Changing Histopathological Diagnostics by Genome-Based Tumor Classification

1
Institute of Pathology, University Hospital Cologne, Kerpener Str. 62, Cologne D-50937, Germany
2
Center for Integrated Oncology Cologne-Bonn, Cologne D-50937, Germany
*
Author to whom correspondence should be addressed.
Genes 2014, 5(2), 444-459; https://doi.org/10.3390/genes5020444
Submission received: 7 January 2014
/
Revised: 5 May 2014
/
Accepted: 8 May 2014
/
Published: 28 May 2014
(This article belongs to the Special Issue Grand Celebration: 10th Anniversary of the Human Genome Project)
Abstract
:Traditionally, tumors are classified by histopathological criteria, i.e., based on their specific morphological appearances. Consequently, current therapeutic decisions in oncology are strongly influenced by histology rather than underlying molecular or genomic aberrations. The increase of information on molecular changes however, enabled by the Human Genome Project and the International Cancer Genome Consortium as well as the manifold advances in molecular biology and high-throughput sequencing techniques, inaugurated the integration of genomic information into disease classification. Furthermore, in some cases it became evident that former classifications needed major revision and adaption. Such adaptations are often required by understanding the pathogenesis of a disease from a specific molecular alteration, using this molecular driver for targeted and highly effective therapies. Altogether, reclassifications should lead to higher information content of the underlying diagnoses, reflecting their molecular pathogenesis and resulting in optimized and individual therapeutic decisions. The objective of this article is to summarize some particularly important examples of genome-based classification approaches and associated therapeutic concepts. In addition to reviewing disease specific markers, we focus on potentially therapeutic or predictive markers and the relevance of molecular diagnostics in disease monitoring.
1. Approaches to a Genome-Based Tumor Classification
1.1. The 2008 WHO Classification of Hematological Malignancies
The 2008 WHO classification of chronic myeloid malignancies is at present the most evolved approach to a taxonomy considering defined molecular aberrations [1]. The malignancies that are included are now classified into five categories:
- (1)
- Acute myeloid leukemia (AML) and related precursor neoplasms;
- (2)
- Myelodysplastic syndromes (MDS);
- (3)
- Myeloproliferative neoplasms (MPN);
- (4)
- Myelodysplastic/Myeloproliferative neoplasms (MDS/MPN);
- (5)
- Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1.
The integration of histology and genetics is particularly visible in the category of myeloproliferative neoplasms (MPN). Classification of specific entities into MPN is dependent on presence or absence of BCR-ABL1, the disease-causing translocation in CML [2]. The first description of the associated karyotype t(9;22)(q34;q11), according to an abnormally short chromosome 22, was published as early as 1960 and is widely known as the Philadelphia (Ph) chromosome [3]. Due to the high specificity of BCR-ABL1, its detection is mandatory for diagnosis of CML and is further underscored by the influence on therapy with the small molecule tyrosine kinase inhibitor (TKI) Imatinib [4]. Nevertheless, the remarkable journey, from the genomic aberration to the specific therapy, required approximately forty years and started long before the elucidation of the human genome (Figure 1).
Figure 1.
Timeline of the elucidation of genomic alterations in myeloproliferative neoplasms. Major breakthroughs in the understanding of the Ph+ neoplasm CML are depicted below the line, those in the understanding of Ph- neoplasms above. Note the significant impact of the human genome project on the elucidation on Ph- specific genomic alterations.
Figure 1.
Timeline of the elucidation of genomic alterations in myeloproliferative neoplasms. Major breakthroughs in the understanding of the Ph+ neoplasm CML are depicted below the line, those in the understanding of Ph- neoplasms above. Note the significant impact of the human genome project on the elucidation on Ph- specific genomic alterations.
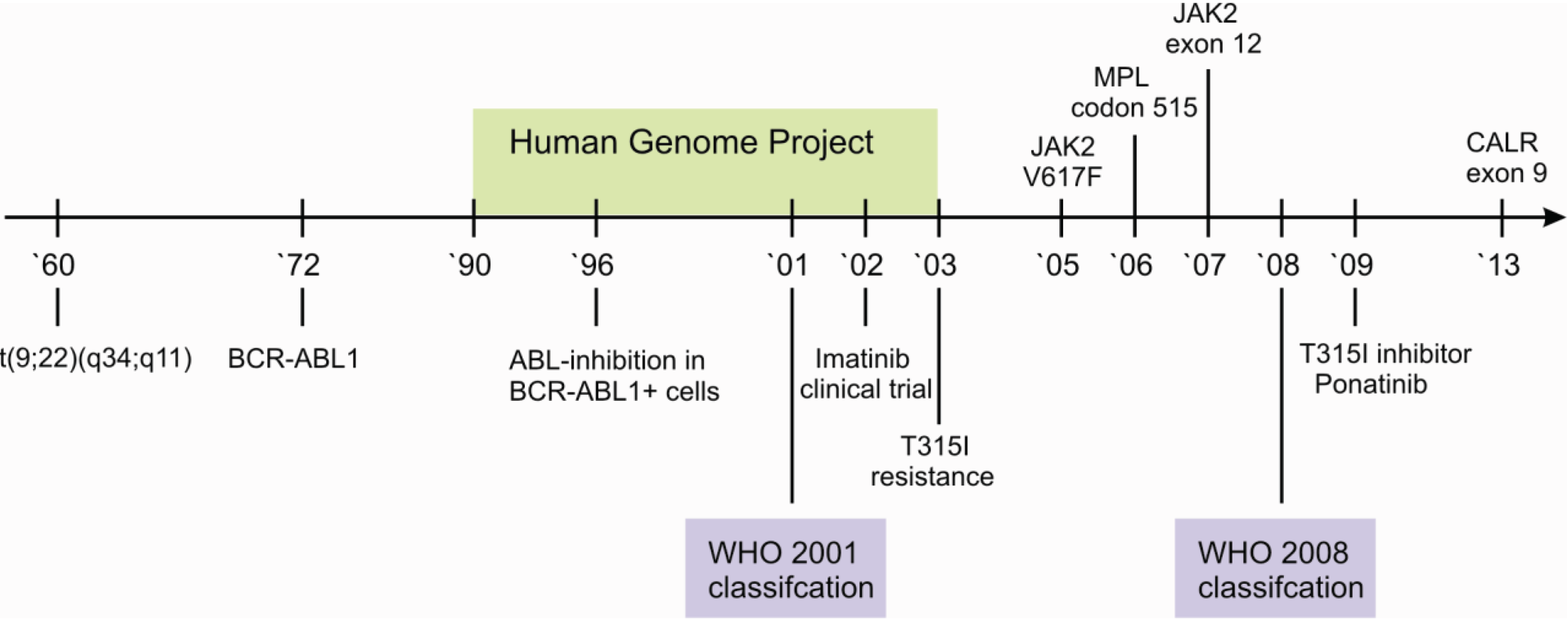
Further myeloproliferative neoplasms are characterized by the absence of BCR-ABL1, including the three Ph-negative classic MPNs—polycythemia vera (PV), essential thrombocythemia (ET) and progressive myelofibrosis (PMF). Despite the absence of BCR-ABL1, it has consecutively been shown that these MPNs are themselves also characterized by additional recurrent aberrations [5,6] (Figure 1).
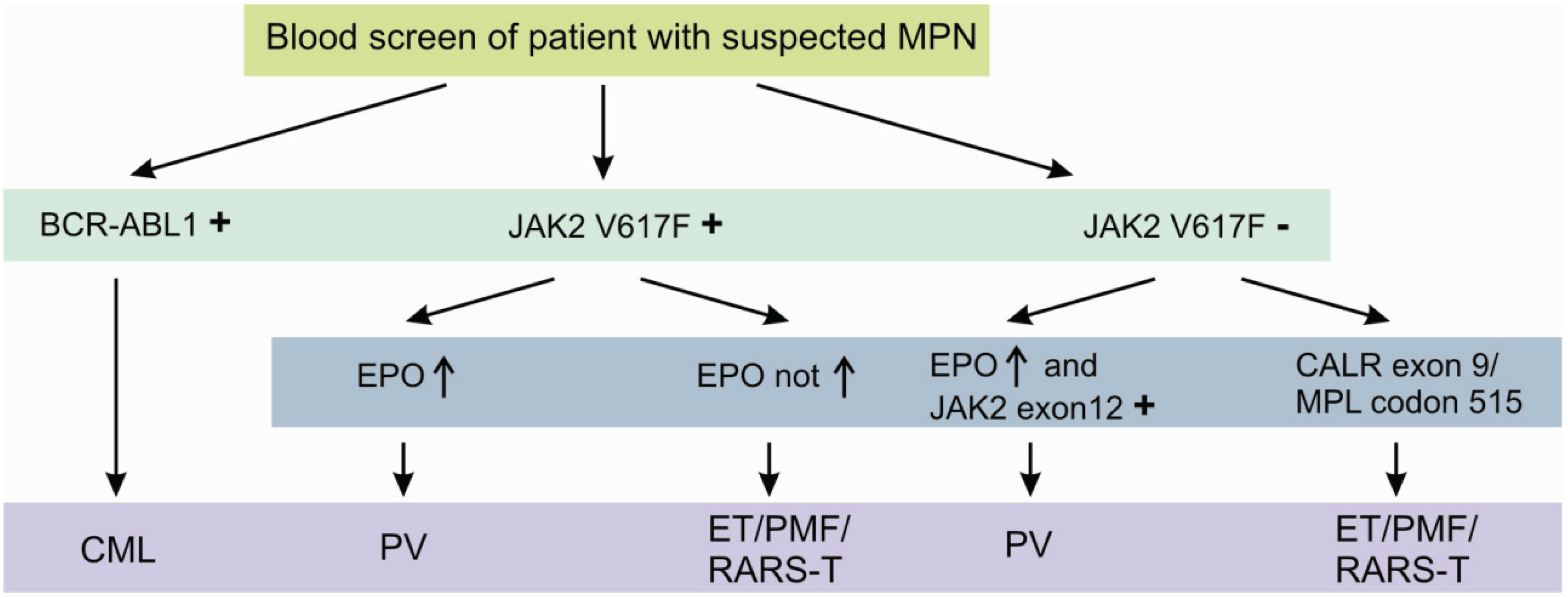
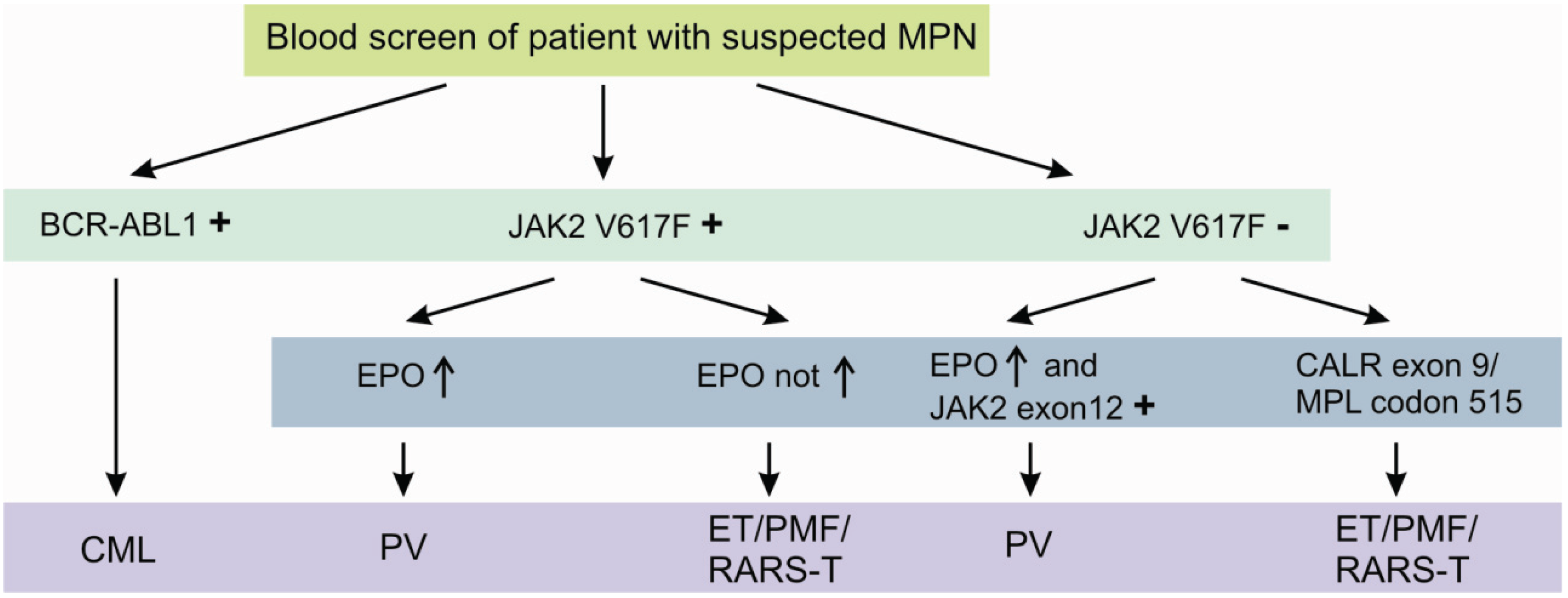
The JAK2 V617F mutation is detectable in approximately 95% of all PV patients and 50% of both ET and PMF patients [7,8,9]. It is further detected in patients with refractory anemia with ring sideroblasts and thrombocytosis (RARS-T), but in less than 5% of patients with acute myeloid leukemia (AML) or myeloid dysplastic syndrome (MDS) and not in solid tumors [10,11]. Although, the diagnostic process is initially dominated by peripheral blood cell count and serum erythropoetin (EPO) levels, JAK2 V617F or less common JAK2 Exon 12 mutations [12] confirm the diagnosis of a suspected PV without the need of a bone marrow biopsy [13]. Beside the two most important genetic aberrations in the diagnostic algorithm of MPNs, namely BCR-ABL1 and JAK2 V617F, several other potentially helpful recurrent aberrations are known. These include the presence of recurrent mutations in CALR exon 9 [14] and MPL codon 515 [15], essentially occurring in JAK2 V617F negative cases (Figure 2).
Figure 2.
Diagnostic algorithm of classic myeloproliferative neoplasms using specific molecular aberrations. Detection of the molecular aberrations depicted above is highly suggestive for the suspected myeloproliferative disorder. Nevertheless, at least in the case of absence of these specific aberrations, a bone marrow biopsy should be performed.
Figure 2.
Diagnostic algorithm of classic myeloproliferative neoplasms using specific molecular aberrations. Detection of the molecular aberrations depicted above is highly suggestive for the suspected myeloproliferative disorder. Nevertheless, at least in the case of absence of these specific aberrations, a bone marrow biopsy should be performed.
Another important example of the integration of molecular aberrations in the 2008 WHO classification of hematological malignancies is the newly introduced group of myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. This reclassification highlights the consideration of the targetable alterations FIP1L1-PDGFRA or PDGFRB-rearrangements and those harboring FGFR1-rearrangements, indicating response or resistance to Imatinib [2,16].
1.2. Lung Cancer as a Paradigm: Advances in the Molecular Characterization of Solid Malignancies
The ongoing comprehensive characterizations of solid tumors, such as those conducted by The Cancer Genome Atlas (TCGA) will significantly impact upcoming tumor classifications. This also includes lung cancer, the leading cause of cancer death worldwide [17] and an example for substantial advances by genome-based therapy approaches [18]. In general, NSCLC is subclassified into adenocarcinoma, squamous cell carcinoma and large cell carcinoma. Future classifications need to characterize clinically relevant subtypes, instead of the traditional distinction of non-small cell lung cancers (NSCLC) and small-cell lung cancer (SSLC). Concepts for a reclassification of lung adenocarcinoma were suggested, particularly with respect to reclassify large cell carcinoma based on genomic aberrations into adenocarcinoma, squamous cell carcinoma and large cell neuroendocrine carcinoma, respectively. Recommendations for (immuno)histological diagnostic work-up, and also for determining specific molecular aberrations in lung adenocarcinoma subtypes have been published [19].
Major rationales for changes in adenocarcinoma histologic variants are that the invasive mucinous type shows frequent mutations in KRAS and hardly any in EGFR, whereas non-mucinous adenocarcinoma of predominant lepidic subtype is characterized by frequent mutations in EGFR and fewer in KRAS [20]. In this context, it is worth noting and described in more detail below, that clinical studies with the TKIs Gefitinib and Erlotinib showed significantly improved survival in patients suffering from lung adenocarcinoma harboring mutations in the kinase domain of EGFR [21,22]. Besides the mutations in EGFR and KRAS, the targetable translocation EML4-ALK recurrently occurs in lung adenocarcinoma [23] and is most frequent in tumours with mucinous signet-ring appearance. Clinical trials using the tyrosine kinase inhibitors Crizotinib and Ceritinib have now shown improved progression-free survival in those patients [24,25].
In contrast, squamous cell carcinoma of the lung is commonly associated with mutations in the NFE2L2/KEAP-axis [26] and often harbors targetable alterations of FGFR1 [27] and in fewer cases mutations in DDR2. Interestingly, DDR2-transformed cell lines maintain SRC phosphorylation and are sensitive to Dasatinib [28], proved by the response in squamous cell lung cancer patients [29].
Beyond the combined loss of RB1 and TP53 in neuroendocrine pulmonary tumors [30], it was shown that low- and intermediate-grade pulmonary cacinoids harbor recurrent mutations in chromatin remodeling complexes [31], whereas the high-grade neuroendocrine tumor, small cell lung cancer, is associated with sequential changes, including the deletion of PTEN [32].
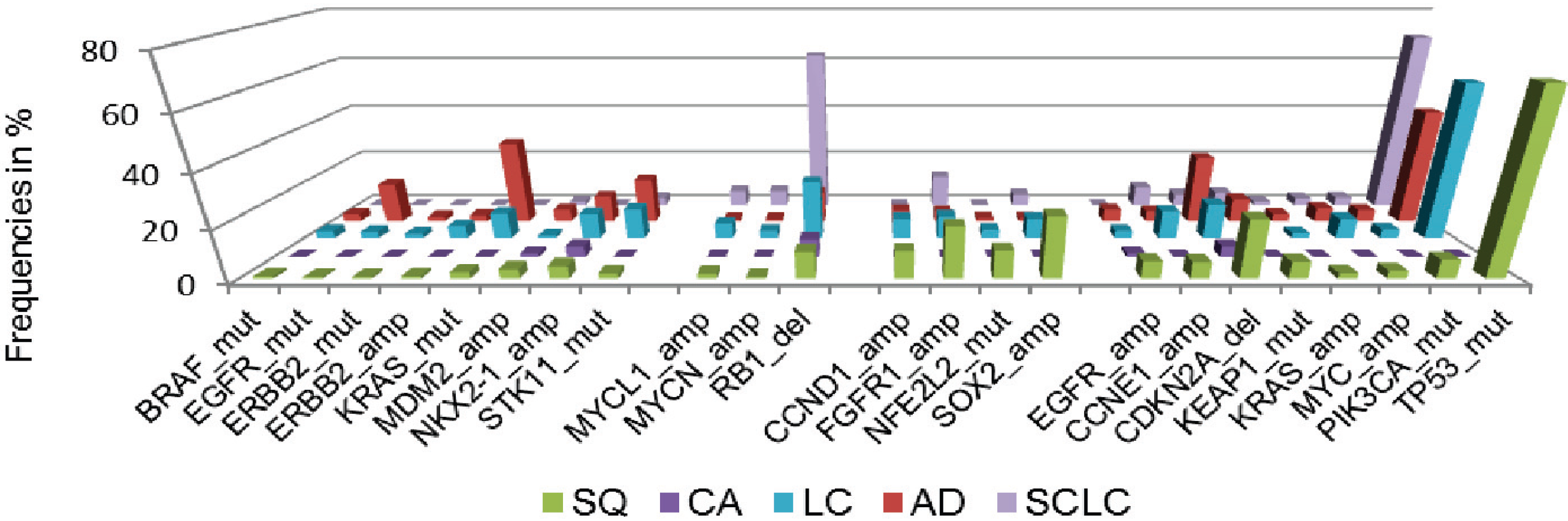
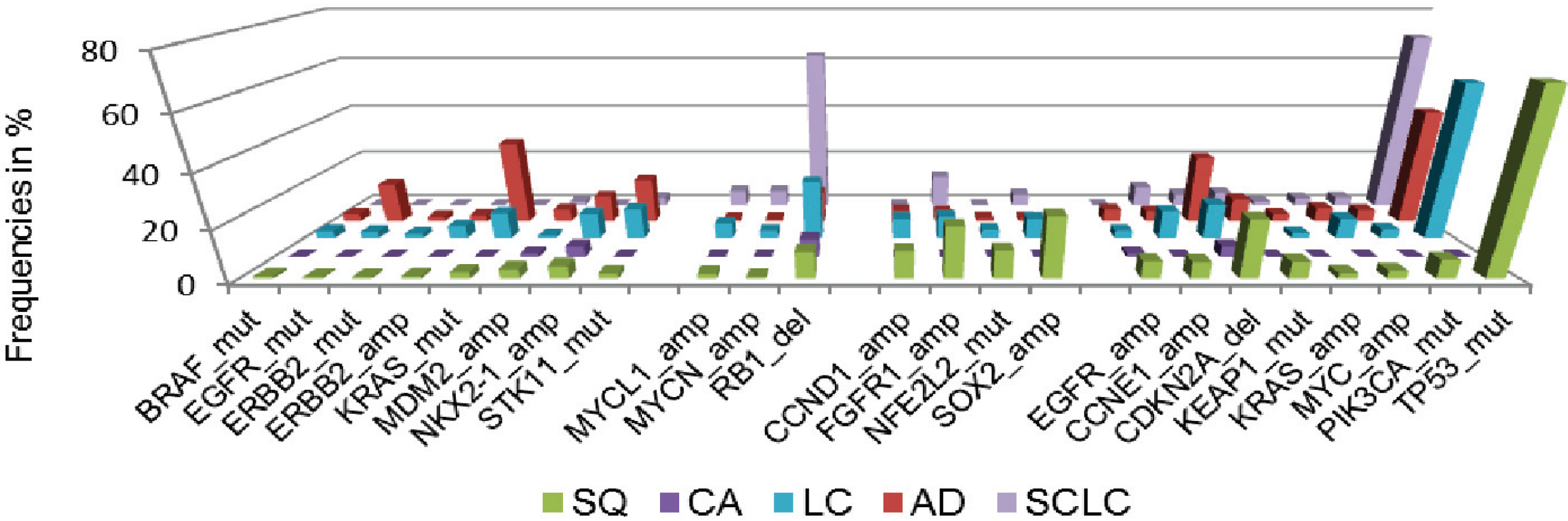
Additionally, we assessed cancer genome alterations linked to histomorphological and immunohistochemical features, considering high therapeutic relevance and improved patient outcome [33] (Figure 3). By this approach, we devised a genomic-based prediction model of lung cancer subtypes. This model shows that the majority of large cell cancers could be reassigned to adenocarcinoma, squamous cell carcinoma or small-cell lung cancer. By the combined analysis of immunohistochemical, genomic and clinical features it becomes further evident that personalized approaches significantly improve the outcome of patients with advanced lung cancer and other solid cancers.
Figure 3.
Frequencies of significant genomic alterations in histological subgroups of lung cancer. Colors of histological subtypes are encoded as follows: green—squamous cell lung cancer (SQ), purple—carcinoid tumor, light blue—large cell lung cancer (LC), red—adenocarcinoma of the lung (AD), dark blue—small cell lung cancer (SCLC). Data adapted from [33].
Figure 3.
Frequencies of significant genomic alterations in histological subgroups of lung cancer. Colors of histological subtypes are encoded as follows: green—squamous cell lung cancer (SQ), purple—carcinoid tumor, light blue—large cell lung cancer (LC), red—adenocarcinoma of the lung (AD), dark blue—small cell lung cancer (SCLC). Data adapted from [33].
Beyond the principally well-characterized situation in lung cancer, we already know of highly recurrent genomic alterations in many other solid malignancies. Prostate cancer is the most common cancer in men and is characterized by fewer mutations in typical cancer genes, when compared to other solid cancers [34,35,36]. However, genomic alterations in androgen signaling, the rearrangement of ETS transcription factors, especially the fusion of TMPRSS2-ERG [37], as well as the deletion of PTEN [38] are known to be highly recurrent in primary cancers [39], early-onset cancers [40] and castration-resistant prostate cancers [41]. Beside the outstanding therapeutic role of androgen deprivation, recent efforts investigate a mechanistic rationale of PARP inhibition in ETS-rearranged prostate cancers [42].
1.3. EWS and the Importance of Translocations in the Diagnostic Workup of Mesenchymal Malignancies
Tumors ascribed to the family of Ewing’s sarcomas or primitive neuroectodermal tumors (PNET) are the second most common bone tumors in children, but can also arise from any other tissue. The recent molecular understanding of these aggressive tumors has greatly advanced new therapeutic approaches. However, whereas chemotherapy improved the survival rate from 10% to 70%–80% in localized disease, the survival of patients with distant metastases is still poor [43]. The WHO classification of Ewing sarcoma (ES) as a single entity is underlined by its cytogenetic signature t(11;12)(q24;q12), according to the translocation EWSR1-FLI1 in approximately 85% of patients. Although, EWSR1 translocations can be found in many other mesenchymal tumors, almost all of the remaining ES cases are characterized by further ES-specific EWSR1 translocations [44,45]. This also includes the second most common cytogenetic aberration t(21;22)(q22;q12), corresponding to EWSR1-ERG [46] (Table 1).
Intriguingly, the well-characterized molecular situation of ES is in contrast to the fact that the cell of origin of the small round cell appearing tumor is not known. A further complication of the histological diagnosis is caused by atypical ES, including large/epithelioid/clear/spindle cell ES, vascular-like ES, adamantinoma-like ES, ES with neuroectodermal features, synovial sarcoma-like PNET and sclerosing PNET [47]. Beyond this difficult histopathological classification, sarcomas may be genetically classified from a near-diploid karyotpe to a more complex genomic instability. The first is characterized by highly recurrent translocations, the latter by numerical and structural abnormalities affecting multiple chromosomes [48]. In the case of small round cell appearing sarcoma, recently, new subtypes were defined by recurrent translocations of BCOR-CCNB3 [49] and CIC-DUX4 [50].
Furthermore, we already know several diagnostically relevant genomic rearrangements in soft tissue malignancies [51]. The recurrently occurring reciprocal translocation t(X;18) is characteristic of synovial sarcoma, and leads to the potentially therapeutic relevant SSX-S18 protein [52]. The translocation FUS-CHOP is detected in myxoid liposarcoma [53] and recurrent amplifications of the E3-Ubiqutitin ligase MDM2, as well as CDK4, in well differentiated and dedifferentiated liposarcoma [54,55]. Interestingly, ongoing efforts investigate the reactivation of p53 by MDM2-p53 interaction inhibitors [56].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histological type | Translocation | EWS-rearrangements |
|---|---|---|
| Ewing’s Sarcoma | t(11;22)(q24;q12) | EWSR1-FLI1 |
| t(21;22)(q22;q12) | EWSR1-ERG | |
| t(7;22)(q22;q12) | EWSR1-ETV1 | |
| t(17;22)(q21;q12) | EWSR1-ETV4 | |
| t(2;22)(q36;q12) | EWSR1-FEV | |
| inv(22)(q12q12) | EWSR1-PATZ1 | |
| t(2;22)(q31;q12) | EWSR1-SP3 | |
| t(20;22)(q13;q12) | EWSR1-NFATC2 | |
| t(4;22)(q31;12) | EWSR1-SMARCA5 | |
| t(17;22)(q12;q12) | EWSR1-E1AF | |
| inv(22)(q21;12) | EWSR1-ZSG | |
| Angiomatoid fibrous histiocytoma | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Malignant gastrointestinal neuroectodermal tumor | t(12;22)(q13;q12) | EWSR1-ATF1 |
| t(2;22)(q33;q12) | EWSR1-CREB1 | |
| Myoepithelial tumor of soft tissue and bone | t(1;22)(q23;q12) | EWSR1-PBX1 |
| t(19;22)(q13;q12) | EWSR1-ZNF444 | |
| t(6;22)(p21;q12) | EWSR1-POU5F1 | |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1-NR4A3 |
| Myxoid liposarcoma | t(12;22)(q13;q12) | EWSR1-DD1T3 |
1.4. Cancer of Unknown Primary Origin (CUP)
Carcinoma of an unknown primary origin (CUP) is descriptive of a metastatic cancer without an identifiable primary tumor site. CUP accounts for 3%–5% of all cancer diagnoses and is usually characterized by an aggressive metastatic growth and a challenging clinical presentation. In theory, CUP could be considered as a unique biological entity, or in the opposite view, as a group of different entities. The classification of CUP is essentially based on the prognostic outcome, thereby distinguishing favorable and unfavorable carcinomas [57,58,59] (Table 2).
| Clinically favorable CUP | Clinically unfavorable CUP |
|---|---|
| Extragonadal germ-cell cancer | Metastatic adenocarcinoma |
| Peritoneal papillary adenocarcinoma | Non papillary malignant ascites |
| Adenocarcinoma in axillary lymph nodes | Multiple cerebral metastases |
| Cervical squamous-cell carcinoma | Squamous-cell carcinoma of the abdominopelvic cavity |
| Neuroendocrine carcinoma | Lytic bone metastases |
| Blastic bone metastases and PSA elevation |
This classification predominantly depends on the morphological and immunohistochemical appearance. At present, the specimen is investigated by the use of specific antibodies (investigated epitopes in parentheses):
- (1)
- Identification of the cancer type:Carcinoma CK AE1/3), mesothelioma (Calretinin, BerEP4), sarcoma (Vimentin), lymphoma (LCA), melanoma (HMB-45, MITF, S100);
- (2)
- Identification of the subtype:Adenocarcinoma CK7, CK20), squamous cell carcinoma (CK5/6, p40, p63), hepatocellular carcinoma (Hepar1), renal cell carcinoma (RCC, PAX8, CA9), urothelial carcinoma (GATA3, S100P, Uroplakin), thyroid cancer (hTG, TTF1), neuroendocrine cancer (CD56, Synaptophysin, ChromoA), germ-cell tumor (PLAP);
- (3)
- Identification of the origin:Lung TTF1, NapsinA), colorectal cancer (CDX2, CK20), breast (ER, PR), pancreas (CDX2, CK7, CK20), ovary (Ca125, ER, WT1), prostate (PSA, PSAP, AR).
However, only 30% of all cases studied can be assigned to a primary tumor and even fewer benefit from a change in therapy regimen. Over the last decade, the molecular characterization by gene-expression profiling in various tumor types has led to the development of several gene signature assays with identification rates of a putative tissue of origin in up to 90% of cases. Furthermore, it was shown that CUP can be treated along actionable genomic alterations and recent whole exome sequencing revealed the existence of known recurrent mutations [60]. These include therapeutically relevant mutations in PIK3CA, MET, FGFR3, IDH1 as well as several others and it has been already demonstrated that targeted therapies can significantly influence a more favorable outcome in CUP patients [61]. As another variation on this theme, a drug-sensitizing genome alteration in one tumor type may not confer drug susceptibility in another histology, as has been observed in the case of BRAF mutations that confer MEK and BRAF dependency in melanomas [62,63] but not in colorectal carcinomas resulting from EGFR activation [64]. These interactions highlight the need for classifications integrating cancer genome alterations, but also histomorphological and immunohistochemical features.
2. Monitoring of Malignancies
Molecular monitoring of CML is the most advanced routinely used surveillance strategy and reflects the need for standardization and quality controls of diagnostic tests. A main objective is the identification of patients, which have a worse response or resistance to therapy with tyrosine kinase inhibitors. The quantification of BCR-ABL1 transcripts in peripheral blood is thereby of outstanding value in the early phase after initial drug administration, corresponding to the molecular response rate (MMR) upon TKI therapy [65,66,67]. The MMR is calculated in relation to a control gene (e.g., BCR or ABL1) and was standardized between laboratories using the international scale (IS) [68]. Essentially, the WHO has undertaken extensive efforts to simplify and standardize the assay by providing reference reagents. The importance of such efforts is reflected by a comparable poor overall survival of patients with an early MMR >10%, in turn leading to universally valid changes in the associated NCCN and ELN guidelines [66].
Despite significant progress in therapy of lung adenocarcinoma, all patients with EGFR mutations and ALK or ROS1 translocations receiving specific tyrosine kinase inhibitors will ultimately experience relapse. Recent work highlights the potential of noninvasive detection and monitoring of resistance mutations in free circulating plasma DNA of lung cancer patients. The most prominent example is the known resistance mechanisms mediated by T790M in EGFR [69]. Beside the potential of directly targeting cancers harboring T790M by new tyrosine kinase inhibitors [70], it is important to note that T790M cells proliferate more slowly, thereby enabling resensitizing of tumors to primarily used TKIs after temporary withdrawal of the drug. Since it is difficult in clinical practice to undertake repeat biopsies at multiple metastatic sites, noninvasive targeted sequencing techniques may further enable additional therapeutic strategies [71]. It thereby becomes evident that new diagnostic techniques will not only lead to major influences in treatment and monitoring guidelines, but will influence therapeutic guidelines and also the classification of tumors. Especially, the opportunity of non-invasive testing seems to be an attractive and emerging field in diagnostic and therapeutic concepts, as further delineated by testing for TMPRSS2-ERG in the case of suspicion of prostate cancer [72].
3. Clinical Success of Targeted Therapeutic Approaches Based on Molecular Biomarkers
3.1. BCR-ABL1
As depicted above, BCR-ABL1 is the driving lesion in CML, leading to the development of the first small molecule TKI, Imatinib [4,73]. Several multicenter studies confirmed that the overall survival of advanced-stage cancer patients in a chronic disease phase climbed from approximately 50% before 2002 up to approximately 90% after introduction of Imatinib in 2002 [74]. The success of the targeted approach in CML patients is further underscored by ongoing studies investigating the discontinuation of Imatinib [75] and the groundbreaking question of a potential cure [76]. Despite these promising developments, several further therapeutic possibilities for second line therapies are underway. These include the use of second generation TKIs in case of therapeutic failure or intolerance, e.g., Dasatinib, Nilotinib, Bosutinib and Ponatinib, the latter particularly used in case of a secondary resistance by T315I mutation [75] (Figure 1).
3.2. BRAF
The most common melanoma mutation in BRAF exon 15, the activating mutation V600E, leads to response rates of more than 50% of all patients treated with the specific TKI Vemurafenib. Further, therapy with Vemurafenib is associated with a relative reduction in death of 63% when compared to standard therapeutic regime with Dacarbazine [62]. In contrast to the activating biology of BRAF V600E, ongoing clinical trials investigate the potency of Dasatinib in tumors harboring inactivating BRAF exon 11 mutations (NCT01514864 clinicaltrials.gov).
3.3. EGFR-Family and KRAS Mutational Status
Genetic aberrations in members of the EGFR-family are well known for targeted therapies, including HER2- and EGFR-targeted inhibition of downstream signaling cascades. HER2/ERBB2 is primary known as being amplified and activated in breast cancer causing high recurrence rates and increased mortality in approximately 15% of all patients [77]. Patients with amplification of HER2/ERBB2 treated with the monoclonal antibody Trastuzumab in combination with chemotherapy showed improved outcome in several studies [78]. Beyond Trastuzumab several ongoing studies are investigating further drug therapies targeting the HER2-axis. These include the combined inhibition of HER2/HER3-heterodimerization and activation by Trastuzumab/Pertuzumab [79] and the use of the covalent immunoconjugate Trastuzumab-Emtansine (T-DM1) [80] (Figure 4). Moreover, several trials evaluate the therapeutic significance of small molecule inhibition in HER2-positive breast cancer, e.g., Lapatinib, Afatinib, Pazopanib and Neratinib [81].
Figure 4.
Predictive Biomarkers for Targeted and Selective Therapies. Signaling of EGFR-family receptors is characterized by homo-/heterodimerization and subsequent activation of the targetable downstream signaling pathways RAS/RAF and PI3K/AKT. Present therapeutic approaches focus on the inhibition of ligand-dependent activation, dimerization and receptor tyrosine kinases. Immunoconjugates, e.g., T-DM1, specifically deliver chemotherapeutic agents by the process of receptor internalization. As described in the text in more detail, ongoing efforts investigate the effectiveness of combined or dual approaches.
Figure 4.
Predictive Biomarkers for Targeted and Selective Therapies. Signaling of EGFR-family receptors is characterized by homo-/heterodimerization and subsequent activation of the targetable downstream signaling pathways RAS/RAF and PI3K/AKT. Present therapeutic approaches focus on the inhibition of ligand-dependent activation, dimerization and receptor tyrosine kinases. Immunoconjugates, e.g., T-DM1, specifically deliver chemotherapeutic agents by the process of receptor internalization. As described in the text in more detail, ongoing efforts investigate the effectiveness of combined or dual approaches.
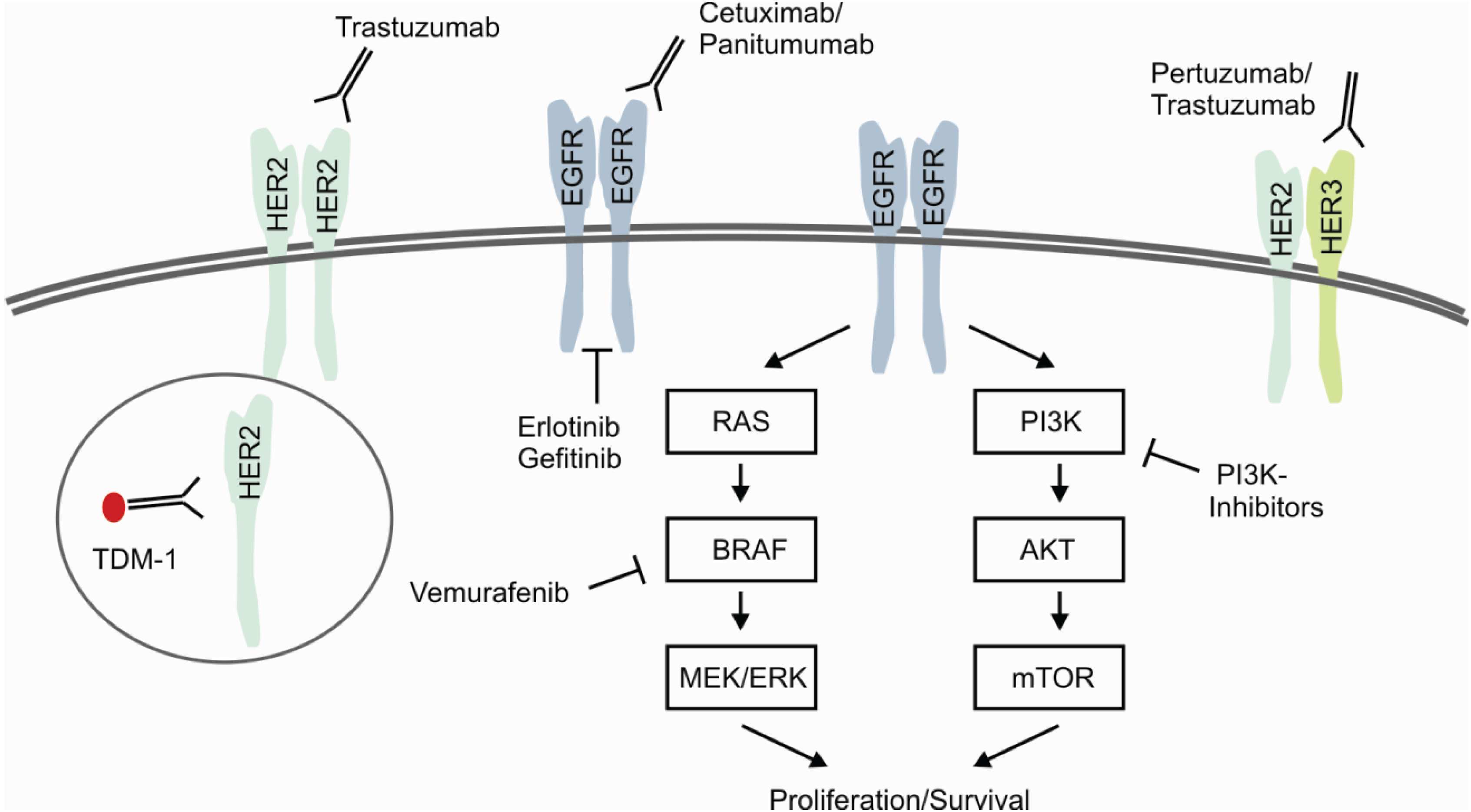
Comparable to the situation in HER2-amplified breast cancer, substantial progress has been made by the introduction of EGFR-targeted therapies in the treatment of lung cancer and colorectal cancer. These efforts become evident by comparing the median overall survival of lung adenocarcinoma patients under standard therapeutic regimes of approximately 12 months with approximately 2 years under EGFR-targeted therapy with Erlotinib or Gefitinib in EGFR-mutated cancers [82]. As depicted above, further efforts are made to overcome primary and secondary therapeutic resistance by next generation TKIs [70]. Similar positive achievements were made for treatment of metastatic colorectal cancer (mCRC) by an improvement of survival from 12 months with fluoruracil monotherapy up to approximately 2 years with EGFR/VEGFR-targeted therapy combined with chemotherapy [83].
Beyond that, it recently became evident that we need to predict therapeutic response to cetuximab/panitumumab in mCRC not only by KRAS mutational status, but also by NRAS mutational status [84], highlighting the increasing importance of mutations in downstream or interacting pathways. As depicted above, it becomes also clear that combined approaches, like the inhibition of the EGFR/BRAF-axis in BRAF V600E mutated colorectal cancers [64], could be used to overcome primary resistance in histological subtypes.
4. Conclusions
The purpose of integrating pathogenetic and molecular information into disease classification systems, exemplified by the 2008 WHO classification of hematological malignancies, reflects the high clinical relevance for predicting therapy outcome and prognosis. The human genome project and emerging technologies in the last decade have led to fundamental pathogenetic breakthroughs, which substantially improved the translation into clinical practice and individual therapeutic possibilities. Altogether, this data underlines the significant influence of cancer genomics and the substantial increase in genomic information on the process of defining tumor entities and effective and selective treatment approaches.
Author Contributions
Wrote the paper: MK and RB.
Conflicts of Interest
MK declares no conflict of interest. RB is serving on Scientific Advisory Boards and has received lecture honoraria from AstraZeneca, Qiagen, Roche, Lilly, MerckSerono, Pfizer and is a co-founder and co-owner of Targos Molecular Pathology, Inc. RB received grant support from the Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe and the Bundesministerium für Bildung und Forschung (BMBF).
References
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the world health organization (who) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Cross, N.C.; Reiter, A. Fibroblast growth factor receptor and platelet-derived growth factor receptor abnormalities in eosinophilic myeloproliferative disorders. Acta Haematol. 2008, 119, 199–206. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Kantarjian, H.; Sawyers, C.; Hochhaus, A.; Guilhot, F.; Schiffer, C.; Gambacorti-Passerini, C.; Niederwieser, D.; Resta, D.; Capdeville, R.; Zoellner, U.; et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002, 346, 645–652. [Google Scholar] [CrossRef]
- Tefferi, A.; Skoda, R.; Vardiman, J.W. Myeloproliferative neoplasms: Contemporary diagnosis using histology and genetics. Nat. Rev. Clin. Oncol. 2009, 6, 627–637. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Guglielmelli, P.; Tefferi, A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J. Clin. 2009, 59, 171–191. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase jak2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of jak2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Szpurka, H.; Tiu, R.; Murugesan, G.; Aboudola, S.; Hsi, E.D.; Theil, K.S.; Sekeres, M.A.; Maciejewski, J.P. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (rars-t), another myeloproliferative condition characterized by jak2 v617f mutation. Blood 2006, 108, 2173–2181. [Google Scholar] [CrossRef]
- Nishii, K.; Nanbu, R.; Lorenzo, V.F.; Monma, F.; Kato, K.; Ryuu, H.; Katayama, N. Expression of the jak2 v617f mutation is not found in de novo aml and mds but is detected in mds-derived leukemia of megakaryoblastic nature. Leukemia 2007, 21, 1337–1338. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. Jak2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Tefferi, A.; Vainchenker, W. Myeloproliferative neoplasms: Molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 2011, 29, 573–582. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. Mpl515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef]
- Gotlib, J. World health organization-defined eosinophilic disorders: 2011 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2011, 86, 677–688. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Buettner, R.; Wolf, J.; Thomas, R.K. Lessons learned from lung cancer genomics: The emerging concept of individualized diagnostics and treatment. J. Clin. Oncol. 2013, 31, 1858–1865. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef]
- Hata, A.; Katakami, N.; Fujita, S.; Kaji, R.; Imai, Y.; Takahashi, Y.; Nishimura, T.; Tomii, K.; Ishihara, K. Frequency of egfr and kras mutations in japanese patients with lung adenocarcinoma with features of the mucinous subtype of bronchioloalveolar carcinoma. J. Thorac. Oncol. 2010, 5, 1197–1200. [Google Scholar] [CrossRef]
- Roberts, P.J.; Stinchcombe, T.E. Kras mutation: Should we test for it, and does it matter? J. Clin. Oncol. 2013, 31, 1112–1121. [Google Scholar]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced egfr mutation-positive non-small-cell lung cancer (eurtac): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Doebele, R.C. Targeted therapies: Time to shift the burden of proof for oncogene-positive cancer? Nat. Rev. Clin. Oncol. 2013, 10, 492–493. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; de Pas, T.; et al. Ceritinib in alk-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal fgfr1 amplification associates with therapeutically tractable fgfr1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar]
- Hammerman, P.S.; Sos, M.L.; Ramos, A.H.; Xu, C.; Dutt, A.; Zhou, W.; Brace, L.E.; Woods, B.A.; Lin, W.; Zhang, J.; et al. Mutations in the ddr2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011, 1, 78–89. [Google Scholar] [CrossRef]
- Pitini, V.; Arrigo, C.; di Mirto, C.; Mondello, P.; Altavilla, G. Response to dasatinib in a patient with sqcc of the lung harboring a discoid-receptor-2 and synchronous chronic myelogenous leukemia. Lung Cancer 2013, 82, 171–172. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretic, L.; Seidel, D.; Zander, T.; Leenders, F.; George, J.; Muller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef]
- The Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci. Transl. Med. 2013, 5, 209ra153. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent spop, foxa1 and med12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. Pten, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(adp-ribose) polymerase in ets gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef]
- Ross, K.A.; Smyth, N.A.; Murawski, C.D.; Kennedy, J.G. The biology of ewing sarcoma. ISRN Oncol. 2013, 2013. [Google Scholar] [CrossRef]
- Riggi, N.; Cironi, L.; Suva, M.L.; Stamenkovic, I. Sarcomas: Genetics, signalling, and cellular origins. Part 1: The fellowship of tet. J. Pathol. 2007, 213, 4–20. [Google Scholar] [CrossRef]
- Demicco, E.G. Sarcoma diagnosis in the age of molecular pathology. Adv. Anat. Pathol. 2013, 20, 264–274. [Google Scholar] [CrossRef]
- Wang, W.L.; Patel, N.R.; Caragea, M.; Hogendoorn, P.C.; Lopez-Terrada, D.; Hornick, J.L.; Lazar, A.J. Expression of erg, an ets family transcription factor, identifies erg-rearranged ewing sarcoma. Mod. Pathol. 2012, 25, 1378–1383. [Google Scholar] [CrossRef]
- Machado, I.; Noguera, R.; Mateos, E.A.; Calabuig-Farinas, S.; Lopez, F.I.; Martinez, A.; Navarro, S.; Llombart-Bosch, A. The many faces of atypical ewing’s sarcoma. A true entity mimicking sarcomas, carcinomas and lymphomas. Virchows Arch. 2011, 458, 281–290. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef]
- Pierron, G.; Tirode, F.; Lucchesi, C.; Reynaud, S.; Ballet, S.; Cohen-Gogo, S.; Perrin, V.; Coindre, J.M.; Delattre, O. A new subtype of bone sarcoma defined by bcor-ccnb3 gene fusion. Nat. Genet. 2012, 44, 461–466. [Google Scholar] [CrossRef]
- Choi, E.Y.; Thomas, D.G.; McHugh, J.B.; Patel, R.M.; Roulston, D.; Schuetze, S.M.; Chugh, R.; Biermann, J.S.; Lucas, D.R. Undifferentiated small round cell sarcoma with t(4;19)(q35;q13.1) cic-dux4 fusion: A novel highly aggressive soft tissue tumor with distinctive histopathology. Am. J. Surg. Pathol. 2013, 37, 1379–1386. [Google Scholar] [CrossRef]
- Tanas, M.R.; Goldblum, J.R. Fluorescence in situ hybridization in the diagnosis of soft tissue neoplasms: A review. Adv. Anat. Pathol. 2009, 16, 383–391. [Google Scholar] [CrossRef]
- Trautmann, M.; Sievers, E.; Aretz, S.; Kindler, D.; Michels, S.; Friedrichs, N.; Renner, M.; Kirfel, J.; Steiner, S.; Huss, S.; et al. Ss18-ssx fusion protein-induced wnt/beta-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene 2013. [Google Scholar] [CrossRef]
- Shing, D.C.; McMullan, D.J.; Roberts, P.; Smith, K.; Chin, S.F.; Nicholson, J.; Tillman, R.M.; Ramani, P.; Cullinane, C.; Coleman, N. Fus/erg gene fusions in ewing’s tumors. Cancer Res. 2003, 63, 4568–4576. [Google Scholar]
- Leach, F.S.; Tokino, T.; Meltzer, P.; Burrell, M.; Oliner, J.D.; Smith, S.; Hill, D.E.; Sidransky, D.; Kinzler, K.W.; Vogelstein, B. P53 mutation and mdm2 amplification in human soft tissue sarcomas. Cancer Res. 1993, 53, 2231–2234. [Google Scholar]
- Pilotti, S.; Della Torre, G.; Lavarino, C.; Sozzi, G.; Minoletti, F.; Vergani, B.; Azzarelli, A.; Rilke, F.; Pierotti, M.A. Molecular abnormalities in liposarcoma: Role of mdm2 and cdk4-containing amplicons at 12q13–22. J. Pathol. 1998, 185, 188–190. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435. [Google Scholar] [CrossRef]
- Massard, C.; Loriot, Y.; Fizazi, K. Carcinomas of an unknown primary origin—Diagnosis and treatment. Nat. Rev. Clin. Oncol. 2011, 8, 701–710. [Google Scholar] [CrossRef]
- Stella, G.M.; Senetta, R.; Cassenti, A.; Ronco, M.; Cassoni, P. Cancers of unknown primary origin: Current perspectives and future therapeutic strategies. J. Transl. Med. 2012, 10, 12. [Google Scholar] [CrossRef]
- Tothill, R.W.; Li, J.; Mileshkin, L.; Doig, K.; Siganakis, T.; Cowin, P.; Fellowes, A.; Semple, T.; Fox, S.; Byron, K.; et al. Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J. Pathol. 2013, 231, 413–423. [Google Scholar] [CrossRef]
- Tan, D.S.; Montoya, J.; Ng, Q.S.; Chan, K.S.; Lynette, O.; Sakktee Krisna, S.; Takano, A.; Lim, W.T.; Tan, E.H.; Lim, K.H. Molecular profiling for druggable genetic abnormalities in carcinoma of unknown primary. J. Clin. Oncol. 2013, 31, e237–e239. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated braf in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to braf(v600e) inhibition through feedback activation of egfr. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Hanfstein, B.; Muller, M.C.; Hehlmann, R.; Erben, P.; Lauseker, M.; Fabarius, A.; Schnittger, S.; Haferlach, C.; Gohring, G.; Proetel, U.; et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (cml). Leukemia 2012, 26, 2096–2102. [Google Scholar] [CrossRef]
- Oehler, V.G. Update on current monitoring recommendations in chronic myeloid leukemia: Practical points for clinical practice. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 176–183. [Google Scholar] [CrossRef]
- Merx, K.; Muller, M.C.; Kreil, S.; Lahaye, T.; Paschka, P.; Schoch, C.; Weisser, A.; Kuhn, C.; Berger, U.; Gschaidmeier, H.; et al. Early reduction of bcr-abl mrna transcript levels predicts cytogenetic response in chronic phase cml patients treated with imatinib after failure of interferon alpha. Leukemia 2002, 16, 1579–1583. [Google Scholar] [CrossRef]
- Cross, N.C.; White, H.E.; Muller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W.; et al. Detection of mutations in egfr in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of egfr that overcomes t790m-mediated resistance in nsclc. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra168. [Google Scholar]
- Tomlins, S.A.; Aubin, S.M.; Siddiqui, J.; Lonigro, R.J.; Sefton-Miller, L.; Miick, S.; Williamsen, S.; Hodge, P.; Meinke, J.; Blase, A.; et al. Urine tmprss2:Erg fusion transcript stratifies prostate cancer risk in men with elevated serum psa. Sci. Transl. Med. 2011, 3, 94ra72. [Google Scholar]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of bcr-abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987. [Google Scholar] [CrossRef]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European leukemianet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef]
- Mahon, F.X. Is going for cure in chronic myeloid leukemia possible and justifiable? Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 122–128. [Google Scholar]
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The her-2 receptor and breast cancer: Ten years of targeted anti-her-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of her2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2012, 9, 16–32. [Google Scholar]
- Swain, S.M.; Kim, S.B.; Cortes, J.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Knott, A.; et al. Pertuzumab, trastuzumab, and docetaxel for her2-positive metastatic breast cancer (cleopatra study): Overall survival results from a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Dirix, L.; Kocsis, J.; Bianchi, G.V.; Lu, J.; Vinholes, J.; Guardino, E.; Song, C.; Tong, B.; Ng, V.; et al. Phase ii randomized study of trastuzumab emtansine versus trastuzumab plus docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer. J. Clin. Oncol. 2013, 31, 1157–1163. [Google Scholar] [CrossRef]
- Incorvati, J.A.; Shah, S.; Mu, Y.; Lu, J. Targeted therapy for her2 positive breast cancer. J. Hematol. Oncol. 2013, 6, 38. [Google Scholar] [CrossRef]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of egfr-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-folfox4 treatment and ras mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Kloth, M.; Buettner, R. Changing Histopathological Diagnostics by Genome-Based Tumor Classification. Genes 2014, 5, 444-459. https://doi.org/10.3390/genes5020444
AMA Style
Kloth M, Buettner R. Changing Histopathological Diagnostics by Genome-Based Tumor Classification. Genes. 2014; 5(2):444-459. https://doi.org/10.3390/genes5020444
Chicago/Turabian StyleKloth, Michael, and Reinhard Buettner. 2014. "Changing Histopathological Diagnostics by Genome-Based Tumor Classification" Genes 5, no. 2: 444-459. https://doi.org/10.3390/genes5020444