Mechanisms of Base Substitution Mutagenesis in Cancer Genomes




Abstract
:1. Introduction
2. Meta-Analyses of Cancer Genomes
2.1. Mutational Signatures in Cancer Genomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
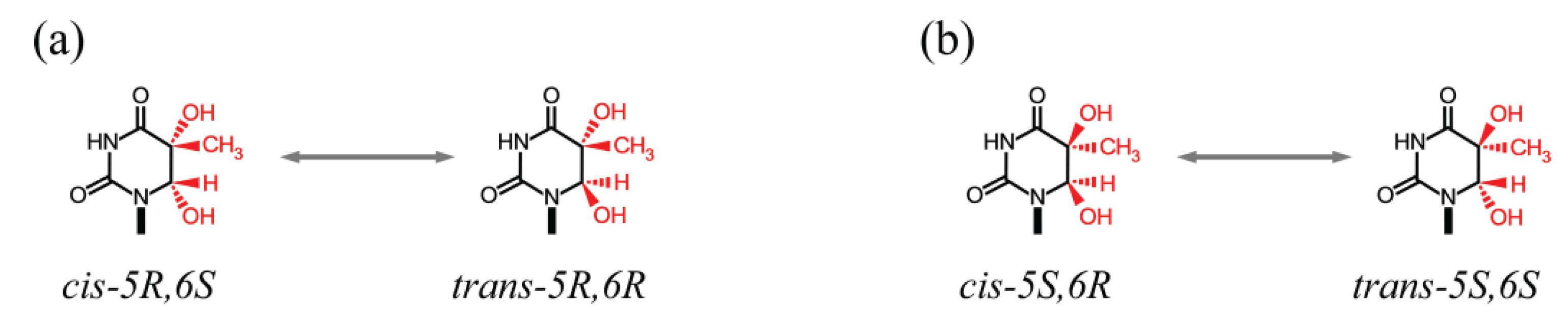{kind=link}
{kind=link}
| A. Main mutational signatures revealed from meta-analyses of cancer genomes | |||||
|---|---|---|---|---|---|
| Total number of mutations | Total number of cancer types | Major SBS signature (% cancer types) | Sequence context | References | |
| 4,938,362 | 30 | C:G→T:A (80%) | NCG:CGN | [13] | |
| C:G→T:A or G:C (50%) | TCN:NGA | ||||
| 1,000,000 | 19 | C:G→any subst. (32%) | TCN:NGA | [15] | |
| 617,354 | 12 | C:G→T:A (33%) | CG:CG | [16] | |
| C:G→G:C (25%) | TC:GA | ||||
| 533,482 | 14 | C:G→any subst. (93%) | NNCG:CGNN | [17] | |
| C:G→any subst. (36%) | NYCH:DGRN | ||||
| 373,909 | 27 | C:G→T:A (30%) | CG:CG | [18] | |
| C:G→any subst. (11%) | TC:GA | ||||
| B. Main cancer type-specific mutational signatures | |||||
| Cancer type | Putative cause | SBS signature | Sequence context | References | |
| Lung cancer | tobacco smoke | C:G→A:T | none | [22,27,28,29,30] | |
| arsenic exposure | T:A→G:C | none | [35] | ||
| Melanoma | UV, APOBEC3A | C:G→T:A | pyrimidine dimers | [36,37,38,39,40,41,42,44] | |
| unknown | G:C→any subst. | NGRA:TYCN | [17] | ||
| Liver carcinoma | carcinogens | T:A→C:G | none | [13,18] | |
| Leukemia | unknown | A:T→T:A | TA:TA | [13,18] | |
| Endometrial cancer | POLEP286R | G:C→T:A | AGA:TCT | ||
2.2. Mismatch Repair and DNA Replicative Polymerase Proofreading Genes
2.3. The APOBEC Family of Cytosine Deaminases
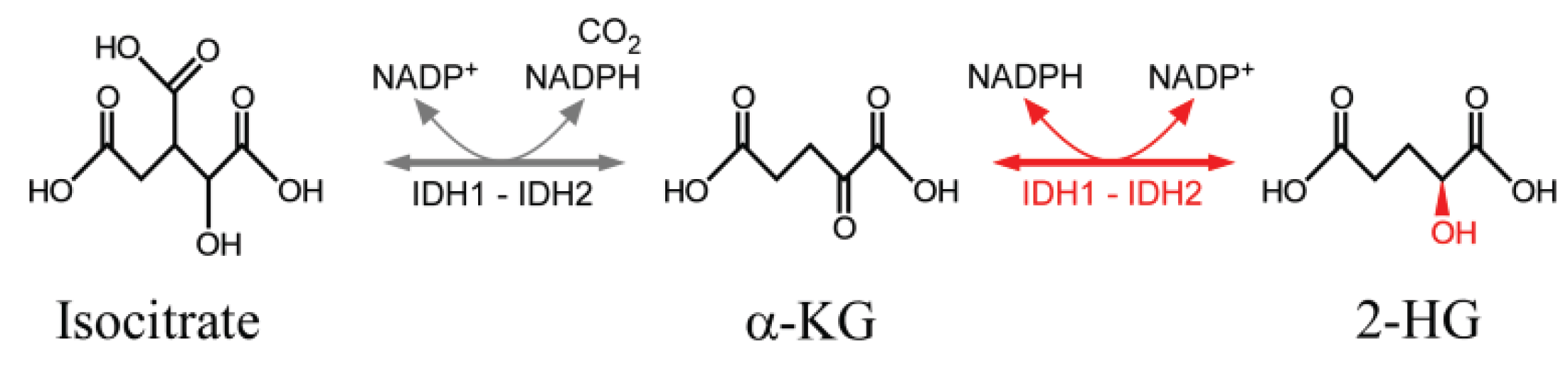
2.4. Electron Transfer in DNA Oxidation
2.5. DNA Replication Timing
2.6. Chromatin Organization
3. Mechanisms of Base Modification
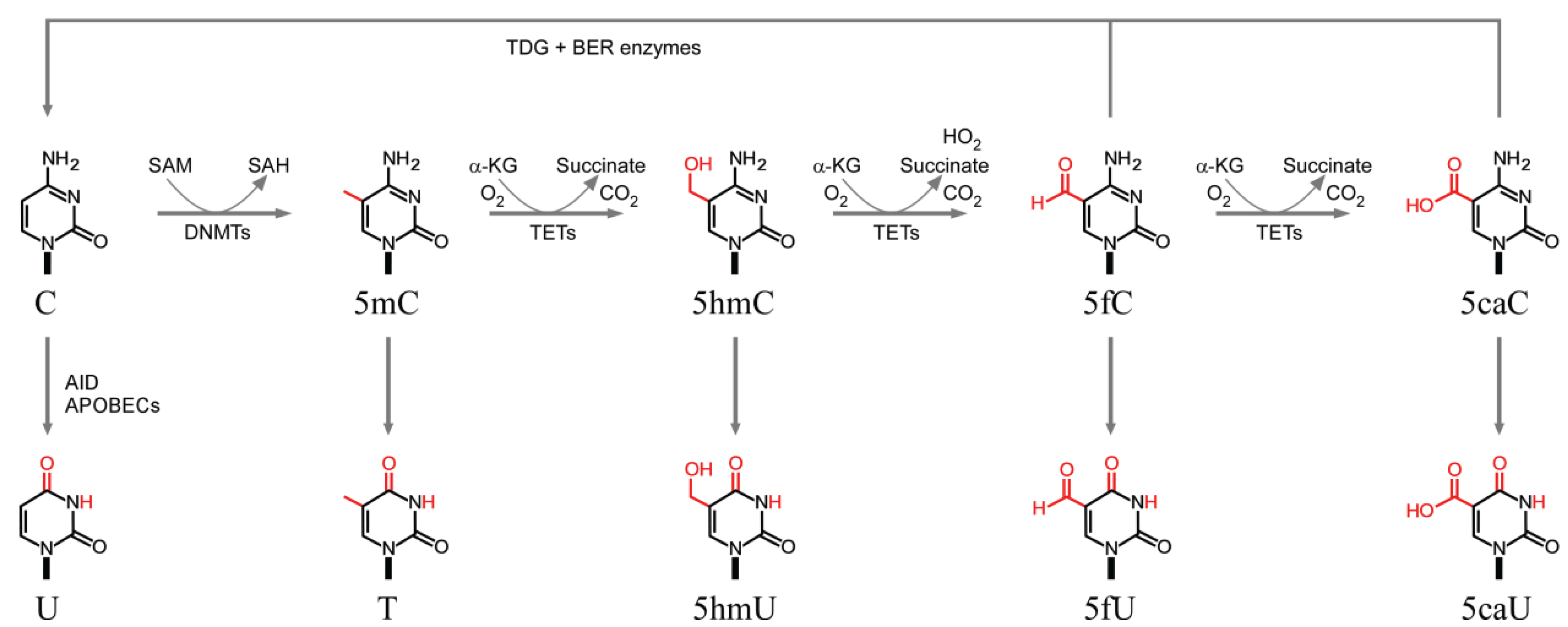
3.1. Cytosine Methylation and Demethylation
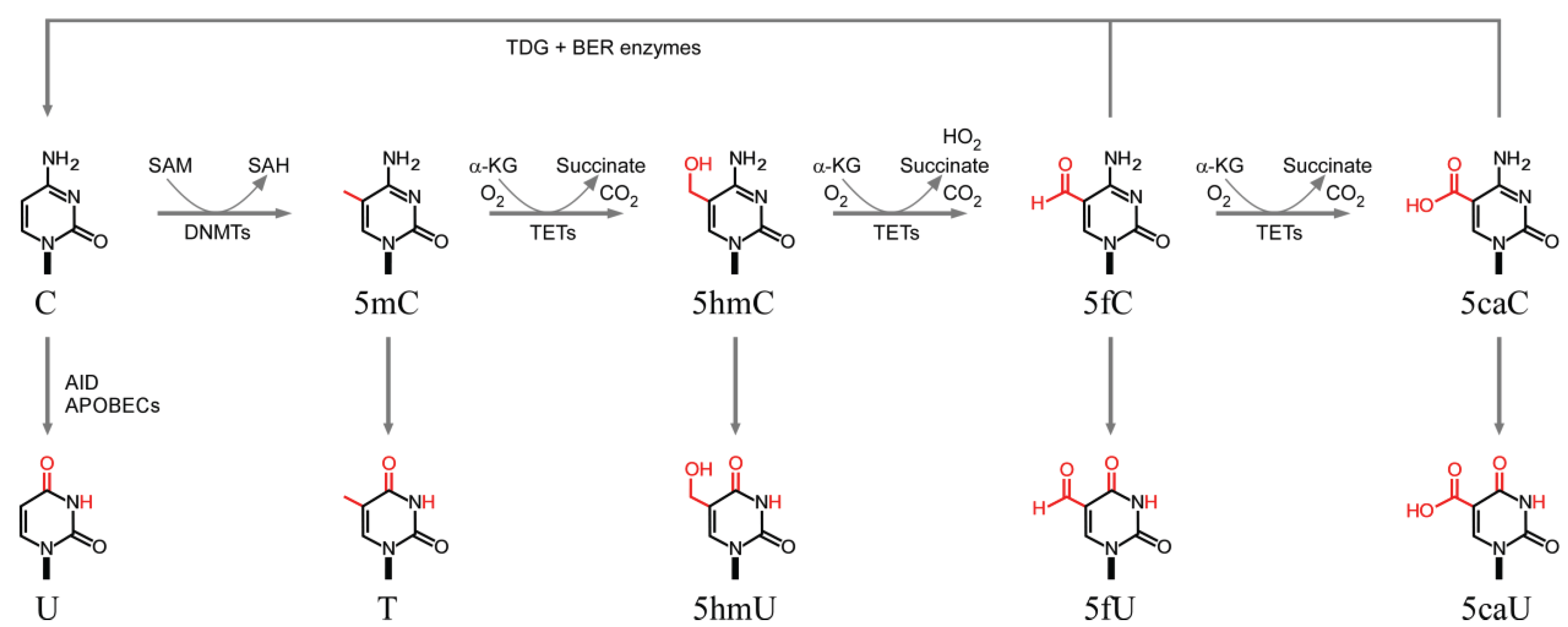
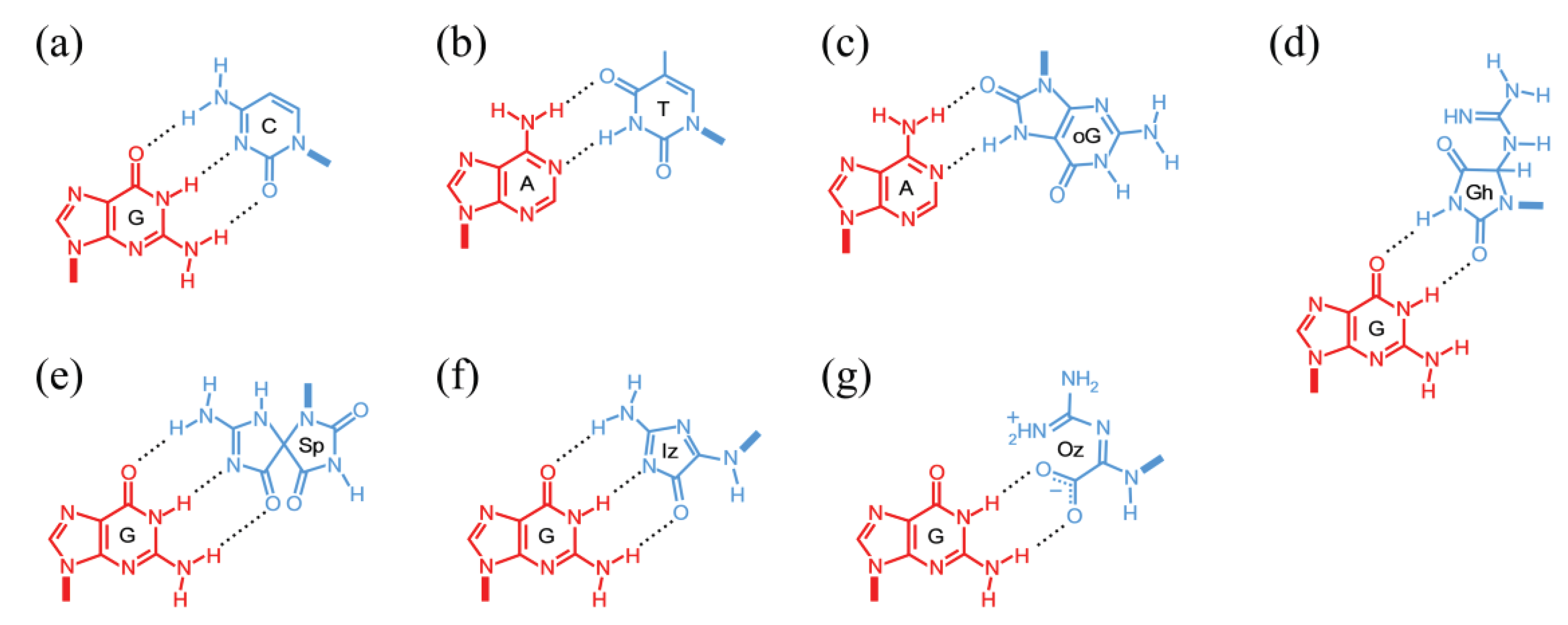
3.2. Deamination of Cytosine Bases
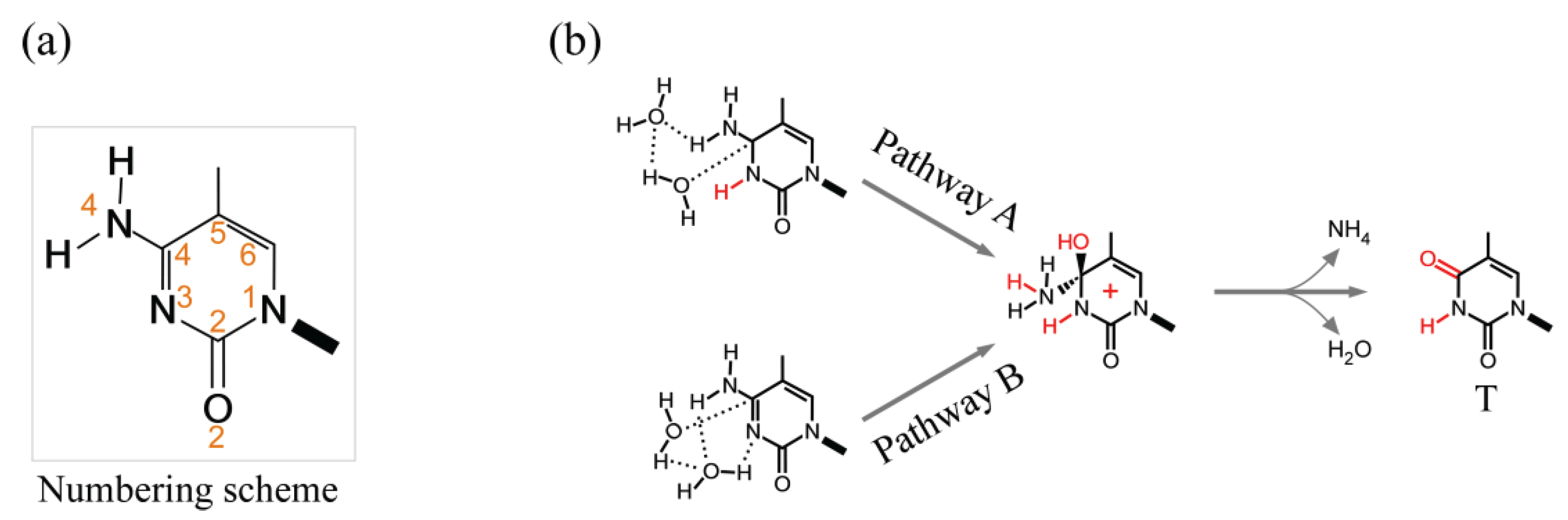
3.2.1. Spontaneous Deamination
3.2.2. ROS-Induced Deamination
| Deaminating base | Sequence context | Deaminated base product | Deamination rate at 37 °C (s−1) | References |
|---|---|---|---|---|
| C | Free nucleoside | U | 9.4 ± 0.5 × 10−10 | [95] |
| 3mC+ | Free nucleoside | 3mU | 5 × 10−7 | [100] |
| 3mC+ | Free nucleotide | 3mU | 13 × 10−7 | [100] |
| 5mC | Free nucleoside | T | 7.8 ± 0.3 × 10−10 | [95] |
| 5hmC | Free nucleoside | 5hmU | 5.8 ± 0.8 × 10−10 | [95] |
| 5fC | Free nucleoside | 5fU | 1.2 ± 0.2 × 10−9 | [95] |
| 5caC | Free nucleoside | 5caU | not detected | [95] |
| 5mCg(5S,6S) | Free nucleoside | Tg | 1.1 × 10−5 | [115] |
| 5mCg(5R,6R) | Free nucleoside | Tg | 8.6 × 10−6 | [115] |
| C | ssDNA | U | 2.1 × 10−10 | [96] |
| C | ssDNA | U | ~1 × 10−10 | [98] |
| 5mC | ssDNA | T | 9.5 × 10−10 | [96] |
| C | dsDNA | U | 2.6 × 10−13 | [97] |
| C | dsDNA | U | 4 × 10−13 | [99] |
| C | dsDNA | U | ~7 × 10−13 | [98] |
| 5mC | dsDNA | T | 5.8 × 10−13 | [97] |
| 5mC | dsDNA | T | 1.5 × 10−11 | [99] |
| 5mCg(5S,6S) | dsDNA | Tg | 5.2 × 10−6 | [109] |
| 5mCg(5R,6R) | dsDNA | Tg | 7.0 × 10−6 | [109] |
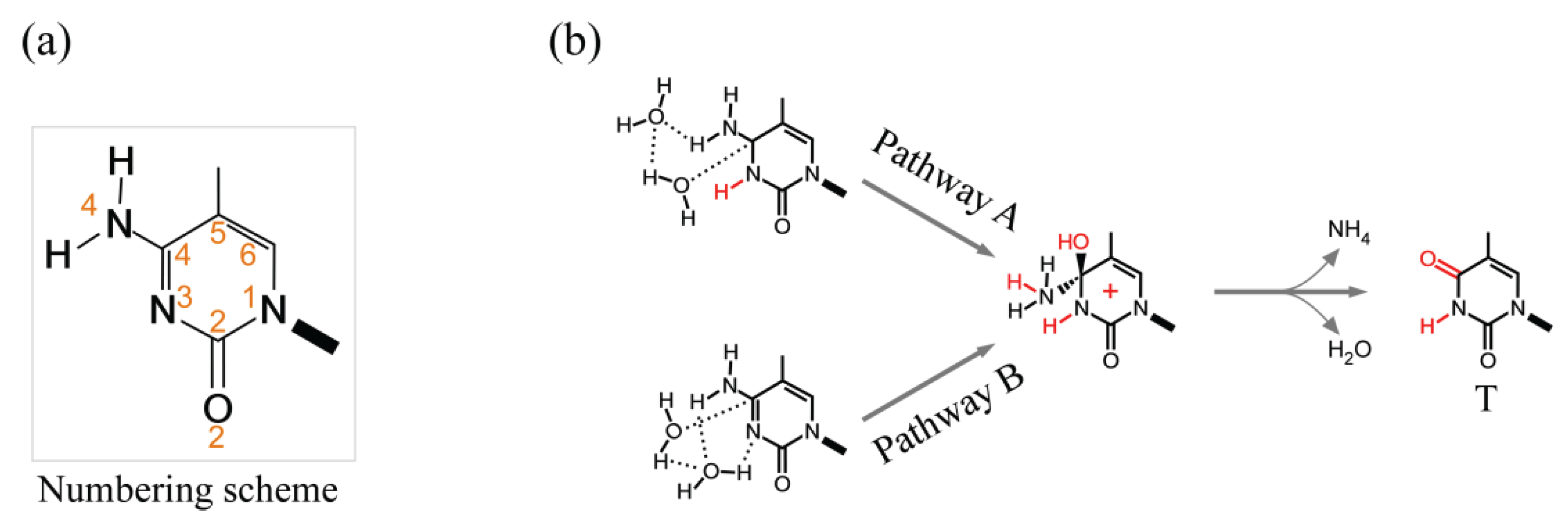
3.2.3. Enzymatic Deamination by Single-Stranded DNA-Specific AID/APOBECs
3.3. Sequence Context-Dependent Guanine Oxidation Products
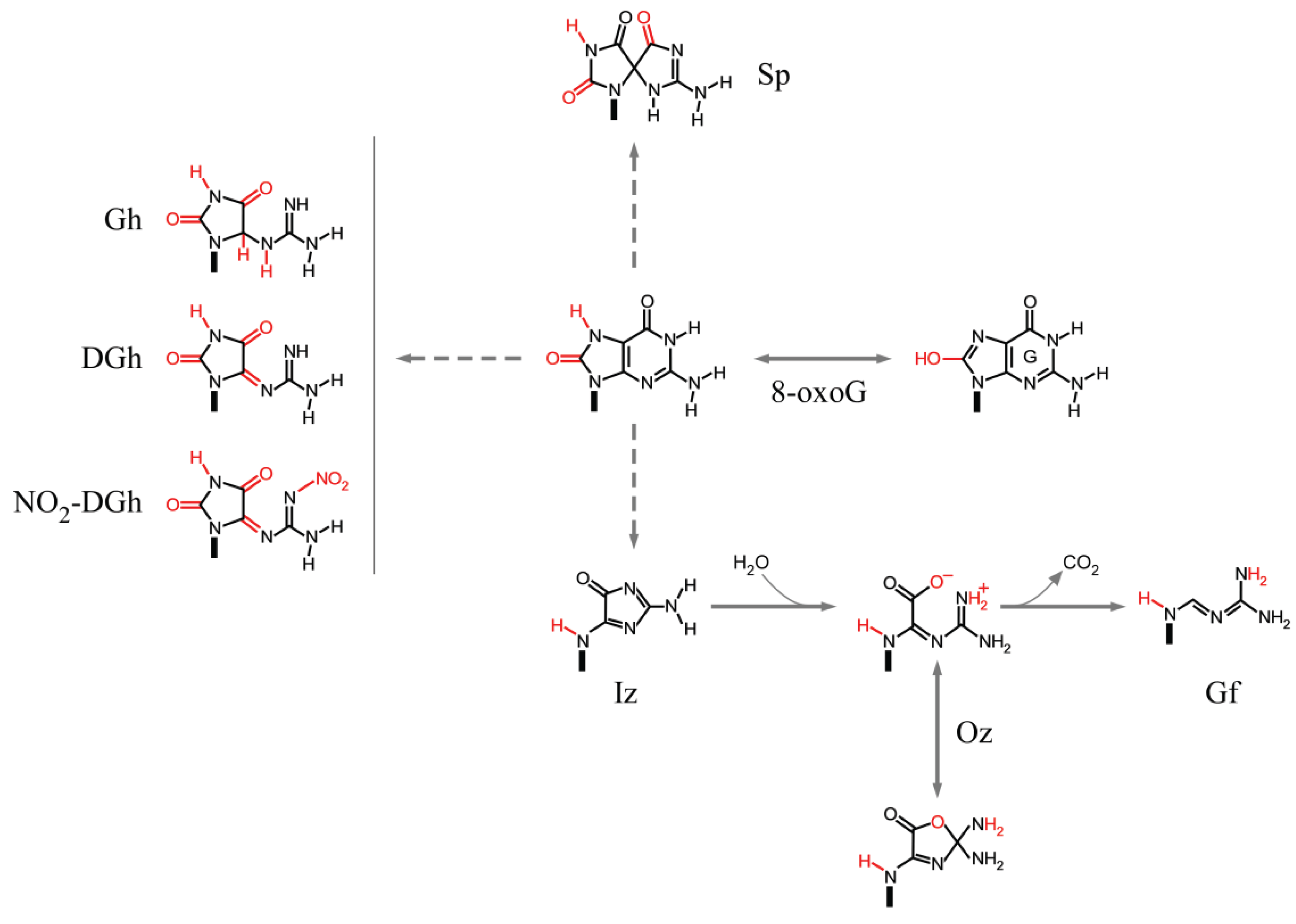
3.3.1. Guanine
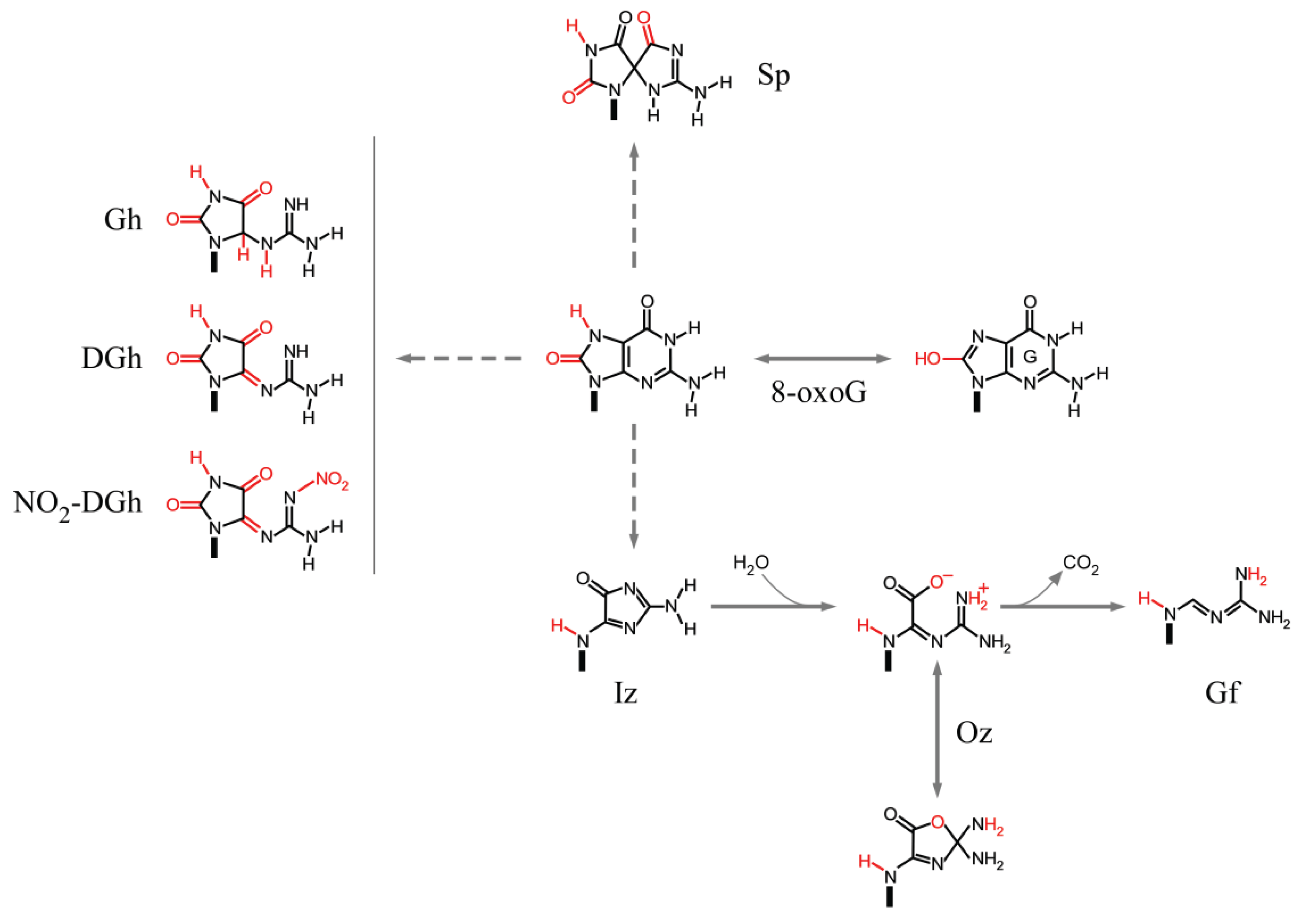
3.3.2. 8-oxoG
3.3.3. Charge Transfer in Nucleosomal DNA
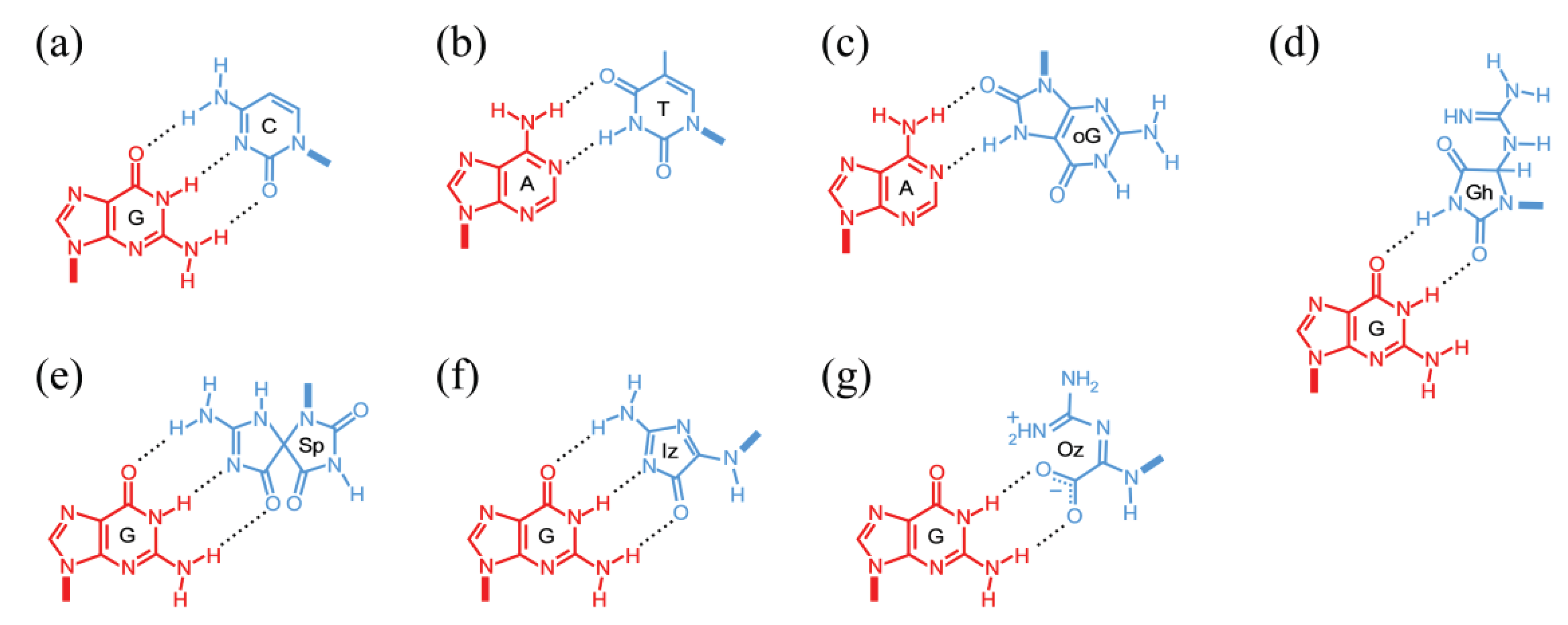
4. DNA Repair Pathways and Synthesis across Modified Bases and Mismatches
4.1. Base Excision Repair
4.1.1. Modified CG:CG Sites

4.1.2. Uracil
4.1.3. Guanine Lesions
4.2. Lesion Bypass
4.3. Transcription-Coupled Repair
5. Proposed Mutational Mechanisms
Acknowledgments
Author Contributions
Conflict of Interest
References
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Yang, L.; Luquette, L.J.; Gehlenborg, N.; Xi, R.; Haseley, P.S.; Hsieh, C.H.; Zhang, C.; Ren, X.; Protopopov, A.; Chin, L.; et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 2013, 153, 919–929. [Google Scholar] [CrossRef]
- Drier, Y.; Lawrence, M.S.; Carter, S.L.; Stewart, C.; Gabriel, S.B.; Lander, E.S.; Meyerson, M.; Beroukhim, R.; Getz, G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013, 23, 228–235. [Google Scholar] [CrossRef]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Lange, S.S.; Takata, K.; Wood, R.D. DNA polymerases and cancer. Nat. Rev. Cancer 2011, 11, 96–110. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell. Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Fischer, A.; Illingworth, C.J.; Campbell, P.J.; Mustonen, V. EMu: Probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol. 2013, 14, R39. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Bacolla, A.; Temiz, N.A.; Yi, M.; Ivanic, J.; Cer, R.Z.; Donohue, D.E.; Ball, E.V.; Mudunuri, U.S.; Wang, G.; Jain, A.; et al. Guanine holes are prominent targets for mutation in cancer and inherited disease. PLoS Genet. 2013, 9, e1003816. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Ivanov, D.; Hamby, S.E.; Stenson, P.D.; Phillips, A.D.; Kehrer-Sawatzki, H.; Cooper, D.N.; Chuzhanova, N. Comparative analysis of germline and somatic microlesion mutational spectra in 17 human tumor suppressor genes. Hum. Mutat. 2011, 32, 620–632. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- TCGARN. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Xiong, D.; Li, G.; Li, K.; Xu, Q.; Pan, Z.; Ding, F.; Vedell, P.; Liu, P.; Cui, P.; Hua, X.; et al. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis 2012, 33, 1797–1805. [Google Scholar] [CrossRef]
- Liu, P.; Morrison, C.; Wang, L.; Xiong, D.; Vedell, P.; Cui, P.; Hua, X.; Ding, F.; Lu, Y.; James, M.; et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012, 33, 1270–1276. [Google Scholar] [CrossRef]
- Lee, W.; Jiang, Z.; Liu, J.; Haverty, P.M.; Guan, Y.; Stinson, J.; Yue, P.; Zhang, Y.; Pant, K.P.; Bhatt, D.; et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 2010, 465, 473–477. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Stephens, P.J.; O’Meara, S.; McBride, D.J.; Meynert, A.; Jones, D.; Lin, M.L.; Beare, D.; Lau, K.W.; Greenman, C.; et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010, 463, 184–190. [Google Scholar] [CrossRef]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Matter, B.; Wang, G.; Jones, R.; Tretyakova, N. Formation of diastereomeric benzo [a] pyrene diol epoxide-guanine adducts in p53 gene-derived DNA sequences. Chem. Res. Toxicol. 2004, 17, 731–741. [Google Scholar] [CrossRef]
- Ziegel, R.; Shallop, A.; Jones, R.; Tretyakova, N. K-ras gene sequence effects on the formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-DNA adducts. Chem. Res. Toxicol. 2003, 16, 541–550. [Google Scholar] [CrossRef]
- Anna, L.; Holmila, R.; Kovacs, K.; Gyorffy, E.; Gyori, Z.; Segesdi, J.; Minarovits, J.; Soltesz, I.; Kostic, S.; Csekeo, A.; et al. Relationship between TP53 tumour suppressor gene mutations and smoking-related bulky DNA adducts in a lung cancer study population from Hungary. Mutagenesis 2009, 24, 475–480. [Google Scholar] [CrossRef]
- Martinez, V.D.; Thu, K.L.; Vucic, E.A.; Hubaux, R.; Adonis, M.; Gil, L.; MacAulay, C.; Lam, S.; Lam, W.L. Whole-genome sequencing analysis identifies a distinctive mutational spectrum in an arsenic-related lung tumor. J. Thorac. Oncol. 2013, 8, 1451–1455. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2012, 44, 133–139. [Google Scholar]
- Stark, M.S.; Woods, S.L.; Gartside, M.G.; Bonazzi, V.F.; Dutton-Regester, K.; Aoude, L.G.; Chow, D.; Sereduk, C.; Niemi, N.M.; Tang, N.; et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet. 2012, 44, 165–169. [Google Scholar]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; Program, N.C.S.; Stemke-Hale, K.; Davies, M.A.; et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011, 43, 442–446. [Google Scholar] [CrossRef]
- Dutton-Regester, K.; Kakavand, H.; Aoude, L.G.; Stark, M.S.; Gartside, M.G.; Johansson, P.; O’Connor, L.; Lanagan, C.; Tembe, V.; Pupo, G.M.; et al. Melanomas of unknown primary have a mutation profile consistent with cutaneous sun-exposed melanoma. Pigment Cell Melanoma Res. 2013, 26, 852–860. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar]
- Pham, P.; Landolph, A.; Mendez, C.; Li, N.; Goodman, M.F. A biochemical analysis linking APOBEC3A to disparate HIV-1 restriction and skin cancer. J. Biol. Chem. 2013, 288, 29294–29304. [Google Scholar] [CrossRef]
- Korona, D.A.; Lecompte, K.G.; Pursell, Z.F. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2011, 39, 1763–1773. [Google Scholar] [CrossRef]
- Agbor, A.A.; Goksenin, A.Y.; LeCompte, K.G.; Hans, S.H.; Pursell, Z.F. Human Pol epsilon-dependent replication errors and the influence of mismatch repair on their correction. DNA Repair 2013, 12, 954–963. [Google Scholar] [CrossRef]
- Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef]
- Pluciennik, A.; Dzantiev, L.; Iyer, R.R.; Constantin, N.; Kadyrov, F.A.; Modrich, P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc. Natl. Acad. Sci. USA 2010, 107, 16066–16071. [Google Scholar]
- Arana, M.E.; Kunkel, T.A. Mutator phenotypes due to DNA replication infidelity. Semin. Cancer Biol. 2010, 20, 304–311. [Google Scholar] [CrossRef]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA replication fidelity and cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef]
- Aquilina, G.; Bignami, M. Mismatch repair in correction of replication errors and processing of DNA damage. J. Cell. Physiol. 2001, 187, 145–154. [Google Scholar] [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capella, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar]
- TCGAN. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- TCGARN; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Nagarajan, N.; Bertrand, D.; Hillmer, A.M.; Zang, Z.J.; Yao, F.; Jacques, P.E.; Teo, A.S.; Cutcutache, I.; Zhang, Z.; Lee, W.H.; et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012, 13, R115. [Google Scholar] [CrossRef]
- Kim, T.M.; Laird, P.W.; Park, P.J. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef]
- Yoshida, R.; Miyashita, K.; Inoue, M.; Shimamoto, A.; Yan, Z.; Egashira, A.; Oki, E.; Kakeji, Y.; Oda, S.; Maehara, Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. Eur. J. Hum. Genet. 2011, 19, 320–325. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef]
- Dai, H.; Hickey, R.J.; Liu, J.; Bigsby, R.M.; Lanner, C.; Malkas, L.H. Error-promoting DNA synthesis in ovarian cancer cells. Gynecol. Oncol. 2013, 131, 198–206. [Google Scholar] [CrossRef]
- Vieira, V.C.; Soares, M.A. The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res. Int. 2013, 2013, 683095. [Google Scholar]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef]
- Koito, A.; Ikeda, T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013, 4, 28. [Google Scholar]
- Bohn, M.F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Kurosawa, T.; Yokomaku, Y.; Yamane, T.; Watanabe, N.; Suzuki, A.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Lewis, P.D.; Harvey, J.S.; Waters, E.M.; Skibinski, D.O.; Parry, J.M. Spontaneous mutation spectra in supF: Comparative analysis of mammalian cell line base substitution spectra. Mutagenesis 2001, 16, 503–515. [Google Scholar] [CrossRef]
- Bacolla, A.; Wang, G.; Jain, A.; Chuzhanova, N.A.; Cer, R.Z.; Collins, J.R.; Cooper, D.N.; Bohr, V.A.; Vasquez, K.M. Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J. Biol. Chem. 2011, 286, 10017–10026. [Google Scholar] [CrossRef]
- Giese, B. Long-distance electron transfer through DNA. Annu. Rev. Biochem. 2002, 71, 51–70. [Google Scholar] [CrossRef]
- Sugiyama, H.; Saito, I. Theoretical studies of GC-specific photocleavage of DNA via electron transfer: Significant lowering of ionization potential and 5'-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar] [CrossRef]
- Saito, I.; Nakamura, T.; Nakatani, K.; Yoshioka, Y.; Yamaguchi, K.; Sugiyama, H. Mapping of the hot spots for DNA damage by one-electron oxidation: Efficacy of GG doublets and GGG triplets as a trap in long-range hole migration. J. Am. Chem. Soc. 1998, 120, 12686–12687. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Guerra, C.F.; Bickelhaupt, F.M.; Siebbeles, L.D. Mapping the sites for selective oxidation of guanines in DNA. J. Am. Chem. Soc. 2003, 125, 13658–13659. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hayano, M.; Masai, H. Replication timing regulation of eukaryotic replicons: Rif1 as a global regulator of replication timing. Trends Genet. 2013, 29, 449–460. [Google Scholar] [CrossRef]
- Aparicio, O.M. Location, location, location: It’s all in the timing for replication origins. Genes Dev. 2013, 27, 117–128. [Google Scholar] [CrossRef]
- Liu, L.; De, S.; Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat. Commun. 2013, 4, 1502. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef]
- Gavin, D.P.; Chase, K.A.; Sharma, R.P. Active DNA demethylation in post-mitotic neurons: A reason for optimism. Neuropharmacology 2013, 75, 233–245. [Google Scholar] [CrossRef]
- Schuster-Bockler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar]
- Wang, G.; Christensen, L.A.; Vasquez, K.M. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2677–2682. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Mertz, T.M.; Zhang, Y.; Northam, M.R.; Sheng, Z.; Lobachev, K.S.; Shcherbakova, P.V.; Kadyrov, F.A. A reversible histone H3 acetylation cooperates with mismatch repair and replicative polymerases in maintaining genome stability. PLOS Genet. 2013, 9, e1003899. [Google Scholar] [CrossRef]
- Kinney, S.R.; Pradhan, S. Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog. Mol. Biol. Transl. Sci. 2011, 101, 311–333. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Thomson, J.P.; Moggs, J.G.; Wolf, C.R.; Meehan, R.R. Epigenetic profiles as defined signatures of xenobiotic exposure. Mutat. Res. 2013. [Google Scholar] [CrossRef]
- Song, C.X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013, 153, 678–691. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar]
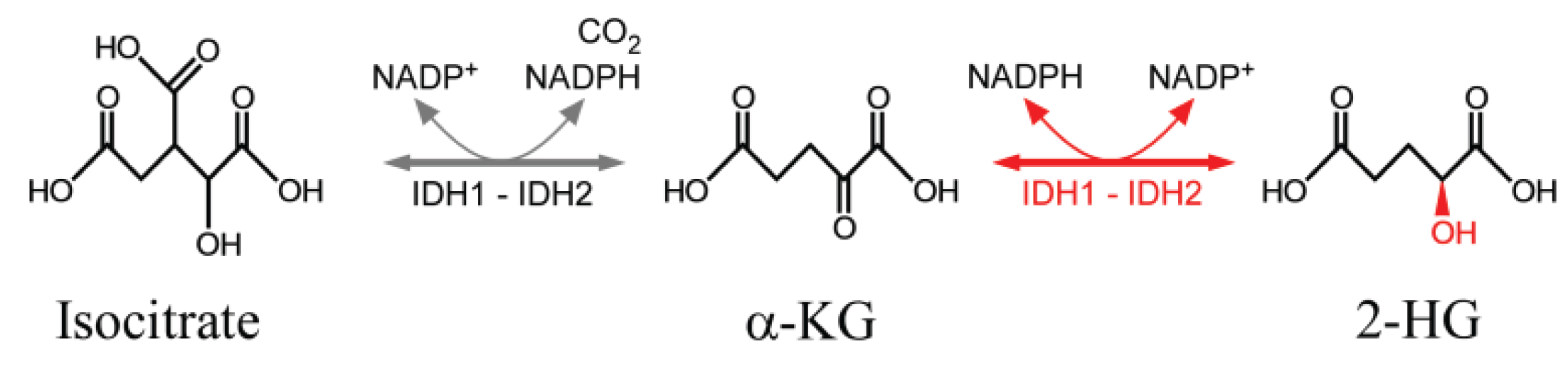
- Losman, J.A.; Kaelin, W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Guilhamon, P.; Eskandarpour, M.; Halai, D.; Wilson, G.A.; Feber, A.; Teschendorff, A.E.; Gomez, V.; Hergovich, A.; Tirabosco, R.; Fernanda Amary, M.; et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat. Commun. 2013, 4, 2166. [Google Scholar]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Sandoval, J.; Mendez-Gonzalez, J.; Nadal, E.; Chen, G.; Carmona, F.J.; Sayols, S.; Moran, S.; Heyn, H.; Vizoso, M.; Gomez, A.; et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4140–4147. [Google Scholar] [CrossRef]
- Schiesser, S.; Pfaffeneder, T.; Sadeghian, K.; Hackner, B.; Steigenberger, B.; Schroder, A.S.; Steinbacher, J.; Kashiwazaki, G.; Hofner, G.; Wanner, K.T.; et al. Deamination, oxidation, and C-C bond cleavage reactivity of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine. J. Am. Chem. Soc. 2013, 135, 14593–14599. [Google Scholar] [CrossRef]
- Ehrlich, M.; Norris, K.F.; Wang, R.Y.; Kuo, K.C.; Gehrke, C.W. DNA cytosine methylation and heat-induced deamination. Biosci. Rep. 1986, 6, 387–393. [Google Scholar] [CrossRef]
- Shen, J.C.; Rideout, W.M., 3rd; Jones, P.A. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994, 22, 927–976. [Google Scholar]
- Frederico, L.A.; Kunkel, T.A.; Shaw, B.R. A sensitive genetic assay for the detection of cytosine deamination: Determination of rate constants and the activation energy. Biochemistry 1990, 29, 2532–2537. [Google Scholar] [CrossRef]
- Zhang, X.; Mathews, C.K. Effect of DNA cytosine methylation upon deamination-induced mutagenesis in a natural target sequence in duplex DNA. J. Biol. Chem. 1994, 269, 7066–7069. [Google Scholar]
- Sowers, L.C.; Sedwick, W.D.; Shaw, B.R. Hydrolysis of N3-methyl-2'-deoxycytidine: Model compound for reactivity of protonated cytosine residues in DNA. Mutat. Res. 1989, 215, 131–138. [Google Scholar] [CrossRef]
- Nikolova, E.N.; Goh, G.B.; Brooks, C.L., 3rd; Al-Hashimi, H.M. Characterizing the protonation state of cytosine in transient G.C Hoogsteen base pairs in duplex DNA. J. Am. Chem. Soc. 2013, 135, 6766–6769. [Google Scholar]
- Fryxell, K.J.; Zuckerkandl, E. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol. Biol. Evol. 2000, 17, 1371–1383. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Z.; Chen, H.; Su, Z.; Yang, J.; Lin, F.; Shi, S.; He, X. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 2012, 335, 1235–1238. [Google Scholar] [CrossRef]
- Labet, V.; Morell, C.; Cadet, J.; Eriksson, L.A.; Grand, A. Hydrolytic deamination of 5-methylcytosine in protic medium—A theoretical study. J. Phys. Chem. A 2009, 113, 2524–2533. [Google Scholar] [CrossRef]
- Patel, J.C.; Rice, M.E. Classification of H(2)O(2) as a neuromodulator that regulates striatal dopamine release on a subsecond time scale. ACS Chem. Neurosci. 2012, 3, 991–1001. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef]
- Cao, H.; Jiang, Y.; Wang, Y. Kinetics of deamination and Cu(II)/H2O2/ascorbate-induced formation of 5-methylcytosine glycol at CpG sites in duplex DNA. Nucleic Acids Res. 2009, 37, 6635–6643. [Google Scholar] [CrossRef]
- Wagner, J.R.; Cadet, J. Oxidation reactions of cytosine DNA components by hydroxyl radical and one-electron oxidants in aerated aqueous solutions. Acc. Chem. Res. 2010, 43, 564–571. [Google Scholar] [CrossRef]
- Liang, Q.; Dedon, P.C. Cu(II)/H2O2-induced DNA damage is enhanced by packaging of DNA as a nucleosome. Chem. Res. Toxicol. 2001, 14, 416–422. [Google Scholar] [CrossRef]
- Tan, S.; Davey, C.A. Nucleosome structural studies. Curr. Opin. Struct. Biol. 2011, 21, 128–136. [Google Scholar] [CrossRef]
- Zavitsanos, K.; Nunes, A.M.; Malandrinos, G.; Hadjiliadis, N. Copper effective binding with 32–62 and 94–125 peptide fragments of histone H2B. J. Inorg. Biochem. 2011, 105, 102–110. [Google Scholar] [CrossRef]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar]
- Bienvenu, C.; Cadet, J. Synthesis and kinetic study of the deamination of the cis diastereomers of 5,6-dihydroxy-5,6-dihydro-5-methyl-2'-deoxycytidine. J. Org. Chem. 1996, 61, 2632–2637. [Google Scholar] [CrossRef]
- Bransteitter, R.; Prochnow, C.; Chen, X.S. The current structural and functional understanding of APOBEC deaminases. Cell. Mol. Life Sci. 2009, 66, 3137–3147. [Google Scholar] [CrossRef]
- Larijani, M.; Martin, A. The biochemistry of activation-induced deaminase and its physiological functions. Semin. Immunol. 2012, 24, 255–263. [Google Scholar] [CrossRef]
- Iwatani, Y.; Takeuchi, H.; Strebel, K.; Levin, J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J. Virol. 2006, 80, 5992–6002. [Google Scholar] [CrossRef]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef]
- Wijesinghe, P.; Bhagwat, A.S. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012, 40, 9206–9217. [Google Scholar] [CrossRef]
- Morgan, H.D.; Dean, W.; Coker, H.A.; Reik, W.; Petersen-Mahrt, S.K. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: Implications for epigenetic reprogramming. J. Biol. Chem. 2004, 279, 52353–52360. [Google Scholar]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Isaksson Vogel, R.; Nikas, J.B.; Law, E.K.; Brown, W.L.; et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef]
- Langlois, M.A.; Beale, R.C.; Conticello, S.G.; Neuberger, M.S. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005, 33, 1913–1923. [Google Scholar] [CrossRef]
- Wiegand, H.L.; Doehle, B.P.; Bogerd, H.P.; Cullen, B.R. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004, 23, 2451–2458. [Google Scholar] [CrossRef]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef]
- Kohli, R.M.; Maul, R.W.; Guminski, A.F.; McClure, R.L.; Gajula, K.S.; Saribasak, H.; McMahon, M.A.; Siliciano, R.F.; Gearhart, P.J.; Stivers, J.T. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J. Biol. Chem. 2010, 285, 40956–40964. [Google Scholar] [CrossRef]
- Krijger, P.H.; Tsaalbi-Shtylik, A.; Wit, N.; van den Berk, P.C.; de Wind, N.; Jacobs, H. Rev1 is essential in generating G to C transversions downstream of the Ung2 pathway but not the Msh2+Ung2 hybrid pathway. Eur. J. Immunol. 2013, 43, 2765–2770. [Google Scholar] [CrossRef]
- Cui, L.; Ye, W.; Prestwich, E.G.; Wishnok, J.S.; Taghizadeh, K.; Dedon, P.C.; Tannenbaum, S.R. Comparative analysis of four oxidized guanine lesions from reactions of DNA with peroxynitrite, singlet oxygen, and gamma-radiation. Chem. Res. Toxicol. 2013, 26, 195–202. [Google Scholar] [CrossRef]
- Kino, K.; Sugiyama, H. UVR-induced G-C to C-G transversions from oxidative DNA damage. Mutat. Res. 2005, 571, 33–42. [Google Scholar] [CrossRef]
- Douki, T.; Angelov, D.; Cadet, J. UV laser photolysis of DNA: Effect of duplex stability on charge-transfer efficiency. J. Am. Chem. Soc. 2001, 123, 11360–11366. [Google Scholar] [CrossRef]
- Giese, B.; Amaudrut, J.; Kohler, A.K.; Spormann, M.; Wessely, S. Direct observation of hole transfer through DNA by hopping between adenine bases and by tunnelling. Nature 2001, 412, 318–320. [Google Scholar]
- Prat, F.; Houk, K.N.; Foote, C.S. Effect of guanine stacking on the oxidation of 8-oxoguanine in B-DNA. J. Am. Chem. Soc. 1998, 120, 845–846. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar]
- Stathis, D.; Lischke, U.; Koch, S.C.; Deiml, C.A.; Carell, T. Discovery and mutagenicity of a guanidinoformimine lesion as a new intermediate of the oxidative deoxyguanosine degradation pathway. J. Am. Chem. Soc. 2012, 134, 4925–4930. [Google Scholar] [CrossRef]
- Hickerson, R.P.; Prat, F.; Muller, J.G.; Foote, C.S.; Burrows, C.J. Sequence and stacking dependence of 8-oxoguanine oxidation: Comparison of one-electron vs. singlet oxygen mechanisms. J. Am. Chem. Soc. 1999, 121, 9423–9428. [Google Scholar]
- Lim, K.S.; Taghizadeh, K.; Wishnok, J.S.; Babu, I.R.; Shafirovich, V.; Geacintov, N.E.; Dedon, P.C. Sequence-dependent variation in the reactivity of 8-Oxo-7,8-dihydro-2'-deoxyguanosine toward oxidation. Chem. Res. Toxicol. 2012, 25, 366–373. [Google Scholar] [CrossRef]
- Lim, K.S.; Cui, L.; Taghizadeh, K.; Wishnok, J.S.; Chan, W.; DeMott, M.S.; Babu, I.R.; Tannenbaum, S.R.; Dedon, P.C. In situ analysis of 8-oxo-7,8-dihydro-2'-deoxyguanosine oxidation reveals sequence- and agent-specific damage spectra. J. Am. Chem. Soc. 2012, 134, 18053–18064. [Google Scholar] [CrossRef]
- McKibbin, P.L.; Fleming, A.M.; Towheed, M.A.; Van Houten, B.; Burrows, C.J.; David, S.S. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc. 2013, 135, 13851–13861. [Google Scholar] [CrossRef]
- Davis, W.B.; Bjorklund, C.C.; Deline, M. Probing the effects of DNA-protein interactions on DNA hole transport: The N-terminal histone tails modulate the distribution of oxidative damage and chemical lesions in the nucleosome core particle. Biochemistry 2012, 51, 3129–3142. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar]
- Sarasin, A. An overview of the mechanisms of mutagenesis and carcinogenesis. Mutat. Res. 2003, 544, 99–106. [Google Scholar] [CrossRef]
- Ray, S.; Menezes, M.R.; Senejani, A.; Sweasy, J.B. Cellular roles of DNA polymerase beta. Yale J. Biol. Med. 2013, 86, 463–469. [Google Scholar]
- Hashimoto, H.; Zhang, X.; Cheng, X. Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: Structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 8276–8284. [Google Scholar] [CrossRef]
- Sjolund, A.B.; Senejani, A.G.; Sweasy, J.B. MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 2013, 743–744, 12–25. [Google Scholar] [CrossRef]
- Millar, C.B.; Guy, J.; Sansom, O.J.; Selfridge, J.; MacDougall, E.; Hendrich, B.; Keightley, P.D.; Bishop, S.M.; Clarke, A.R.; Bird, A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 2002, 297, 403–405. [Google Scholar] [CrossRef]
- Wong, E.; Yang, K.; Kuraguchi, M.; Werling, U.; Avdievich, E.; Fan, K.; Fazzari, M.; Jin, B.; Brown, A.M.; Lipkin, M.; et al. Mbd4 inactivation increases C→T transition mutations and promotes gastrointestinal tumor formation. Proc. Natl. Acad. Sci. USA 2002, 99, 14937–14942. [Google Scholar] [CrossRef]
- Sansom, O.J.; Bishop, S.M.; Bird, A.; Clarke, A.R. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene 2004, 23, 5693–5696. [Google Scholar] [CrossRef]
- Ishibashi, T.; So, K.; Cupples, C.G.; Ausio, J. MBD4-mediated glycosylase activity on a chromatin template is enhanced by acetylation. Mol. Cell. Biol. 2008, 28, 4734–4744. [Google Scholar] [CrossRef]
- Vasovcak, P.; Krepelova, A.; Menigatti, M.; Puchmajerova, A.; Skapa, P.; Augustinakova, A.; Amann, G.; Wernstedt, A.; Jiricny, J.; Marra, G.; et al. Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA Repair 2012, 11, 616–623. [Google Scholar] [CrossRef]
- Ocampo-Hafalla, M.T.; Altamirano, A.; Basu, A.K.; Chan, M.K.; Ocampo, J.E.; Cummings, A., Jr.; Boorstein, R.J.; Cunningham, R.P.; Teebor, G.W. Repair of thymine glycol by hNth1 and hNeil1 is modulated by base pairing and cis-trans epimerization. DNA Repair 2006, 5, 444–454. [Google Scholar] [CrossRef]
- Chan, M.K.; Ocampo-Hafalla, M.T.; Vartanian, V.; Jaruga, P.; Kirkali, G.; Koenig, K.L.; Brown, S.; Lloyd, R.S.; Dizdaroglu, M.; Teebor, G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 2009, 8, 786–794. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Popanda, O.; Warta, R.; Dyckhoff, G.; Herpel, E.; Geiselhart, L.; Claus, R.; Lasitschka, F.; Campos, B.; Oakes, C.C.; et al. Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma. Oncogene 2012, 31, 5108–5116. [Google Scholar] [CrossRef]
- Doseth, B.; Ekre, C.; Slupphaug, G.; Krokan, H.E.; Kavli, B. Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair 2012, 11, 587–593. [Google Scholar] [CrossRef]
- Slupianek, A.; Falinski, R.; Znojek, P.; Stoklosa, T.; Flis, S.; Doneddu, V.; Pytel, D.; Synowiec, E.; Blasiak, J.; Bellacosa, A.; et al. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia 2013, 27, 629–634. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef]
- Ruggieri, V.; Pin, E.; Russo, M.T.; Barone, F.; Degan, P.; Sanchez, M.; Quaia, M.; Minoprio, A.; Turco, E.; Mazzei, F.; et al. Loss of MUTYH function in human cells leads to accumulation of oxidative damage and genetic instability. Oncogene 2013, 32, 4500–4508. [Google Scholar] [CrossRef]
- Bjoras, M.; Seeberg, E.; Luna, L.; Pearl, L.H.; Barrett, T.E. Reciprocal “flipping” underlies substrate recognition and catalytic activation by the human 8-oxo-guanine DNA glycosylase. J. Mol. Biol. 2002, 317, 171–177. [Google Scholar] [CrossRef]
- Luncsford, P.J.; Chang, D.Y.; Shi, G.; Bernstein, J.; Madabushi, A.; Patterson, D.N.; Lu, A.L.; Toth, E.A. A structural hinge in eukaryotic MutY homologues mediates catalytic activity and Rad9-Rad1-Hus1 checkpoint complex interactions. J. Mol. Biol. 2010, 403, 351–370. [Google Scholar] [CrossRef]
- Amouroux, R.; Campalans, A.; Epe, B.; Radicella, J.P. Oxidative stress triggers the preferential assembly of base excision repair complexes on open chromatin regions. Nucleic Acids Res. 2010, 38, 2878–2890. [Google Scholar] [CrossRef]
- Menoni, H.; Shukla, M.S.; Gerson, V.; Dimitrov, S.; Angelov, D. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res. 2012, 40, 692–700. [Google Scholar] [CrossRef]
- Kino, K.; Takao, M.; Miyazawa, H.; Hanaoka, F. A DNA oligomer containing 2,2,4-triamino-5(2H)-oxazolone is incised by human NEIL1 and NTH1. Mutat Res. 2012, 734, 73–77. [Google Scholar] [CrossRef]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar] [CrossRef]
- Liu, M.; Doublie, S.; Wallace, S.S. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. 2013, 743–744, 4–11. [Google Scholar]
- Regnell, C.E.; Hildrestrand, G.A.; Sejersted, Y.; Medin, T.; Moldestad, O.; Rolseth, V.; Krokeide, S.Z.; Suganthan, R.; Luna, L.; Bjoras, M.; et al. Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep. 2012, 2, 503–510. [Google Scholar] [CrossRef]
- Rolseth, V.; Krokeide, S.Z.; Kunke, D.; Neurauter, C.G.; Suganthan, R.; Sejersted, Y.; Hildrestrand, G.A.; Bjoras, M.; Luna, L. Loss of Neil3, the major DNA glycosylase activity for removal of hydantoins in single stranded DNA, reduces cellular proliferation and sensitizes cells to genotoxic stress. Biochim. Biophys. Acta 2013, 1833, 1157–1164. [Google Scholar] [CrossRef]
- Krokeide, S.Z.; Laerdahl, J.K.; Salah, M.; Luna, L.; Cederkvist, F.H.; Fleming, A.M.; Burrows, C.J.; Dalhus, B.; Bjoras, M. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair 2013, 12, 1159–1164. [Google Scholar] [CrossRef]
- Wardle, J.; Burgers, P.M.; Cann, I.K.; Darley, K.; Heslop, P.; Johansson, E.; Lin, L.J.; McGlynn, P.; Sanvoisin, J.; Stith, C.M.; et al. Uracil recognition by replicative DNA polymerases is limited to the archaea, not occurring with bacteria and eukarya. Nucleic Acids Res. 2008, 36, 705–711. [Google Scholar]
- Aller, P.; Duclos, S.; Wallace, S.S.; Doublie, S. A crystallographic study of the role of sequence context in thymine glycol bypass by a replicative DNA polymerase serendipitously sheds light on the exonuclease complex. J. Mol. Biol. 2011, 412, 22–34. [Google Scholar] [CrossRef]
- Fischhaber, P.L.; Gerlach, V.L.; Feaver, W.J.; Hatahet, Z.; Wallace, S.S.; Friedberg, E.C. Human DNA polymerase kappa bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J. Biol. Chem. 2002, 277, 37604–37611. [Google Scholar]
- Yoon, J.H.; Bhatia, G.; Prakash, S.; Prakash, L. Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases kappa and zeta in human cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14116–14121. [Google Scholar] [CrossRef]
- Takata, K.; Shimizu, T.; Iwai, S.; Wood, R.D. Human DNA polymerase N (POLN) is a low fidelity enzyme capable of error-free bypass of 5S-thymine glycol. J. Biol. Chem. 2006, 281, 23445–23455. [Google Scholar] [CrossRef]
- Takata, K.; Arana, M.E.; Seki, M.; Kunkel, T.A.; Wood, R.D. Evolutionary conservation of residues in vertebrate DNA polymerase N conferring low fidelity and bypass activity. Nucleic Acids Res. 2010, 38, 3233–3244. [Google Scholar] [CrossRef]
- Kusumoto, R.; Masutani, C.; Iwai, S.; Hanaoka, F. Translesion synthesis by human DNA polymerase eta across thymine glycol lesions. Biochemistry 2002, 41, 6090–6099. [Google Scholar] [CrossRef]
- Hogg, M.; Seki, M.; Wood, R.D.; Doublie, S.; Wallace, S.S. Lesion bypass activity of DNA polymerase theta (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J. Mol. Biol. 2011, 405, 642–652. [Google Scholar] [CrossRef]
- Seki, M.; Masutani, C.; Yang, L.W.; Schuffert, A.; Iwai, S.; Bahar, I.; Wood, R.D. High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 2004, 23, 4484–4494. [Google Scholar] [CrossRef]
- Shibutani, S.; Takeshita, M.; Grollman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef]
- Fazlieva, R.; Spittle, C.S.; Morrissey, D.; Hayashi, H.; Yan, H.; Matsumoto, Y. Proofreading exonuclease activity of human DNA polymerase delta and its effects on lesion-bypass DNA synthesis. Nucleic Acids Res. 2009, 37, 2854–2866. [Google Scholar] [CrossRef]
- Markkanen, E.; Castrec, B.; Villani, G.; Hubscher, U. A switch between DNA polymerases delta and lambda promotes error-free bypass of 8-oxo-G lesions. Proc. Natl. Acad. Sci. USA 2012, 109, 20401–20406. [Google Scholar] [CrossRef]
- Markkanen, E.; Hubscher, U.; van Loon, B. Regulation of oxidative DNA damage repair: The adenine:8-oxo-guanine problem. Cell Cycle 2012, 11, 1070–1075. [Google Scholar] [CrossRef]
- Brown, J.A.; Duym, W.W.; Fowler, J.D.; Suo, Z. Single-turnover kinetic analysis of the mutagenic potential of 8-oxo-7,8-dihydro-2'-deoxyguanosine during gap-filling synthesis catalyzed by human DNA polymerases lambda and beta. J. Mol. Biol. 2007, 367, 1258–1269. [Google Scholar] [CrossRef]
- Freudenthal, B.D.; Beard, W.A.; Wilson, S.H. DNA polymerase minor groove interactions modulate mutagenic bypass of a templating 8-oxoguanine lesion. Nucleic Acids Res. 2013, 41, 1848–1858. [Google Scholar] [CrossRef]
- Avkin, S.; Livneh, Z. Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutat. Res. 2002, 510, 81–90. [Google Scholar] [CrossRef]
- Maga, G.; Crespan, E.; Markkanen, E.; Imhof, R.; Furrer, A.; Villani, G.; Hubscher, U.; van Loon, B. DNA polymerase delta-interacting protein 2 is a processivity factor for DNA polymerase lambda during 8-oxo-7,8-dihydroguanine bypass. Proc. Natl. Acad. Sci. USA 2013, 110, 18850–18855. [Google Scholar]
- Aller, P.; Ye, Y.; Wallace, S.S.; Burrows, C.J.; Doublie, S. Crystal structure of a replicative DNA polymerase bound to the oxidized guanine lesion guanidinohydantoin. Biochemistry 2010, 49, 2502–2509. [Google Scholar]
- Beckman, J.; Wang, M.; Blaha, G.; Wang, J.; Konigsberg, W.H. Substitution of Ala for Tyr567 in RB69 DNA polymerase allows dAMP and dGMP to be inserted opposite Guanidinohydantoin. Biochemistry 2010, 49, 8554–8563. [Google Scholar] [CrossRef]
- Kino, K.; Ito, N.; Sugasawa, K.; Sugiyama, H.; Hanaoka, F. Translesion synthesis by human DNA polymerase eta across oxidative products of guanine. Nucleic Acids Symp. Ser. (Oxf.) 2014, 48, 171–172. [Google Scholar]
- Henderson, P.T.; Delaney, J.C.; Muller, J.G.; Neeley, W.L.; Tannenbaum, S.R.; Burrows, C.J.; Essigmann, J.M. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry 2003, 42, 9257–9262. [Google Scholar] [CrossRef]
- Suzuki, M.; Kino, K.; Morikawa, M.; Kobayashi, T.; Komori, R.; Miyazawa, H. Calculation of the stabilization energies of oxidatively damaged guanine base pairs with guanine. Molecules 2012, 17, 6705–6715. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar]
- Vermeulen, W.; Fousteri, M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012625. [Google Scholar]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 2011, 6, 70. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Tornaletti, S.; Maeda, L.S.; Lloyd, D.R.; Reines, D.; Hanawalt, P.C. Effect of thymine glycol on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. J. Biol. Chem. 2001, 276, 45367–45371. [Google Scholar]
- Tornaletti, S.; Maeda, L.S.; Kolodner, R.D.; Hanawalt, P.C. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair 2004, 3, 483–494. [Google Scholar] [CrossRef]
- Hoang, M.L.; Chen, C.H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar]
- Poon, S.L.; Pang, S.T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar]
- Sidorenko, V.S.; Yeo, J.E.; Bonala, R.R.; Johnson, F.; Scharer, O.D.; Grollman, A.P. Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res. 2012, 40, 2494–2505. [Google Scholar] [CrossRef]
- Charlet-Berguerand, N.; Feuerhahn, S.; Kong, S.E.; Ziserman, H.; Conaway, J.W.; Conaway, R.; Egly, J.M. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006, 25, 5481–5491. [Google Scholar] [CrossRef]
- Khobta, A.; Epe, B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 253–260. [Google Scholar] [CrossRef]
- D’Errico, M.; Pascucci, B.; Iorio, E.; van Houten, B.; Dogliotti, E. The role of CSA and CSB protein in the oxidative stress response. Mech. Ageing Dev. 2013, 134, 261–269. [Google Scholar] [CrossRef]
- Aamann, M.D.; Muftuoglu, M.; Bohr, V.A.; Stevnsner, T. Multiple interaction partners for Cockayne syndrome proteins: Implications for genome and transcriptome maintenance. Mech. Ageing Dev. 2013, 134, 212–224. [Google Scholar] [CrossRef]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef]
- Inukai, N.; Yamaguchi, Y.; Kuraoka, I.; Yamada, T.; Kamijo, S.; Kato, J.; Tanaka, K.; Handa, H. A novel hydrogen peroxide-induced phosphorylation and ubiquitination pathway leading to RNA polymerase II proteolysis. J. Biol. Chem. 2004, 279, 8190–8195. [Google Scholar]
- Pfeifer, G.P.; Besaratinia, A. Mutational spectra of human cancer. Hum. Genet. 2009, 125, 493–506. [Google Scholar] [CrossRef]
- Walser, J.C.; Furano, A.V. The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res. 2010, 20, 875–882. [Google Scholar] [CrossRef]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef]
- Galashevskaya, A.; Sarno, A.; Vagbo, C.B.; Aas, P.A.; Hagen, L.; Slupphaug, G.; Krokan, H.E. A robust, sensitive assay for genomic uracil determination by LC/MS/MS reveals lower levels than previously reported. DNA Repair 2013, 12, 699–706. [Google Scholar] [CrossRef]
- Mangerich, A.; Knutson, C.G.; Parry, N.M.; Muthupalani, S.; Ye, W.; Prestwich, E.; Cui, L.; McFaline, J.L.; Mobley, M.; Ge, Z.; et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E1820–E1829. [Google Scholar] [CrossRef]
- Chan, S.W.; Dedon, P.C. The biological and metabolic fates of endogenous DNA damage products. J. Nucleic Acids 2010, 929047. [Google Scholar]
- Gaikwad, N.W. Metabolomic profiling unravels DNA adducts in human breast that are formed from peroxidase mediated activation of estrogens to quinone methides. PLoS One 2013, 8, e65826. [Google Scholar] [CrossRef]
- Tretyakova, N.; Goggin, M.; Sangaraju, D.; Janis, G. Quantitation of DNA adducts by stable isotope dilution mass spectrometry. Chem. Res. Toxicol. 2012, 25, 2007–2035. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bacolla, A.; Cooper, D.N.; Vasquez, K.M. Mechanisms of Base Substitution Mutagenesis in Cancer Genomes. Genes 2014, 5, 108-146. https://doi.org/10.3390/genes5010108
Bacolla A, Cooper DN, Vasquez KM. Mechanisms of Base Substitution Mutagenesis in Cancer Genomes. Genes. 2014; 5(1):108-146. https://doi.org/10.3390/genes5010108
Chicago/Turabian StyleBacolla, Albino, David N. Cooper, and Karen M. Vasquez. 2014. "Mechanisms of Base Substitution Mutagenesis in Cancer Genomes" Genes 5, no. 1: 108-146. https://doi.org/10.3390/genes5010108
APA StyleBacolla, A., Cooper, D. N., & Vasquez, K. M. (2014). Mechanisms of Base Substitution Mutagenesis in Cancer Genomes. Genes, 5(1), 108-146. https://doi.org/10.3390/genes5010108