Genetic Profiling for Risk Reduction in Human Cardiovascular Disease

Abstract
:1. Introduction
2. Genetic Variation in CVD
2.1. The Overlap between GWAS Hits and Monogenic Disease in CVD
2.2. The Missing Heritability of GWAS
2.3. Rare Variation as a Cause of CVD
3. The Genetics of Dilated Cardiomyopathy
3.1. Next Generation Sequencing Identifies TTN as a Major Contributor to DCM
3.2. Beyond Panel Based Sequencing for Cardiomyopathy and Beyond

{kind=link}
{kind=link}
| Panel | WES | WGS | |
|---|---|---|---|
| Variation in Known Genes | yes | yes | yes |
| Novel Gene Identification | no | yes | yes |
| Structural Variation | no | limited | yes |
| Non-coding Variation | no | limited | yes |
| Repeat testing required if first pass negative | yes | yes | no |
3.3. Exome Sequencing of Multiple Family Members Improves Identification of Pathogenic Variation
3.4. Identifying Cardiomyopathy Modifier Loci Using Broad Based Sequencing
3.5. WES/WGS Can Identify New Genes for Cardiomyopathy
3.6. Limitations of WES
4. WGS as a Tool to Investigate Non-Coding Variation for CVD
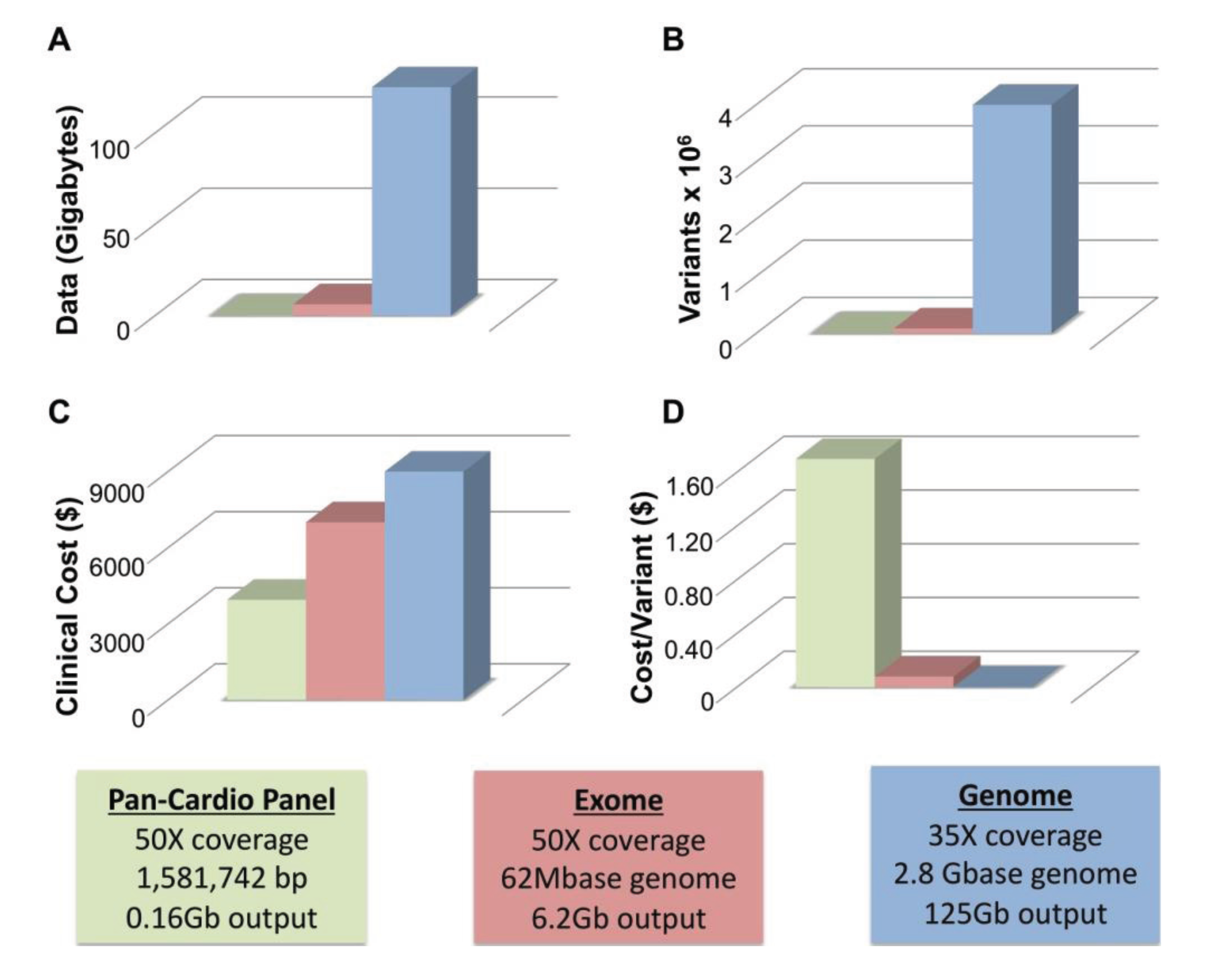
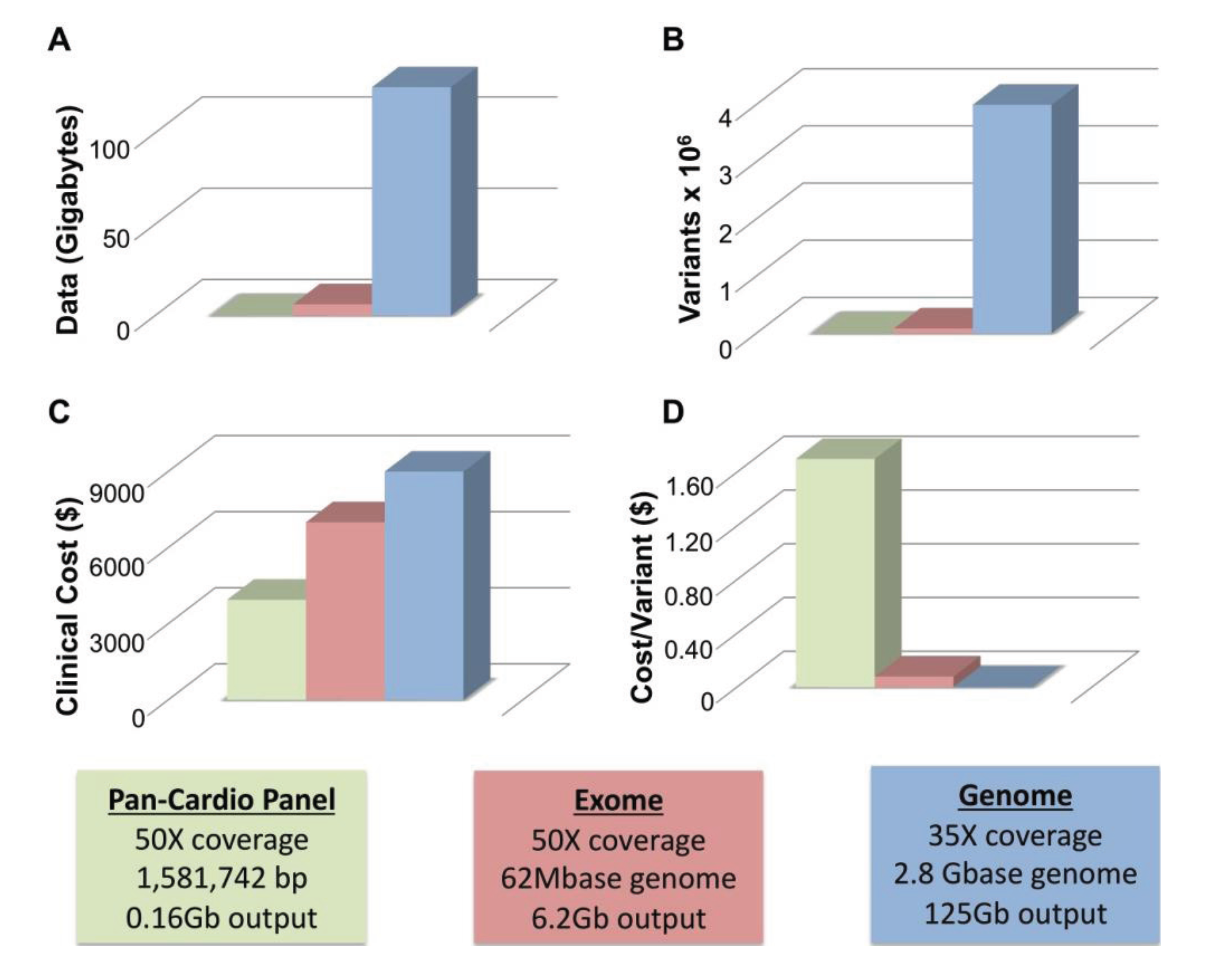
4.1. WGS Has Greater Sensitivity than WES
4.2. Limitations of WGS
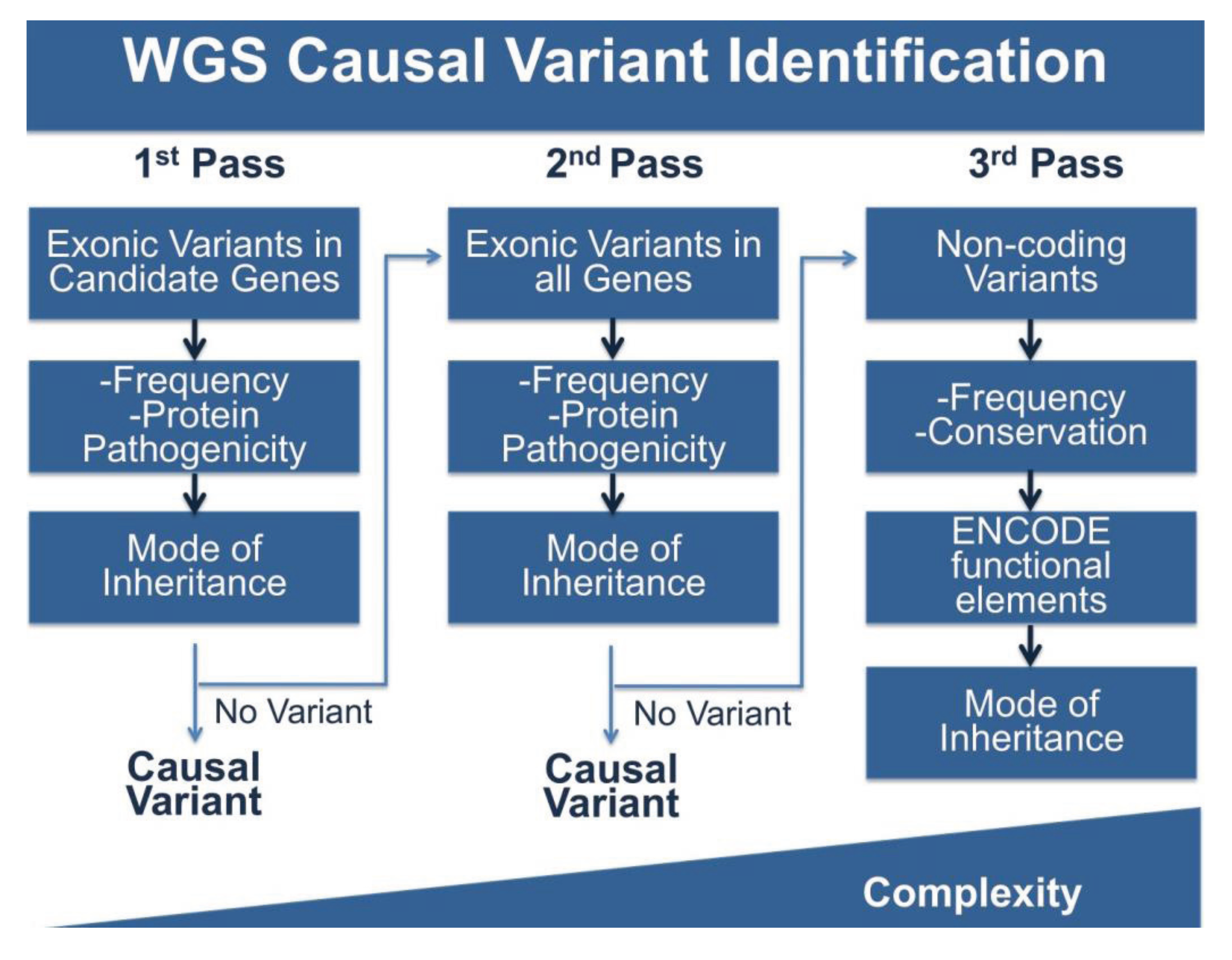
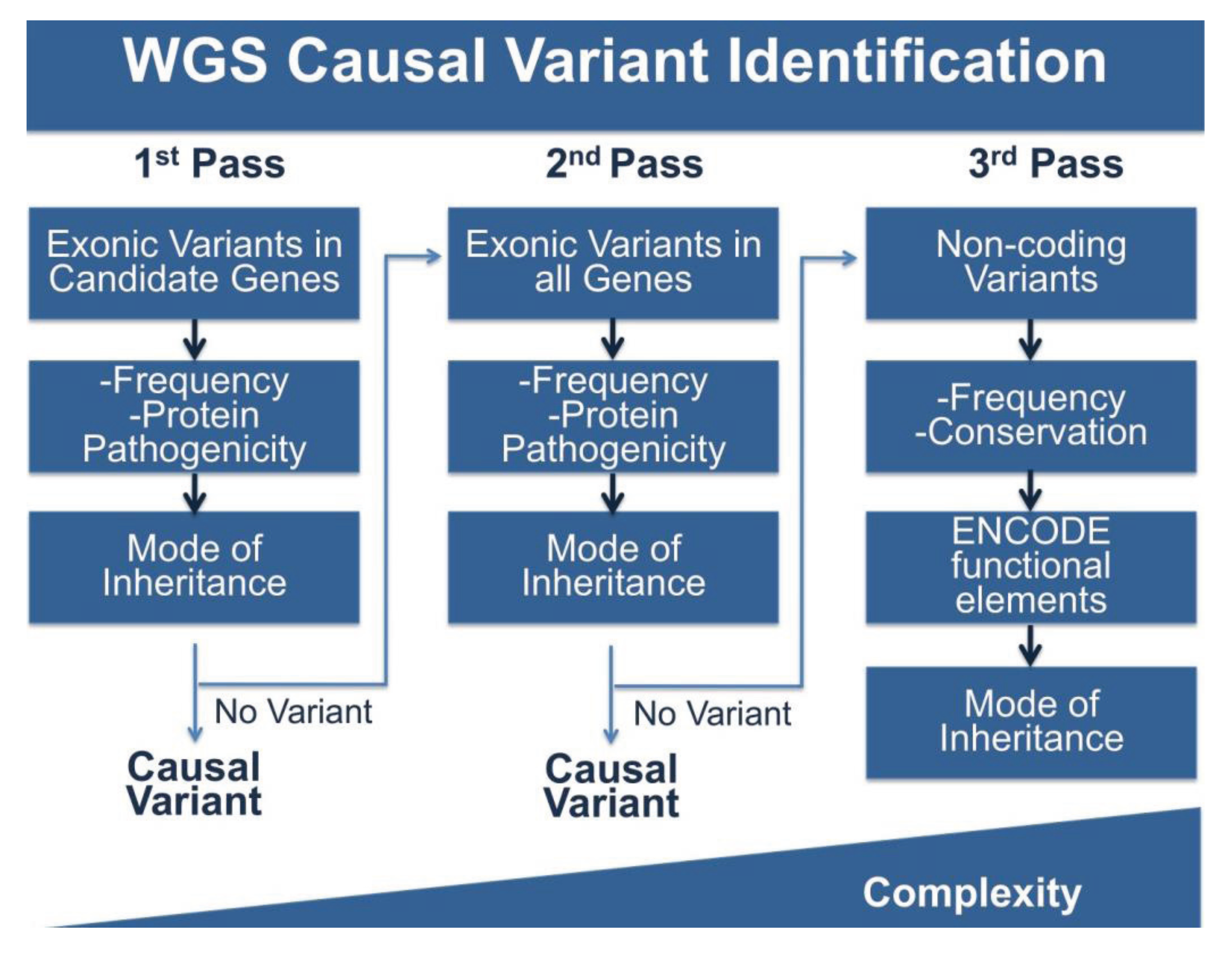
4.3. Multi-Pass Filtering Methods Allow for More Efficient Variant Identification
5. Incidental Findings and Their Importance for CVD Related Phenotypes
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Executive summary: Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar] [CrossRef]
- Marenberg, M.E.; Risch, N.; Berkman, L.F.; Floderus, B.; de Faire, U. Genetic susceptibility to death from coronary heart disease in a study of twins. N. Engl. J. Med. 1994, 330, 1041–1046. [Google Scholar] [CrossRef]
- Post, W.S.; Larson, M.G.; Myers, R.H.; Galderisi, M.; Levy, D. Heritability of left ventricular mass: The Framingham Heart Study. Hypertension 1997, 30, 1025–1028. [Google Scholar] [CrossRef]
- Adams, T.D.; Yanowitz, F.G.; Fisher, A.G.; Ridges, J.D.; Nelson, A.G.; Hagan, A.D.; Williams, R.R.; Hunt, S.C. Heritability of cardiac size: An echocardiographic and electrocardiographic study of monozygotic and dizygotic twins. Circulation 1985, 71, 39–44. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Jarcho, J.A.; McKenna, W.; Pare, J.A.; Solomon, S.D.; Holcombe, R.F.; Dickie, S.; Levi, T.; Donis-Keller, H.; Seidman, J.G.; Seidman, C.E. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 1989, 321, 1372–1378. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef]
- Lander, E.; Kruglyak, L. Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results. Nat. Genet. 1995, 11, 241–247. [Google Scholar] [CrossRef]
- International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef]
- National Human Genome Research Institute (NHGRI) Catalog of Published Genome-Wide Association Studies. Available online: http://www.genome.gov/gwasstudies/ (accessed on 25 February 2014).
- Newton-Cheh, C.; Eijgelsheim, M.; Rice, K.M.; de Bakker, P.I.; Yin, X.; Estrada, K.; Bis, J.C.; Marciante, K.; Rivadeneira, F.; Noseworthy, P.A.; et al. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat. Genet. 2009, 41, 399–406. [Google Scholar] [CrossRef]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef]
- Rampersaud, E.; Kinnamon, D.D.; Hamilton, K.; Khuri, S.; Hershberger, R.E.; Martin, E.R. Common susceptibility variants examined for association with dilated cardiomyopathy. Ann. Hum. Genet. 2010, 74, 110–116. [Google Scholar] [CrossRef]
- Tiret, L.; Mallet, C.; Poirier, O.; Nicaud, V.; Millaire, A.; Bouhour, J.B.; Roizes, G.; Desnos, M.; Dorent, R.; Schwartz, K.; et al. Lack of association between polymorphisms of eight candidate genes and idiopathic dilated cardiomyopathy: The CARDIGENE study. J. Am. Coll. Cardiol. 2000, 35, 29–35. [Google Scholar]
- Takayama, S.; Xie, Z.; Reed, J.C. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Sasse-Klaassen, S.; Probst, S.; Gerull, B.; Shin, J.T.; Toeppel, A.; Heuser, A.; Michely, B.; Yoerger, D.M.; Song, B.S.; et al. A novel locus for dilated cardiomyopathy, diffuse myocardial fibrosis, and sudden death on chromosome 10q25–26. J. Am. Coll. Cardiol. 2006, 48, 106–111. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. 2001, 17, 502–510. [Google Scholar] [CrossRef]
- Wang, W.Y.; Barratt, B.J.; Clayton, D.G.; Todd, J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005, 6, 109–118. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; Masson, G.; et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef]
- Lander, E.S. The new genomics: Global views of biology. Science 1996, 274, 536–539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Cox, N.J. The allelic architecture of human disease genes: Common disease-common variant … or not? Hum. Mol. Genet. 2002, 11, 2417–2423. [Google Scholar] [CrossRef]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2011, 13, 135–145. [Google Scholar] [CrossRef]
- Manolio, T.A. Cohort studies and the genetics of complex disease. Nat. Genet. 2009, 41, 5–6. [Google Scholar] [CrossRef]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdottir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161–1165. [Google Scholar] [CrossRef]
- Marian, A.J.; Belmont, J. Strategic approaches to unraveling genetic causes of cardiovascular diseases. Circ. Res. 2011, 108, 1252–1269. [Google Scholar] [CrossRef]
- Ng, P.C.; Levy, S.; Huang, J.; Stockwell, T.B.; Walenz, B.P.; Li, K.; Axelrod, N.; Busam, D.A.; Strausberg, R.L.; Venter, J.C. Genetic variation in an individual human exome. PLoS Genet. 2008, 4, e1000160. [Google Scholar] [CrossRef]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef]
- Johansen, C.T.; Wang, J.; Lanktree, M.B.; Cao, H.; McIntyre, A.D.; Ban, M.R.; Martins, R.A.; Kennedy, B.A.; Hassell, R.G.; Visser, M.E.; et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010, 42, 684–687. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; Ripatti, S.; Lindqvist, I.; Boomsma, D.; Heid, I.M.; Pramstaller, P.P.; Penninx, B.W.; Janssens, A.C.; Wilson, J.F.; Spector, T.; et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009, 41, 47–55. [Google Scholar] [CrossRef]
- Kathiresan, S.; Willer, C.J.; Peloso, G.M.; Demissie, S.; Musunuru, K.; Schadt, E.E.; Kaplan, L.; Bennett, D.; Li, Y.; Tanaka, T.; et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009, 41, 56–65. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinforma. 2010, 11, 473–483. [Google Scholar] [CrossRef]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Jabbari, J.; Jabbari, R.; Nielsen, M.W.; Holst, A.G.; Nielsen, J.B.; Haunso, S.; Tfelt-Hansen, J.; Svendsen, J.H.; Olesen, M.S. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ. Cardiovasc. Genet. 2013, 6, 481–489. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef]
- Golbus, J.R.; Puckelwartz, M.J.; Fahrenbach, J.P.; Dellefave-Castillo, L.M.; Wolfgeher, D.; McNally, E.M. Population-based variation in cardiomyopathy genes. Circ. Cardiovasc. Genet. 2012, 5, 391–399. [Google Scholar] [CrossRef]
- Risgaard, B.; Jabbari, R.; Refsgaard, L.; Holst, A.G.; Haunso, S.; Sadjadieh, A.; Winkel, B.G.; Olesen, M.S.; Tfelt-Hansen, J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin. Genet. 2013, 84, 489–495. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Landstrom, A.P.; Salisbury, B.A.; Callis, T.E.; Pollevick, G.D.; Tester, D.J.; Cox, M.G.; Bhuiyan, Z.; Bikker, H.; Wiesfeld, A.C.; et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 2011, 57, 2317–2327. [Google Scholar] [CrossRef]
- Andreasen, C.; Refsgaard, L.; Nielsen, J.B.; Sajadieh, A.; Winkel, B.G.; Tfelt-Hansen, J.; Haunso, S.; Holst, A.G.; Svendsen, J.H.; Olesen, M.S. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can. J. Cardiol. 2013, 29, 1104–1109. [Google Scholar] [CrossRef]
- Petretta, M.; Pirozzi, F.; Sasso, L.; Paglia, A.; Bonaduce, D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am. J. Cardiol. 2011, 108, 1171–1176. [Google Scholar] [CrossRef]
- Meder, B.; Haas, J.; Keller, A.; Heid, C.; Just, S.; Borries, A.; Boisguerin, V.; Scharfenberger-Schmeer, M.; Stahler, P.; Beier, M.; et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ. Cardiovasc. Genet. 2011, 4, 110–122. [Google Scholar] [CrossRef]
- Zimmerman, R.S.; Cox, S.; Lakdawala, N.K.; Cirino, A.; Mancini-DiNardo, D.; Clark, E.; Leon, A.; Duffy, E.; White, E.; Baxter, S.; et al. A novel custom resequencing array for dilated cardiomyopathy. Genet. Med. 2010, 12, 268–278. [Google Scholar] [CrossRef]
- Partners Healthcare. Available online: http://pcpgm.partners.org/ (accessed on 25 February 2014).
- Opitz, C.A.; Kulke, M.; Leake, M.C.; Neagoe, C.; Hinssen, H.; Hajjar, R.J.; Linke, W.A. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc. Natl. Acad. Sci. USA 2003, 100, 12688–12693. [Google Scholar] [CrossRef]
- Granzier, H.L.; Irving, T.C. Passive tension in cardiac muscle: Contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 1995, 68, 1027–1044. [Google Scholar] [CrossRef]
- Horowits, R.; Kempner, E.S.; Bisher, M.E.; Podolsky, R.J. A physiological role for titin and nebulin in skeletal muscle. Nature 1986, 323, 160–164. [Google Scholar] [CrossRef]
- Muhle-Goll, C.; Habeck, M.; Cazorla, O.; Nilges, M.; Labeit, S.; Granzier, H. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1—A possible role in the Frank-Starling mechanism of the heart. J. Mol. Biol. 2001, 313, 431–447. [Google Scholar] [CrossRef]
- Cazorla, O.; Wu, Y.; Irving, T.C.; Granzier, H. Titin-based modulation of calcium sensitivity of active tension in mouse skinned cardiac myocytes. Circ. Res. 2001, 88, 1028–1035. [Google Scholar] [CrossRef]
- Siu, B.L.; Niimura, H.; Osborne, J.A.; Fatkin, D.; MacRae, C.; Solomon, S.; Benson, D.W.; Seidman, J.G.; Seidman, C.E. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999, 99, 1022–1026. [Google Scholar] [CrossRef]
- Gerull, B.; Atherton, J.; Geupel, A.; Sasse-Klaassen, S.; Heuser, A.; Frenneaux, M.; McNabb, M.; Granzier, H.; Labeit, S.; Thierfelder, L. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J. Mol. Med. 2006, 84, 478–483. [Google Scholar] [CrossRef]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Majewski, J.; Schwartzentruber, J.; Lalonde, E.; Montpetit, A.; Jabado, N. What can exome sequencing do for you? J. Med. Genet. 2011, 48, 580–589. [Google Scholar] [CrossRef]
- Campbell, N.; Sinagra, G.; Jones, K.L.; Slavov, D.; Gowan, K.; Merlo, M.; Carniel, E.; Fain, P.R.; Aragona, P.; di Lenarda, A.; et al. Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One 2013, 8, e78104. [Google Scholar] [CrossRef]
- Wells, Q.S.; Becker, J.R.; Su, Y.R.; Mosley, J.D.; Weeke, P.; D’Aoust, L.; Ausborn, N.L.; Ramirez, A.H.; Pfotenhauer, J.P.; Naftilan, A.J.; et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 317–326. [Google Scholar] [CrossRef]
- Yandell, M.; Huff, C.; Hu, H.; Singleton, M.; Moore, B.; Xing, J.; Jorde, L.B.; Reese, M.G. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011, 21, 1529–1542. [Google Scholar] [CrossRef]
- Li, D.; Morales, A.; Gonzalez-Quintana, J.; Norton, N.; Siegfried, J.D.; Hofmeyer, M.; Hershberger, R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010, 3, 90–97. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Sebbag, L.; Dulac, A.; Dauphin, C.; Jouk, P.S.; Delrue, M.A.; Thambo, J.B.; Le Metayer, P.; et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur. J. Med. Genet. 2011, 54, e570–e575. [Google Scholar] [CrossRef]
- Lakdawala, N.K.; Dellefave, L.; Redwood, C.S.; Sparks, E.; Cirino, A.L.; Depalma, S.; Colan, S.D.; Funke, B.; Zimmerman, R.S.; Robinson, P.; et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 320–329. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef]
- Robinson, P.N.; Kohler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef]
- Perrot, A.; Hussein, S.; Ruppert, V.; Schmidt, H.H.; Wehnert, M.S.; Duong, N.T.; Posch, M.G.; Panek, A.; Dietz, R.; Kindermann, I.; et al. Identification of mutational hot spots in LMNA encoding lamin A/C in patients with familial dilated cardiomyopathy. Basic Res. Cardiol. 2009, 104, 90–99. [Google Scholar] [CrossRef]
- Theis, J.L.; Sharpe, K.M.; Matsumoto, M.E.; Chai, H.S.; Nair, A.A.; Theis, J.D.; de Andrade, M.; Wieben, E.D.; Michels, V.V.; Olson, T.M. Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 585–594. [Google Scholar] [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef]
- Tuzun, E.; Sharp, A.J.; Bailey, J.A.; Kaul, R.; Morrison, V.A.; Pertz, L.M.; Haugen, E.; Hayden, H.; Albertson, D.; Pinkel, D.; et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005, 37, 727–732. [Google Scholar] [CrossRef]
- Kidd, J.M.; Cooper, G.M.; Donahue, W.F.; Hayden, H.S.; Sampas, N.; Graves, T.; Hansen, N.; Teague, B.; Alkan, C.; Antonacci, F.; et al. Mapping and sequencing of structural variation from eight human genomes. Nature 2008, 453, 56–64. [Google Scholar] [CrossRef]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef]
- Magi, A.; Tattini, L.; Cifola, I.; D’Aurizio, R.; Benelli, M.; Mangano, E.; Battaglia, C.; Bonora, E.; Kurg, A.; Seri, M.; et al. EXCAVATOR: Detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013, 14, R120. [Google Scholar] [CrossRef]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009, 119, 2772–2786. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res. 2009, 105, 585–594. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Wang, K.; Kim, C.; Bradfield, J.; Guo, Y.; Toskala, E.; Otieno, F.G.; Hou, C.; Thomas, K.; Cardinale, C.; Lyon, G.J.; et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med. 2013, 5, 67. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef]
- Sirmaci, A.; Edwards, Y.J.; Akay, H.; Tekin, M. Challenges in whole exome sequencing: An example from hereditary deafness. PLoS One 2012, 7, e32000. [Google Scholar]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kejariwal, A.; Campbell, M.J.; Mi, H.; Diemer, K.; Guo, N.; Ladunga, I.; Ulitsky-Lazareva, B.; Muruganujan, A.; Rabkin, S.; et al. PANTHER: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003, 31, 334–341. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Bookman, E.B.; Langehorne, A.A.; Eckfeldt, J.H.; Glass, K.C.; Jarvik, G.P.; Klag, M.; Koski, G.; Motulsky, A.; Wilfond, B.; Manolio, T.A.; et al. Reporting genetic results in research studies: Summary and recommendations of an NHLBI working group. Am. J. Med. Genet. A 2006, 140, 1033–1040. [Google Scholar]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Puckelwartz, M.J.; McNally, E.M. Genetic Profiling for Risk Reduction in Human Cardiovascular Disease. Genes 2014, 5, 214-234. https://doi.org/10.3390/genes5010214
Puckelwartz MJ, McNally EM. Genetic Profiling for Risk Reduction in Human Cardiovascular Disease. Genes. 2014; 5(1):214-234. https://doi.org/10.3390/genes5010214
Chicago/Turabian StylePuckelwartz, Megan J., and Elizabeth M. McNally. 2014. "Genetic Profiling for Risk Reduction in Human Cardiovascular Disease" Genes 5, no. 1: 214-234. https://doi.org/10.3390/genes5010214
APA StylePuckelwartz, M. J., & McNally, E. M. (2014). Genetic Profiling for Risk Reduction in Human Cardiovascular Disease. Genes, 5(1), 214-234. https://doi.org/10.3390/genes5010214