using phase contrast optics. Fluorescent DNA was visualized using a standard Texas Red filter set. Equal thresholds were set to detect only the brightest fluorescent signals. These signals were then outlined and overlaid onto the phase image. The co-localization of fluorescence and mitochondria is indicated by colored outlines in the overlay image.
2.5. Transcription, RT-PCR, and PCR
Transcription reactions with unlabeled nucleotides were carried out in 0.3–0.33 M sucrose/20 mM Tris (pH 7.5)/20 mM MgCl
2/10 mM KCl/2 mM DTT/500 μM NTPs. Mitochondria were pelleted and resuspended in Trizol Reagent (Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) and RNAs were isolated as specified by the supplier. The resulting RNA pellets were resuspended in 10 mM Tris (pH 7.5)/1 mM EDTA, extracted with 24:1 chloroform: isoamyl alcohol (CIA), and ethanol precipitated. To avoid background signal from residual exogenous DNA in subsequent PCR reactions, RNA preparations were subsequently incubated with
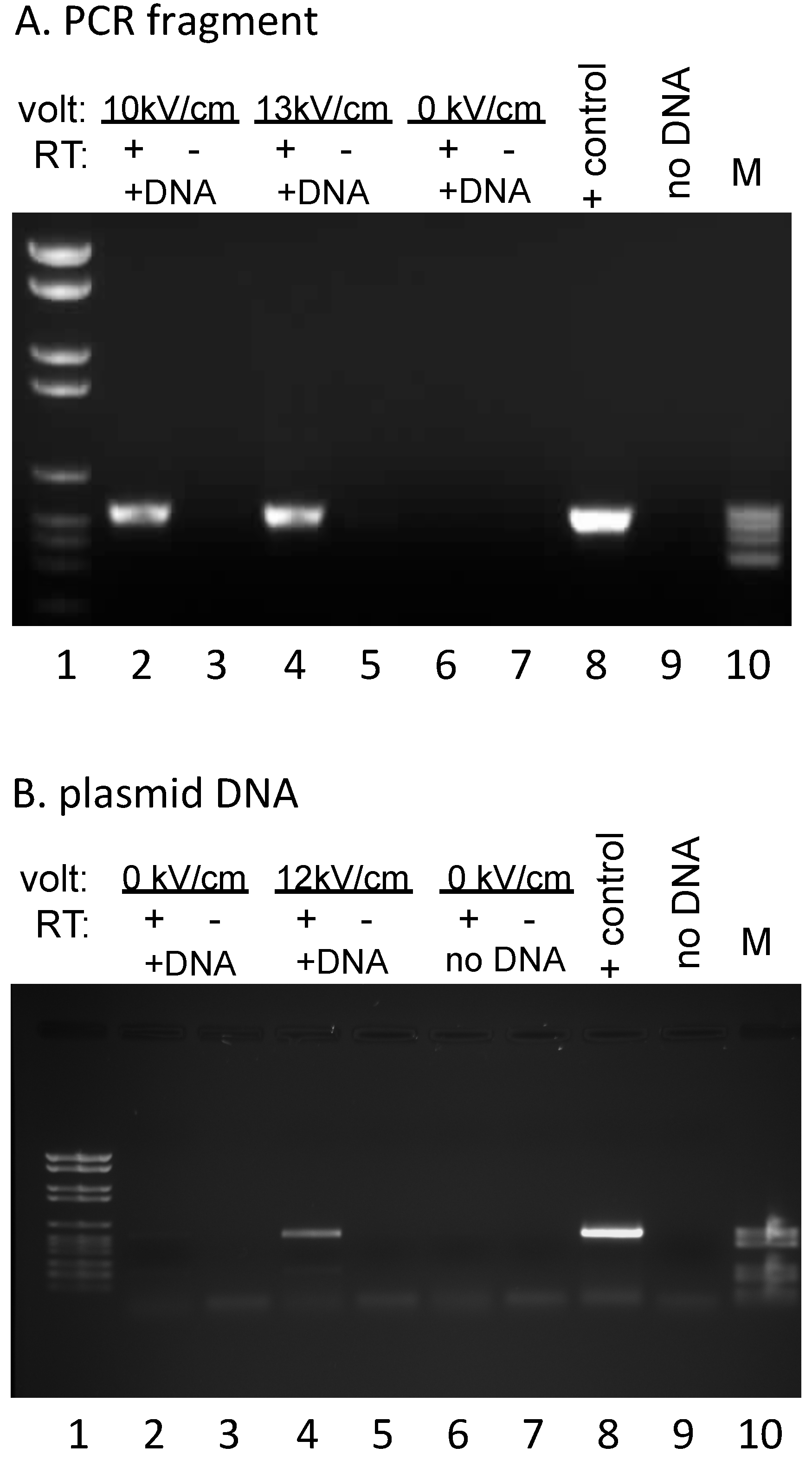
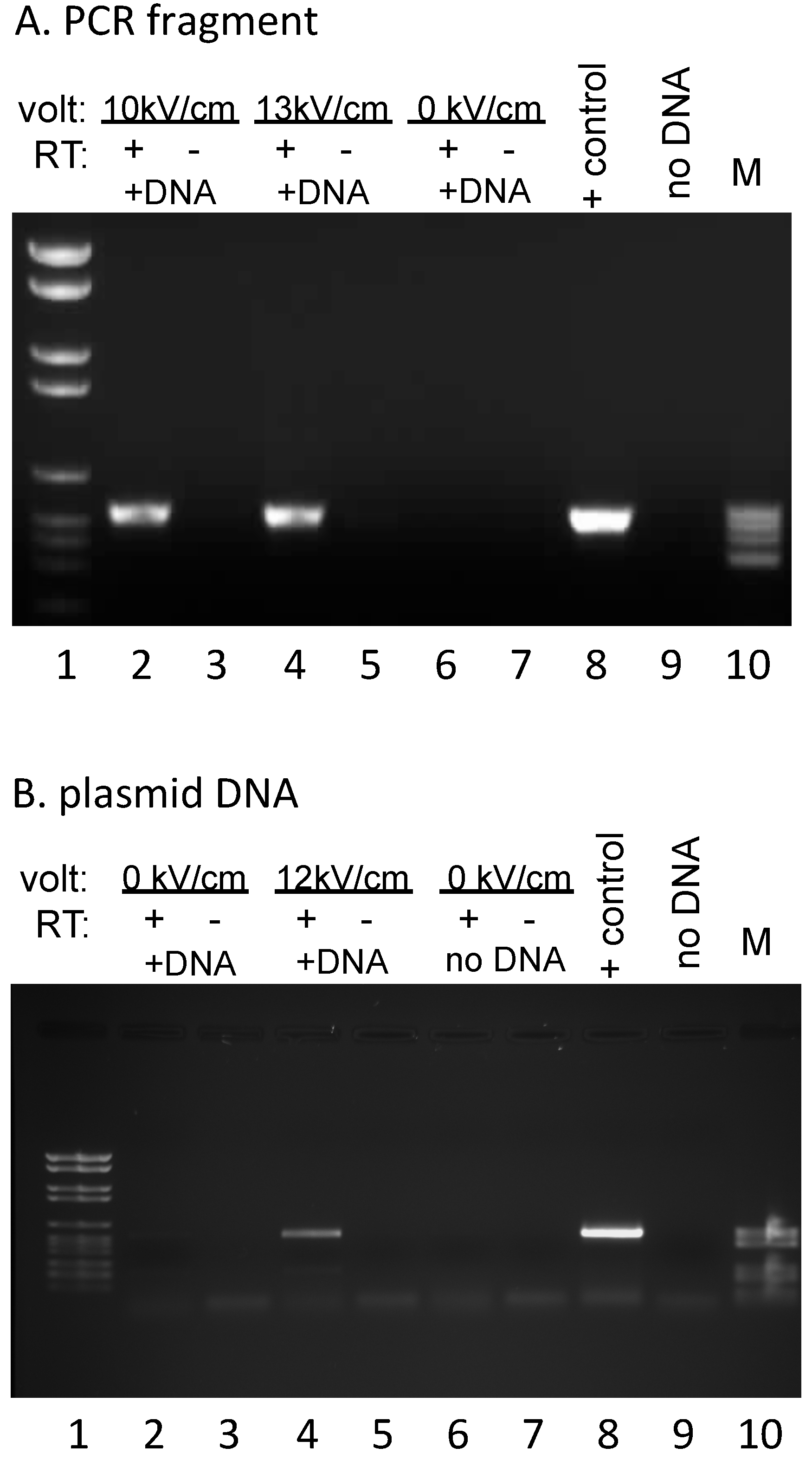
EcoRI (New England Biolabs, Ipswich, MA, USA) to remove the primer binding sites for the reverse transcription and PCR reactions, CIA extracted, and ethanol precipitated prior to two rounds of DNase I (Roche, Indianapolis, IN, USA) treatment. Parallel complementary DNA (cDNA) synthesis reactions were carried out by annealing a primer complementary to sequences specific for the exogenous DNA (#508: GGTTTTCCCAGTCACGAC), then splitting each annealed RNA/primer mix into two equal aliquots, incubating under reaction conditions that were identical except for the presence or absence of AMV reverse transcriptase (Life Sciences, Saint Petersburg, FL, USA). Subsequent PCR reactions were carried out with primers #494 (TGTAAAACGACGGCCAGTG) and #505 (CTAGATCTGGGTCGTTGTC), yielding a 538 bp fragment (
Figure 2). No signal was observed in the absence of added DNA, indicating that endogenous RNA does not yield a product with these primers (
Figure 2B). The 847 bp PCR product used for electroporation (
Figure 1 and
Figure 2A) was generated from plasmid #809 using primers #508 (GGTTTTCCCAGTCACGAC) and #509 (5′-cy3–GAAACAGCTATGACCATG). PCR fragments for dot blots were generated using the primers listed in
Table S1. All PCR products were generated with Taq DNA polymerase (New England Biolabs) under conditions specified by the supplier.
2.6. Dot Blot Experiments
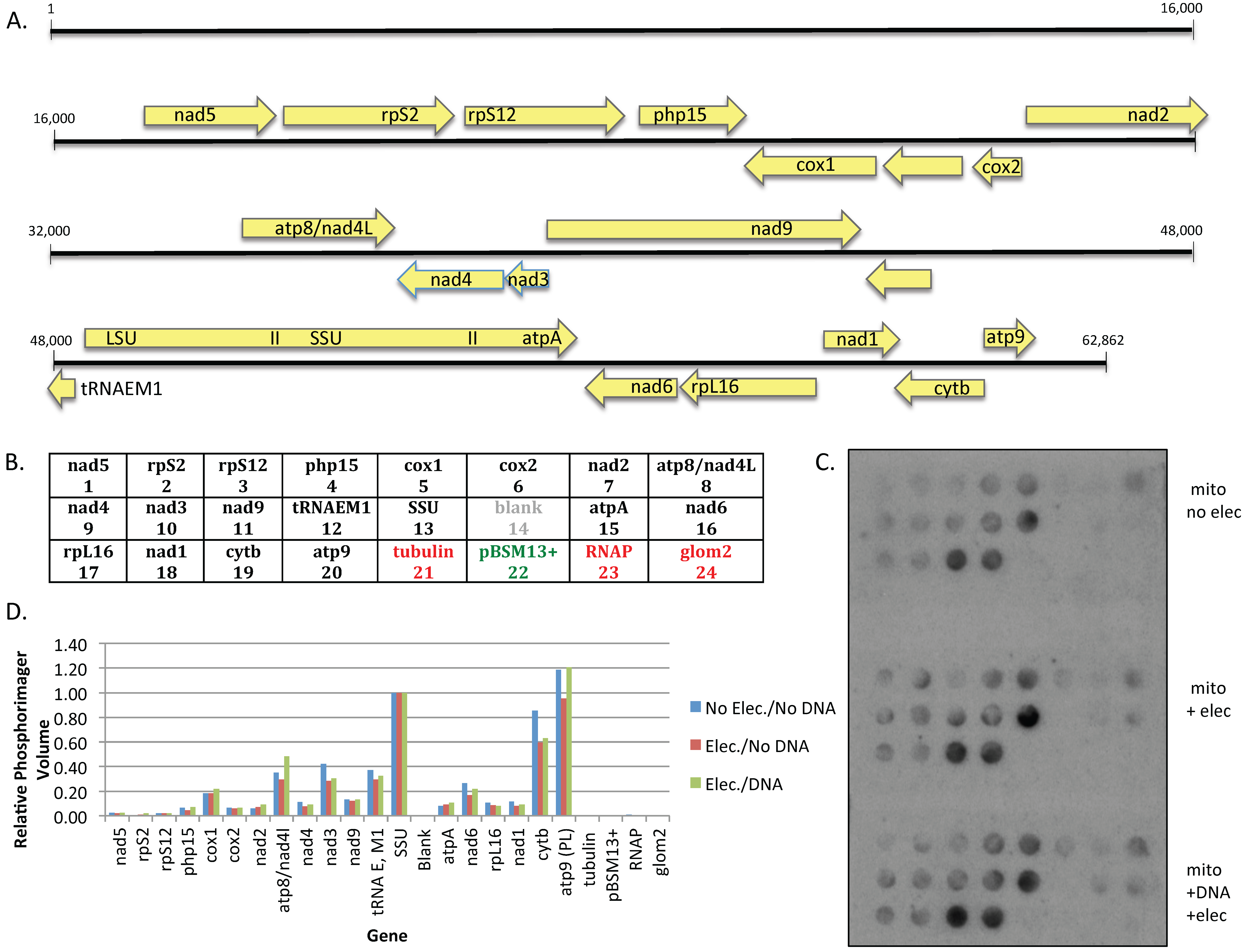
PCR fragments to be immobilized on nitrocellulose filters were heated to 95 °C for 5 min, snap-cooled on ice, then incubated in 0.5 M NaOH for 10 min on ice. Samples were neutralized by the addition of one-half volume of neutralization solution [0.5 M Tris-HCl (pH8)/217 mM Na citrate/1 M NaCl/1 M HCl] and 20 μL aliquots of denatured DNAs were spotted onto nitrocellulose filters using a vacuum manifold. Filters were air-dried, washed twice with 6X SSC (0.9 M NaCl/90 mM Na citrate), air-dried, UV cross-linked at 254 nm for 3 min, and baked for 2 hours at 80 °C.
Mitochondria at a protein concentration of 8.25 μg/μL were split into three 50 μL aliquots; one sample was kept on ice, while the remaining aliquots were electroporated either in the absence or presence of plasmid #819 at a field strength of 10 kV/cm as noted. Transcription reactions were carried out at 30 °C in 0.33 M sucrose/20 mM Tris (pH 7.5)/20 mM MgCl
2/10 mM KCl/2 mM DTT/250 μM ATP, CTP, and UTP/ 2 μM α
32P-GTP for 15 min, then chased for 5 min with cold GTP as previously described [
19]. Mitochondria were pelleted and total RNA was isolated with Zymo Spin IIC columns under conditions specified by the supplier to remove unincorporated nucleotides. After a 3 h pre-incubation of dot blot filters in hybridization buffer (0.5 M sodium phosphate (pH 7.2)/1% BSA/15% formamide/7% SDS/I mM EDTA), dot blots were incubated overnight with a fresh solution of hybridization buffer containing 4 × 10
5 cpm labeled RNA per blot. Filters were washed once with 5X SSC/0.1% SDS, and twice with 2X SSC/0.1% SDS prior to visualization using a Typhoon phosphorimager (GE Healthcare, Life Sciences, Pittsburgh, PA, USA). Relative signal intensities were measured using ImageQuant software (Molecular Dynamics version 5.2, GE Healthcare, Life Sciences, Pittsburgh, PA, USA).
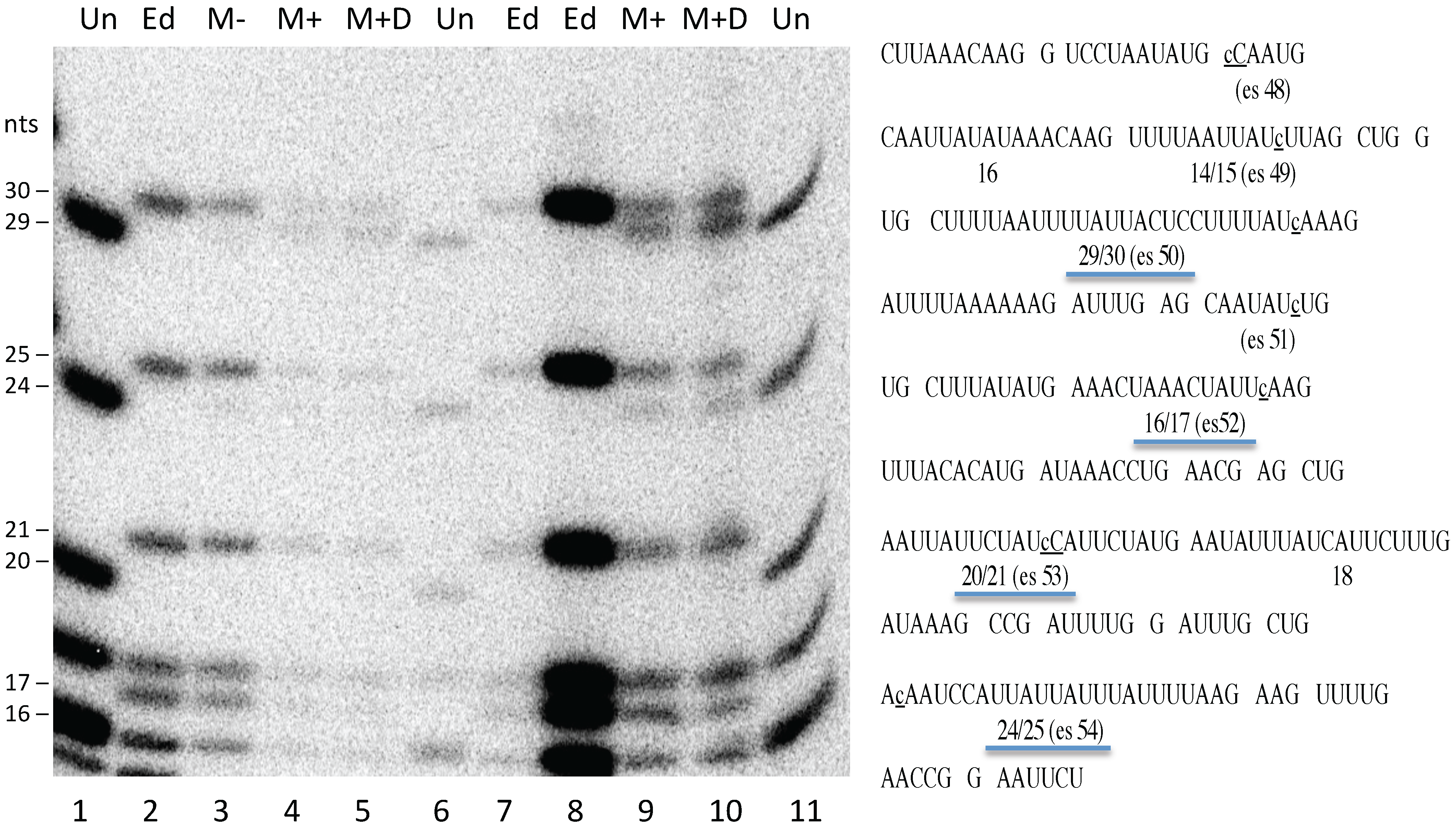
2.7. RNA Editing Assay
The presence of fully edited mitochondrial transcripts precludes assaying editing of newly synthesized RNA from endogenous genes via bulk RNA analysis, necessitating the use of labeling strategies. Transcripts were labeled under the same conditions used for the dot blot experiments, mitochondria were pelleted and total RNA was isolated with Zymo Spin IIC (Zymo Research, Irvine, CA, USA) columns. S1 nuclease protection and RNase T1 digestions were carried out as described by [
34], using single stranded DNA complementary to the 3′ portion of the
atpA mRNA as described in [
17].
4. Discussion
Run-on transcripts synthesized in
P. polycephalum mitochondria isolated via differential centrifugation with or without subsequent gradient purification are completely edited at most insertion sites under a range of transcription conditions [
19,
35,
37,
38]. However, mitochondrial yields are quite low upon secondary purification on Percoll gradients. In order to obtain sufficient quantities of mitochondria for electroporation experiments, a new purification scheme was developed. This involved the use of gentler lysis conditions, direct layering of filtered lysates onto Percoll step gradients, and minimization of applied centrifugal force (see Materials and Methods). Mitochondria purified in this way are free of contaminating nuclei, membranes, and pigment granules (
Figure 1), are capable of taking up both linear and circular DNAs (
Figure 1 and
Figure 2), express the normal complement of endogenous genes (
Figure 3), and synthesize edited transcripts (
Figure 4).
Electroporation methods have been developed for plant mitochondria from both monocots and dicots. Plasmid constructs introduced into mitochondria isolated from wheat embryos [
23], etiolated maize seedlings [
29], and potato tubers [
31] support transcription, splicing, and C to U editing, providing a means of investigating editing signals [
22,
25,
26] and the interplay between different forms of RNA processing [
27,
29,
30,
31]. The studies in plant mitochondria utilized two distinct sets of electroporation and expression conditions, and variations of each were tested using isolated
P. polycephalum mitochondria. None of these conditions supported editing of transcripts derived from DNA constructs introduced into
P. polycephalum mitochondria. Switching to conditions known to support insertion editing in
P. polycephalum mitochondria also failed to result in editing of transcripts from exogenous genes (
Figure S1), despite the fact that native transcripts were largely edited under the same conditions (
Figure 4). Curiously, although transcription from the mitochondrial genome was unaffected (
Figure 3), electroporation reduced the level of editing at some sites but not others within the endogenous transcripts, although all sites examined were edited to at least 50%. The reason for this is not clear, given that all sites were fully edited in untreated mitochondria processed in parallel (
Figure 4). The finding that cells electroporated in the absence or presence of DNA both yielded the same editing pattern, with some sites being fully edited, argues against the possibility that editing is only affected in the ~85% of mitochondria that have taken up DNA.
The inability of DNA introduced into isolated
P. polycephalum mitochondria to support editing may be due to the lack of associated editing factors. We have previously demonstrated that insertion editing requires a factor or factors associated with the
P. polycephalum mitochondrial genome [
39]. Fractionation of mitochondrial lysates over a gel filtration column yields crude mitochondrial transcription elongation complexes (mtTECs) that are editing competent [
18]. The DNA in these complexes can be digested with restriction enzymes and the resulting fragments can be ligated to create chimeric templates. RNAs produced from intra- and inter-genic fusions are edited to the same extent as RNAs produced by mtTECs. However, when exogenous DNA fragments are ligated to digested mtTEC fragments, the portion of the run-on RNAs produced from the native template is edited, but the portion derived from the added DNA is not. Importantly, the same is true when deproteinized fragments of
P. polycephalum mitochondrial DNA are fused to mtTEC templates, strongly suggesting a role for a DNA-bound factor(s) [
39].
The lack of editing in transcripts derived from exogenous templates is unlikely to be due to context effects based on our previous work with chimeric templates, which utilized dozens of intra- and intergenic chimeras, including rRNA–rRNA, mRNA–mRNA, and rRNA–mRNA fusions [
20,
39,
40]. Using chimeric templates with junctions very close to editing sites, we demonstrated that the
cis-acting elements required for the insertion of non-encoded nucleotides are limited to template sequences within 9 bp upstream and 9–10 bp downstream of an editing site [
20]. Template changes outside of this critical region had no effect on the level of editing at any site within the chimeric RNAs, many of which contained sequences derived from the
LSU gene. Thus, even if inclusion of
LSU sequences upstream of the
cox1 region resulted in inhibitory RNA structures, there is no reason to believe that all 20 of the downstream editing sites would be negatively affected.
The work presented here was initiated to determine whether a lack of editing factors could be overcome by expressing exogenous DNA templates within the confines of isolated mitochondria. The finding that editing was not observed when either linear or circular DNA was introduced into mitochondria and transcribed under a variety of conditions may be due to the absence of some component needed for de novo assembly of editing complexes. Additional conditions, such as inclusion of amino acids to support new protein synthesis, will be explored in future studies.


{kind=link}
{kind=link}
{kind=link}
{kind=link}