1. Introduction
Somewhere between 2% and 5% of all colorectal cancers (CRCs) are classified as hereditary non-polyposis colorectal cancer (HNPCC). Families with germline mutations or complex genomic changes (without structural gene alterations) that render one of four DNA mismatch repair (MMR) genes ineffective compose a subset of HNPCC known as Lynch Syndrome (LS).
The clinical diagnosis of HNPCC is defined by any one of several reiterations of the Amsterdam Criteria, first established in 1990 to enable the identification of the genetic basis of the disease [
1]. As such mutations in
MLH1,
MSH2,
MSH6 and
PMS2 have been identified to account for all LS families [
2,
3,
4]. Recently, loss of
EPCAM, has been associated with transcriptional silencing of
MSH2, and rare epimutations in
MLH1 have also been implicated in LS [
5,
6].
Despite the definition of HNPCC up to 50% of clinically tested patients with tumours demonstrating microsatellite instability (MSI), the hallmark phenotype of HNPCC, will fail to have any germline mutation identified in any one of the four MMR genes responsible for LS [
7,
8,
9]. This suggests that there are either other genes associated with this disorder or different mechanisms of gene silencing responsible for HNPCC.
Since the sequencing of the human genome it has become apparent that genomic rearrangements are ubiquitous in the population. Genomic duplication or deletion have been shown to encompass large stretches of contiguous DNA and are commonly termed copy number variants (CNVs). As CNVs range from kilobase (Kb) to megabase in size, they may encompass or disrupt functional DNA sequences, result in gene amplification or loss, or alter epigenetic patterning [
10]. As such, CNVs have been well documented in their contribution to disease development and variation in disease phenotype [
11,
12,
13,
14,
15,
16].
CNVs have been implicated in the development of many forms of CRC, e.g., germline deletion of two genes,
PTEN and
BMPR1A have been identified to be the cause of Juvenile Polyposis (JP) in four unrelated children [
17], while genomic deletions in the genes
SMAD4,
BMPR1A and
PTEN result in JP [
18] and furthermore, the Leiden Open Variation Database (LOVD) database lists nearly 3,000 mutations in four MMR genes associated with HNPCC, of which many are gains and losses of genomic material [
19]. Recent reports specifically examining the association between genomic rearrangements and LS have revealed that loss of a region on chromosome 2 encompassing
EPCAM appears to be associated with LS [
6,
20]. The loss of
EPCAM appears to re-write the epigenetic programming in the region such that the
MSH2 becomes silenced as a result of CpG methylation of the 5' promoter region. This evidence suggests that a proportion of HNPCC families may be accounted for by genomic rearrangements that may not be readily identified using more traditional gene mutation searches.
CNVs are detected using DNA arrays that comprise a series of oligonucleotides that represent evenly distributed markers across the entire genome. As the number of oligonucleotide markers has increased from a few hundred thousand to over five million, CNV resolution has improved such that ever smaller rearrangements can be detected in a single experiment. In this study we have used the Affymetrix Cytogenetic Whole Genome2.7M (Cyto2.7M) array which contains over 400,000 SNP probes and greater than 2.1 million CNV probes (average spacing 1,395 base pairs) to examine the CNV landscape in HNPCC patients and search for CN gains or CN losses which may reside in or in the vicinity of the 22 genes associated with DNA MMR. We also investigated genes and gene expression regulatory elements (microRNAs or miRs) associated with CNVs unique to the HNPCC patients using pathway analysis to determine if they may contribute to disease development.
2. Experimental Section
2.1. Samples
Genomic DNA samples for the current study were obtained from HNPCC patients who had given informed consent for their DNA to be used for studies into their disease and control DNA samples from the Hunter Community Study (HCS) [
21]. DNA was extracted from whole blood by the salt precipitation method [
22]. The study was approved by the University of Newcastle’s Human Research Ethics Committee (HREC) and the Hunter New England Human Research Ethics Committee (HNEHREC).
A sample size of 125 HNPCC patients was used for the current study. All HNPCC patients were clinically diagnosed as per the Amsterdam Criteria II [
1,
23] or the Bethesda Guidelines [
24]. All patients had been diagnosed with CRC and were the first individual (proband) of their family to seek genetic testing. The samples were referred for routine clinical diagnostic testing involving screening for mutations in:
MLH1,
MSH2,
MSH6 and/or
PMS2. The mutation screening was performed using Sanger Sequencing and/or Multiplex ligation-dependant probe amplification (MLPA). No mutations were identified in any of the patients used for the current study and are thus considered to be MMR mutation negative. The average age of patients recruited for this study was 52 years of age.
A sample size of 40 controls from the Hunter Community Study (HCS) [
21] was used in the current study. Theses samples were from healthy individuals aged >55 years who were cancer free at the time of sample collection.
2.2. Genomic Array Analysis
The DNA from the 165 patients and controls was processed on the Affymetrix Cyto2.7M array according to manufacturer’s protocols. CEL files obtained from scanning the Cyto2.7M array were analysed in the proprietary software from Affymetrix, the Chromosome Analysis Suite (ChAS) (Version CytoB-N1.2.0.232; r4280) using NetAffx Build 30.2 (Hg18) annotation. Quality control parameters were optimized and validated using a training set of 20 randomly selected samples (patients and controls). Identified CNV regions within the training set were assessed according to CNV call confidence, probe count, size, wavinessSd and by visual inspection for distinction from normal CN state. In addition, data was visually inspected to identify regions with low density of markers including centromeric and telomeric regions (
Supplementary Table 1) which were excluded from analysis across all samples. The resultant thresholds were applied to all samples. Most of the thresholds were more stringent than default settings, aiming to minimize the number of false-positive CNVs being included in the analysis. Briefly, all samples were subject to a series of quality cut-off measures: snpQC >1.1 (assesses quality of SNP probes with respect distances between the distribution of alleles AA, AB and BB alleles and larger differences are associated with an increased ability to identify a genotype; default), mapdQC <0.27 (Median Absolute Pair-wise Difference; assesses quality of CN probes with respect to a reference model file; default) and wavinessSd <0.1 (measure of standard deviation in data waviness; the GC content across the genome correlates with average probe intensities
i.e., high GC probes are brighter than low GC probes on average, which creates waves in the data). CNV regions within all samples were then filtered using a set of CNV calling parameters: >90% confidence, autosomes only and a minimum number of 24 probes.
2.3. Statistical, Pathway and Annotation Analysis
Refined ChAS CNV counts and CNV size for the patients were compared to the controls using a two tailed un-paired t-test in Graphpad Prism (Version 6) [
25]. Gene enrichment analysis was performed using WebGestalt analysis software (Version 2013) [
26]. This software was used to assess gene lists derived from the refined CNV results obtained from ChAS according to Gene Ontology (GO) categories, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and miR targets. Analysis was performed using hypergeometric statistical method, Benjamini and Hochberg (BH) correction for multiple testing (both default settings) and a biological significance threshold of <0.05 with a minimum of two genes per category required to assess any enrichment. TAM (Tool for Annotations of miRs) (Version 2) [
27] software was used to annotate miRs according to miR family, cluster, function, Human miR associated disease categories (HMDD) and tissue specificity. Annotations were performed using the following parameters: all miRs in the TAM database were used as a background; to identify meaningful categories we looked at miR over-representation in all categories and analysis was limited to at least one miR in a given category. Enrichment analysis for miRs categories was conducted using hypergeometric testing and
p values were corrected according to Bonferroni correction for multiple testing.
3. Results and Discussion
Recent studies have reported CNV’s as relevant contributors to human diversity and cancer susceptibility [
28,
29,
30]. This study further defines the contribution of CNVs to disease risk in HNPCC.
3.1. Resolution
Refinement of ChAS thresholds resulted in the final analysis of CNVs ranging from a minimum of 8.4 Kb to a maximum of 2,722.5 Kb in size (see
Figure 1 for examples). CN gains ranged from 8.4 Kb to 2,722 Kb in patients and 14.8 Kb to 1,076.2 Kb in controls while CN losses ranged from 17.8 Kb to 529.2 Kb in patients and 16.8 Kb to 1,205.7 Kb in controls. As such we cannot rule out the potential involvement of CNVs below the level of detection of the Cyto2.7M array, in the aetiology of HNPCC.
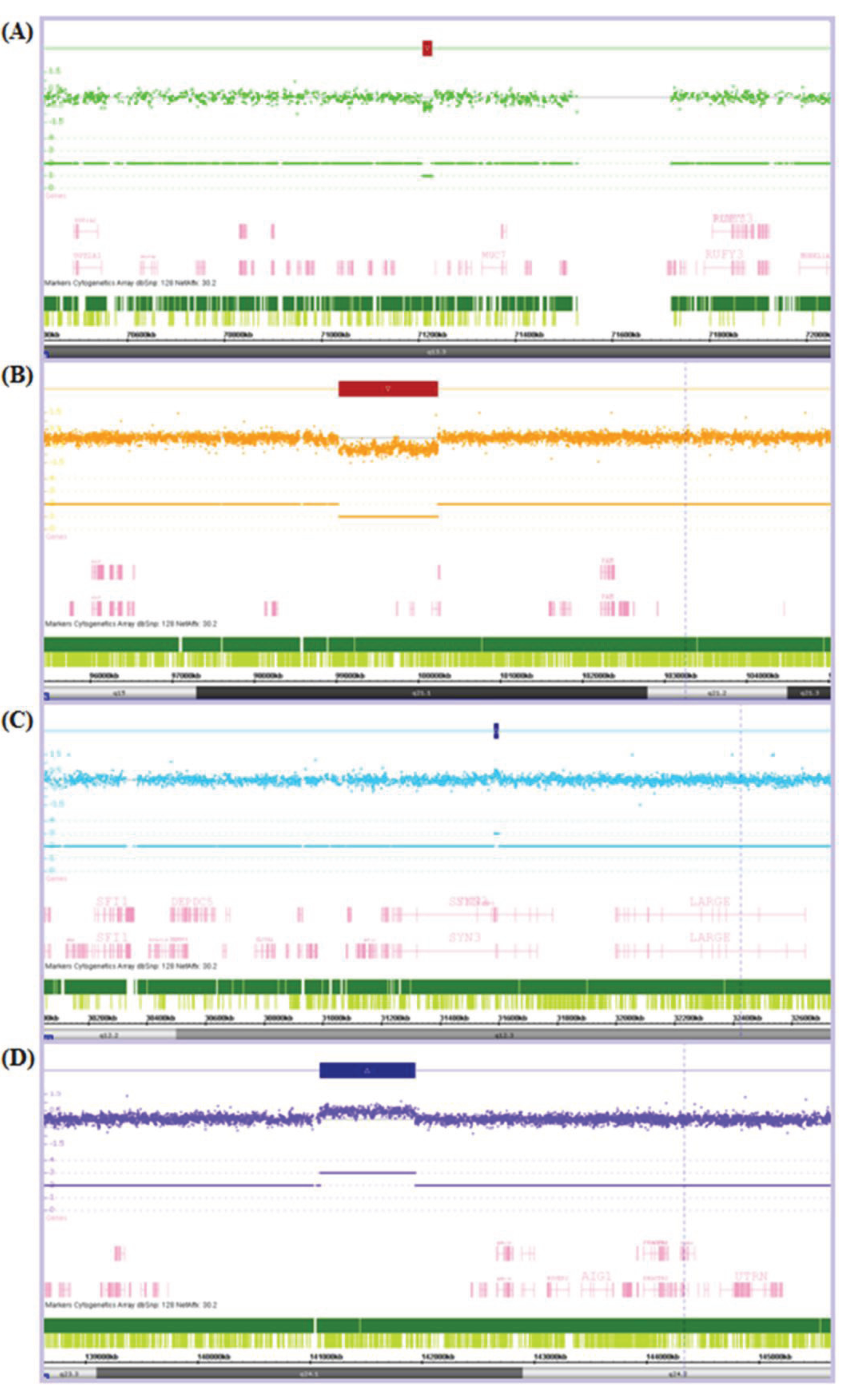
Figure 1.
Chromosome Analysis Suite (ChAS) output showing examples of (A) a small copy number (CN) loss of 17.8 Kb; (B) a large CN loss of 1,205.7 Kb; (C) a small CN gain of 14.8 Kb; and (D) a large CN gain of 843.3 Kb. The Log2Ratio represents the relative fluorescence of each probe (dot) across the genome (from left to right). The fluorescence is reduced in regions of CN loss and increased in regions of CN gain. This is indicated by a CN loss (red box) or CN gain (blue box) over the affected region and the resultant CN state noted below e.g., there is only one of the two alleles present in each CN loss and an extra allele present in each of the CN gains.
Figure 1.
Chromosome Analysis Suite (ChAS) output showing examples of (A) a small copy number (CN) loss of 17.8 Kb; (B) a large CN loss of 1,205.7 Kb; (C) a small CN gain of 14.8 Kb; and (D) a large CN gain of 843.3 Kb. The Log2Ratio represents the relative fluorescence of each probe (dot) across the genome (from left to right). The fluorescence is reduced in regions of CN loss and increased in regions of CN gain. This is indicated by a CN loss (red box) or CN gain (blue box) over the affected region and the resultant CN state noted below e.g., there is only one of the two alleles present in each CN loss and an extra allele present in each of the CN gains.
3.2. CNV Detection
Analysis of Cyto2.7M array data identified a total of 543 CNVs in the 165 patients and controls utilized in this study (
Table 1). Total counts of CNVs observed in the 125 HNPCC patients corresponded to 439 CNV events compared to 104 events in the 40 controls. The mean number of CNVs identified per sample did not significantly differ between patients and controls (3.51 CNVs per patient and 2.60 CNVs per control,
p = 0.2980). Consistent with a recent report looking at CNVs in hereditary breast cancer, similar counts of CNVs detected between patients and controls have been suggested to reflect a lack of genomic instability in the genomes of patients screened [
31,
32]. The mean CNV affected genome per sample did not differ between patients and controls either (284.07 Kb patients and 295.52 Kb controls,
p = 0.9121). However, the mean size of a CNV differed significantly between patients and controls (70.08 Kb in patients and 106.57 Kb in controls,
p = 0.0165). The exact reason why we observe this difference is unclear however it may be a function of the number of samples in each group.
Table 1.
Summary of copy number variants (CNV) results obtained from the Cyto2.7M array analysed in ChAS.
Table 1.
Summary of copy number variants (CNV) results obtained from the Cyto2.7M array analysed in ChAS.
| | CNV Count | CNV Size (Kb) |
|---|
| Participants | | Total CNVs per group | Median CNVs per sample | Mean CNVs per sample | Total CNV affected genome per group | Mean total CNV affected genome per sample | Mean size of a CNV |
|---|
| Patients | 125 | 439 | 2 | 3.51 | 35,508.53 | 284.07 | 70.08 |
| Controls | 40 | 104 | 2 | 2.60 | 11,820.75 | 295.52 | 106.57 |
| p | − | − | − | 0.2980 | − | 0.9121 | 0.0165 * |
3.3. MMR Gene Interrogation
CNVs in patients and controls were interrogated for CN gains and losses residing in or in the vicinity of (50 Kb upstream to 50 Kb downstream) the 22 genes (
EXO1,
LIG1,
MLH1,
MLH3,
MSH2,
MSH3,
MSH6,
PCNA,
PMS1,
PMS2,
POLD1,
POLD2,
POLD3,
POLD4,
RFC1,
RFC2,
RFC3,
RFC4,
RFC5,
RPA1,
RPA2 and
RPA3) in the MMR pathway (see
Table 2). We aimed to identify CNVs which could potentially contribute to disease development directly (e.g., disruption of functional gene sequences or promoter region inactivation) and via other mechanisms, including the alteration to epigenetic marks (as seen with the transcriptional silencing of
MSH2 through a CN loss in
EPCAM in several LS patients, described previously [
6,
20]).
No CN gains or losses were identified within the defined search region for any of the 22 genes in the MMR pathway for all samples utilized in this study, patients and controls. We cannot however rule of the possibility for CNVs residing in these regions which are smaller than the resolution of detection provided by this array (<8.4 Kb).
Table 2.
Regions searched for CN gains and CN losses in and in the vicinity of (±50 Kb) of the 22 genes in the mismatch repair (MMR) pathway. Chromosomal position of gene (start and end), gene size and search region (search start and search end) is noted.
Table 2.
Regions searched for CN gains and CN losses in and in the vicinity of (±50 Kb) of the 22 genes in the mismatch repair (MMR) pathway. Chromosomal position of gene (start and end), gene size and search region (search start and search end) is noted.
| Gene | Chr. | Start (bp) | End (bp) | Size (Kb) | Search start (bp) | Search end (bp) |
|---|
| EXO1 | 1 | 240,078,157 | 240,119,671 | 42 | 240,028,157 | 240,169,671 |
| RPA2 | 1 | 28,090,635 | 28,113,823 | 23 | 28,040,635 | 28,163,823 |
| MSH2 | 2 | 47,783,766 | 47,563,864 | 80 | 47,733,766 | 47,613,864 |
| MSH6 | 2 | 47,863,724 | 47,887,596 | 24 | 47,813,724 | 47,937,596 |
| PMS1 | 2 | 190,357,055 | 190,450,600 | 94 | 190,307,055 | 190,500,600 |
| MLH1 | 3 | 37,009,982 | 37,067,341 | 57 | 36,959,982 | 37,117,341 |
| RFC4 | 3 | 187,990,375 | 188,007,178 | 17 | 187,940,375 | 188,057,178 |
| RFC1 | 4 | 38,965,470 | 39,044,390 | 79 | 38,915,470 | 39,094,390 |
| MSH3 | 5 | 79,986,049 | 80,208,390 | 222 | 79,936,049 | 80,258,390 |
| PMS2 | 7 | 5,979,395 | 6,015,263 | 36 | 5,929,395 | 6,065,263 |
| POLD2 | 7 | 44,120,810 | 44,129,672 | 9 | 44,070,810 | 44,179,672 |
| RFC2 | 7 | 73,283,767 | 73,306,674 | 23 | 73,233,767 | 73,356,674 |
| RPA3 | 7 | 7,643,099 | 7,724,763 | 82 | 7,593,099 | 7,774,763 |
| POLD3 | 11 | 73,981,276 | 74,031,413 | 50 | 73,931,276 | 74,081,413 |
| POLD4 | 11 | 66,875,594 | 66,877,593 | 2 | 66,825,594 | 66,927,593 |
| RFC5 | 12 | 116,938,890 | 116,954,422 | 16 | 116,888,890 | 117,004,422 |
| RFC3 | 13 | 33,290,205 | 33,438,695 | 148 | 33,240,205 | 33,488,695 |
| MLH3 | 14 | 74,550,219 | 74,587,988 | 38 | 74,500,219 | 74,637,988 |
| RPA1 | 17 | 1,680,022 | 1,749,598 | 70 | 1,630,022 | 1,799,598 |
| LIG1 | 19 | 53,310,514 | 53,365,372 | 55 | 53,260,514 | 53,415,372 |
| POLD1 | 19 | 55,579,404 | 55,613,083 | 34 | 55,529,404 | 55,663,083 |
| PCNA | 20 | 5,043,598 | 5,055,268 | 12 | 4,993,598 | 5,105,268 |
3.4. Occurrence and Distribution of CNVs in Patients and Controls
Of the total 104 CNVs identified in controls, 34 CNVs contained genomic regions that were common to genomic regions identified in patients (
Supplementary Table 2). A total of 70 CNVs were unique to the controls of which 47 (67.14%) were associated with genes (
Supplementary Table 3).
Of 439 CNVs identified in patients, 53 CNVs contained genomic regions that were common to genomic regions identified in controls (
Supplementary Table 4). Three hundred and eighty six CNVs were unique to the patients population of which 207 (53.63%) were associated with genes (
Supplementary Table 5).
From the 207 unique CNVs associated with genes identified in the patients, 9 were identified in patients that did not overlap any CNVs in controls but affected the same gene even in multiple patients (
ARPP-21,
C7orf10,
KIAA1217,
LINGO2,
MACROD2 and
NKAIN2). A total of 60 genes associated with 131 CNVs were identified in multiple individuals (as shown in
Table 3). Fifty two genes were affected by a CNV in two individuals; five genes were affected by a CNV in three individuals (
IGSF11,
GK5,
XRN1,
NAMPT and
LCP1); and three genes were affected by a CNV in four individuals (
CTNNA3,
NRG3 and
LOC642597).
Table 3.
Genes associated with unique CNVs (compared to controls) identified across multiple patients. Number of CNV events in which gene (s) have been identified and if they were a CN gain or loss.
Table 3.
Genes associated with unique CNVs (compared to controls) identified across multiple patients. Number of CNV events in which gene (s) have been identified and if they were a CN gain or loss.
| Type | 2 CNV events | | | | 3 CNV events | 4 CNV events |
|---|
| Gains | ADARB2 | DEFB125 | ITGA1 | PSG8 | GK5 | LOC642597 |
| APC | DEFB126 | KIAA1680 | RBCK1 | IGSF11 | |
| ARHGAP19 | DEFB127 | LATS2 | RNF125 | LCP1 | |
| B2M | DEFB128 | MLL | RNF138 | XRN1 | |
| BBOX1 | DEFB129 | MSI2 | SOX12 | NAMPT | |
| C10orf139 | DEFB132 | NRSN2 | TBC1D20 | | |
| C14orf23 | EPHA7 | NXPH1 | TFG | | |
| C20orf96 | FAM134B | ODZ4 | TRIB3 | | |
| C3orf33 | FOXG1 | PELO | TRIM69 | | |
| CNTN5 | GPR128 | PHC3 | WDR37 | | |
| CNTNAP2 | GPR160 | PRKCI | ZCCHC3 | | |
| CSNK2A1 | GYPE | PSG10 | ZMYND11 | | |
| Losses | CNTN4 | | | | | CTNNA3 |
| DCDC1 | | | | | |
| PPP2R3C | | | | | |
| Both | | | | | | NRG3 |
While this study has not investigated further the contribution of any one of these CNVs to disease development, previous studies have reported the involvement of several of the genes influenced by one or more CNVs in CRC: L-plastin (subunit
LCP1) has been shown to be unregulated in various solid human tumours and is also known to contribute to CRC progression via its involvement in cell proliferation and invasion and consequently metastasis [
33,
34,
35]; alpha-catenin (subunit
CTNNA3) has been reported to show reduced expression in CRC cell lines which has been suggested to facilitate metastasis [
36], while another study has reported increased expression of alpha-catenin during adenoma formation via the negative regulation of beta-catenin signalling [
37]; the tumour suppressor gene
APC has been unequivocally associated with the CRC and Familial adenomatous polypsis (FAP) [
38,
39,
40]; and furthermore, expression of
IGSF11 has been reported to be elevated in CRC cells lines and may represent a target for cancer immunotherapy [
41]. Future studies are required to validate and investigate the role of the CNVs identified in our study for their potential contribution in the development of HNPCC.
Of the 386 CNVs identified unique to the patients, of these regions 56.5% of them have been previously reported in the Database of Genomic Variance (DGV). Fifty nine CNVs contained genomic regions which were identified in multiple patients (
Table 4). A total of 15 genomic regions were identified in two patients; five common genomic regions were identified in three patients, located on chromosomes 3, 5, 9, 11 and 12; and one genomic region was identified in four patients on chromosome 16. Two other CNVs were also shown to be common to five patients on chromosomes 3 and 5. Additional studies are required to investigate the sequence content of these regions to identify if novel contributors to disease development may reside in these regions.
Table 4.
Genomic regions associated with unique CNVs (compared to controls) identified across multiple among patients. Note CNV frequency and CNV type; CNV location (chromosome, start bp and end bp) and size; as well as the confidence score associated with CNV call and the number of probes used to call the CNV are also noted.
Table 4.
Genomic regions associated with unique CNVs (compared to controls) identified across multiple among patients. Note CNV frequency and CNV type; CNV location (chromosome, start bp and end bp) and size; as well as the confidence score associated with CNV call and the number of probes used to call the CNV are also noted.
| Chr. | Start (bp) | End (bp) | Size (Kb) | Conf | Probes |
|---|
| 2 CNV gains |
| 3 | 189,058,439 | 189,098,718 | 40.28 | 0.93 | 31 |
| 3 | 189,069,317 | 189,088,009 | 18.69 | 0.94 | 26 |
| 4 | 44,664,798 | 44,699,744 | 34.95 | 0.92 | 41 |
| 4 | 44,664,798 | 44,699,744 | 34.95 | 0.90 | 41 |
| 8 | 120,414,388 | 120,438,172 | 23.78 | 0.91 | 27 |
| 8 | 120,419,721 | 120,451,773 | 32.05 | 0.91 | 30 |
| 11 | 29,547,229 | 29,593,722 | 46.49 | 0.90 | 39 |
| 11 | 29,547,756 | 29,593,722 | 45.97 | 0.91 | 37 |
| 16 | 25,330,672 | 25,438,375 | 107.70 | 0.92 | 46 |
| 16 | 25,330,672 | 25,438,375 | 107.70 | 0.92 | 46 |
| 3 CNV gains |
| 3 | 19,014,033 | 19,041,376 | 27.34 | 0.90 | 31 |
| 3 | 19,016,875 | 19,041,376 | 24.50 | 0.91 | 28 |
| 3 | 19,016,875 | 19,041,376 | 24.50 | 0.91 | 28 |
| 5 | 59,744,695 | 59,807,906 | 63.21 | 0.92 | 52 |
| 5 | 59,744,695 | 59,811,770 | 67.08 | 0.93 | 54 |
| 5 | 59,749,693 | 59,807,906 | 58.21 | 0.92 | 51 |
| 9 | 103,982,826 | 104,016,588 | 33.76 | 0.91 | 27 |
| 9 | 103,982,826 | 104,017,715 | 34.89 | 0.90 | 28 |
| 9 | 103,991,205 | 104,017,715 | 26.51 | 0.91 | 26 |
| 11 | 15,765,333 | 15,791,331 | 26.00 | 0.90 | 30 |
| 11 | 15,770,233 | 15,796,302 | 26.07 | 0.92 | 30 |
| 11 | 15,776,946 | 15,795,665 | 18.72 | 0.91 | 24 |
| 12 | 16,469,855 | 16,503,960 | 34.11 | 0.91 | 33 |
| 12 | 16,469,855 | 16,503,960 | 34.11 | 0.91 | 33 |
| 12 | 16,476,470 | 16,506,851 | 30.38 | 0.92 | 33 |
| 4 CNV gains |
| 16 | 63,364,955 | 63,389,659 | 24.70 | 0.91 | 33 |
| 16 | 63,369,029 | 63,389,029 | 20.00 | 0.92 | 30 |
| 16 | 63,369,960 | 63,388,189 | 18.23 | 0.90 | 28 |
| 16 | 63,371,038 | 63,397,352 | 26.31 | 0.92 | 38 |
| 5 CNV gains |
| 5 | 116,651,923 | 116,698,621 | 46.70 | 0.91 | 36 |
| 5 | 116,655,439 | 116,692,153 | 36.71 | 0.92 | 27 |
| 5 | 116,656,039 | 116,695,730 | 39.69 | 0.91 | 29 |
| 5 | 116,660,694 | 116,697,347 | 36.65 | 0.93 | 28 |
| 5 | 116,660,694 | 116,693,035 | 32.34 | 0.91 | 24 |
| 2 CNV losses |
| 1 | 82,801,000 | 82,821,932 | 20.93 | 0.93 | 31 |
| 1 | 82,801,000 | 82,821,932 | 20.93 | 0.94 | 31 |
| 2 | 22,087,558 | 22,261,901 | 174.34 | 0.91 | 110 |
| 2 | 22,087,558 | 22,261,901 | 174.34 | 0.93 | 110 |
| 2 | 215,167,158 | 215,204,595 | 37.44 | 0.93 | 49 |
| 2 | 215,167,158 | 215,204,595 | 37.44 | 0.91 | 49 |
| 3 | 6,562,398 | 6,603,706 | 41.31 | 0.94 | 42 |
| 3 | 6,562,398 | 6,603,706 | 41.31 | 0.93 | 42 |
| 3 | 166,523,809 | 166,565,186 | 41.38 | 0.95 | 39 |
| 3 | 166,525,250 | 166,565,186 | 39.94 | 0.93 | 38 |
| 5 | 61,460,851 | 61,504,678 | 43.83 | 0.95 | 31 |
| 5 | 61,460,851 | 61,504,678 | 43.83 | 0.93 | 31 |
| 7 | 92,319,307 | 92,343,906 | 24.60 | 0.94 | 26 |
| 7 | 92,319,307 | 92,343,906 | 24.60 | 0.94 | 26 |
| 9 | 104,331,902 | 104,396,632 | 64.73 | 0.96 | 35 |
| 9 | 104,331,902 | 104,396,632 | 64.73 | 0.96 | 35 |
| 5 CNV losses |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.93 | 26 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.94 | 26 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.96 | 26 |
| 3 | 177,370,126 | 177,399,625 | 29.50 | 0.93 | 27 |
| 3 | 177,370,126 | 177,396,832 | 26.71 | 0.93 | 26 |
| 2 CNV gain and loss |
| 7 * | 110,748,452 | 111,047,157 | 298.71 | 0.93 | 291 |
| 7 ** | 111,007,466 | 111,052,498 | 45.03 | 0.94 | 25 |
| 3 ** | 21,228,980 | 21,313,310 | 84.33 | 0.90 | 88 |
| 3 * | 21,273,619 | 21,339,035 | 65.42 | 0.92 | 62 |
3.5. Pathway Analysis
WebGestalt [
26] pathway analysis software was then used to compare a list of 317 genes associated with CNVs uniquely identified across all patients (compared to controls) to all genes in the human genome (Supplementary Table 6). Enrichment analysis of KEGG pathways and miR targets was conducted. In relation to the control population we also undertook a similar analysis which revealed three pathways, a tight junction pathway and two related pathways, an oocyte meiosis and a progesterone mediated oocyte maturation related pathway. Of particular note was the absence of any cancer related pathway in the control population suggests that those identified in the patient population are likely to be involved in some aspect of malignancy.
KEGG analysis revealed a total of 18 significant pathways in which genes uniquely identified in the patients were enriched (
Table 5). The most significant pathways identified included those of the carbohydrate digestion and absorption (
p = 0.0012); starch and sucrose metabolism (
p = 0.0017); and metabolic pathways (
p = 0.0023) affecting a total of 11 patients. Previous studies have suggested that changes occurring in metabolic pathways are commonly observed during carcinogenesis and tumour growth [
42,
43]. In the context of this study, these results suggest the potential existence of a germline predisposition in the affected patients which lead to metabolic conditions that promote disease development. The tight junction pathway (
p = 0.0058) and neurotrophin signalling pathway (
p = 0.0058) were also identified to be enriched and have been shown to play a role in gut permeability and motility [
44,
45,
46]. These pathways have been well documented for their contribution to CRC [
47,
48,
49,
50]. It is interesting to also note among the enriched KEGG pathways the prostate cancer pathway (
p = 0.0251) and endometrial cancer pathway (
p = 0.0251) also featured and represent two cancers commonly arising in the general population and in the setting of HNPCC/LS [
51,
52,
53]. Overall, our KEGG results suggest the existence of genetic risk factors which may act to promote the development of cancer.
Table 5.
Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways from genes identified from CNVs unique to patients.
Table 5.
Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways from genes identified from CNVs unique to patients.
| KEGG Pathway | Genes in Pathway | Observed | Expected | p |
|---|
| Carbohydrate digestion and absorption | 44 | 5 | 0.32 | 0.0012 |
| Starch and sucrose metabolism | 54 | 5 | 0.4 | 0.0017 |
| Metabolic pathways | 1,130 | 21 | 8.31 | 0.0023 |
| Salivary secretion | 89 | 5 | 0.65 | 0.0058 |
| Tight junction | 132 | 6 | 0.97 | 0.0058 |
| Neurotrophin signaling pathway | 127 | 6 | 0.93 | 0.0058 |
| Propanoate metabolism | 32 | 3 | 0.24 | 0.017 |
| Valine, leucine and isoleucine degradation | 44 | 3 | 0.32 | 0.0251 |
| Prostate cancer | 89 | 4 | 0.65 | 0.0251 |
| Ribosome biogenesis in eukaryotes | 80 | 4 | 0.59 | 0.0251 |
| ErbB signaling pathway | 87 | 4 | 0.64 | 0.0251 |
| mRNA surveillance pathway | 83 | 4 | 0.61 | 0.0251 |
| Terpenoid backbone biosynthesis | 15 | 2 | 0.11 | 0.0285 |
| Malaria | 51 | 3 | 0.37 | 0.0293 |
| Endometrial cancer | 52 | 3 | 0.38 | 0.0293 |
| Glycerolipid metabolism | 50 | 3 | 0.37 | 0.0293 |
| Olfactory transduction | 388 | 8 | 2.85 | 0.0346 |
| beta-Alanine metabolism | 22 | 2 | 0.16 | 0.0439 |
Enrichment analysis for targets of miRs identified 65 significant regions within the 3' UTR of the CNV impacted genes unique in the patients. We identified
in silico 114 miRs (Supplementary Table 7) that target these genes regions with over 35% of these having previously been reported to have associations with CRC [
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76]. Of the top 10 most significant regions, 40% of the miRs we identified using our approach have been associated with CRC (mi
R-141, mi
R-15A, mi
R-15B, mi
R-18A, mi
R-200A, mi
R-200B, mi
R-203, mi
R-32, mi
R-429 and mi
R-92). Overall, our miR enrichment analysis supports reported findings on miR involvement in CRC. Confirmation of the respective miRs by functional analysis is required to unequivocally demonstrate their role in HNPCC.
In summary the results obtained from the pathway analysis suggest that many of the genes associated with CNVs uniquely identified in patients are associated with carcinogenesis, tumour growth and disease susceptibility and may be factors in the development of CRC.
3.6. MicroRNA Annotation
From the 317 genes affected by a CNV in the patient cohort we identified, using pathway analysis, 65 genes that enriched 3' UTR microRNA target regions. These 65 binding regions were associated with 114 proposed regulatory microRNAs. TAM (a Tool for Annotations of miRs) software [
27] was then used to identify meaningful miR categories among the 114 miRs that target significantly enriched 3' UTR regions identified in the patients from previous pathway analysis (
Table 6). We identified a total of 261 miR categories: 22 families, 33 clusters, 39 functional categories, 162 HMDD and 5 tissue specificity categories. It was identified that miRs were enriched in the family category miR-17 (
p = 0.0011). A total of 10 functional categories were enriched including those associated with onco-miRs (
p = 0.0264), processes of apoptosis (
p = 0.0291) and cell-cycle (
p = 0.0406). For the HMDD category miRs were enriched in various forms of cancer, with cancer enrichment alone accounting for 80% of the most significant findings.
Table 6.
Summary of significant findings from miR annotation analysis in TAM (A Tool for Annotations of miRs) software.
Table 6.
Summary of significant findings from miR annotation analysis in TAM (A Tool for Annotations of miRs) software.
| Category | # sub-categories | # of Sig. Categories | Significant Findings |
|---|
| Family | 22 | 1 | miR-17 |
| Cluster | 33 | 0 | − |
| Function | 39 | 8 | Onco-miRs, Apoptosis, Cell cycle related |
| HMDD | 162 | 20 | Cancer (80%), Cardiovascular (4%), Infection (4%) and Psychological disorders (10%) |
| Tissue | 5 | 1 | Placenta |
In the context of our study it was reassuring to note the presence of adenocarcinoma (
p = 0.0000155) and colorectal neoplasm’s (
p = 0.0253) among the cancers enriched in miR annotation. Cardiovascular diseases (4%), psychological disorders (10%) and infection (4%) represented the minority of other significant finds (all with
p < 0.0047). According to tissue specificity, the placenta represented the most significant miR enriched tissue (
p = 0.00001284). Previous studies have suggested that processes of angiogenesis and vascularisation occurring during placental development in pregnancy are also present during tumour development and this has been observed in CRC [
77,
78,
79].
Overall the results obtained from the TAM analysis suggest that the 114 miRs associated with the 3' UTR regions significantly identified in the patients may stimulate processes leading to carcinogenesis which is consistent with what we expect to find in these cancer patients [
80].
3.7. CNV Burden
A study by Girirajan and Eichler [
81] has suggested that the severity of disease may be explained by the overall burden CNVs place on an individual’s genome where increased sensitivity to developing disease is correlated with increased CNV burden and furthermore that variation in CNV burden will result in phenotype variation in patients. In a recent study looking at both HNPCC MMR mutation negative and MMR mutation positive patients, we observed an increased average size of CNVs in patients tested and suggested that this was related to an increased genomic burden [
82]. An increased CNV burden was not observed among the patients utilized in the current study, though we did detect a decreased mean size of CNVs in patients compared to controls. Importantly, the current study compared 125 patients and 40 controls whereas the recent HNPCC study compared 96 patients and 384 controls [
82]. We suggest that discrepancies in these findings are likely to be related to the inequity of sample populations between studies, the limited number of controls used in the current study, the type of array used (noting differences in both the array coverage and density), as well as the algorithm used by analysis software may all contribute to variation in the observed results [
83,
84,
85].
3.8. CNV Bias
CNV analysis has suffered from a lack of standardization in analytical techniques used for data mining. The Hidden Markov Model (HMM) and Circular Binary Segmentation (CBS) represent the algorithms utilized to develop CNV calling programs that have been reported to be the most efficient [
83,
84,
85]. Furthermore, using algorithms developed for a specific data type has been shown to perform better in CNV calling compared to platform-independent software algorithms [
86]. The robustness of software algorithms, batch effects, and population stratification will therefore influence the accuracy of calls made to segmented data and hence the reliability of CNV calls and CNV boundary descriptors derived from arrays [
83,
84,
85]. The Cyto2.7M array was chosen for use in the current study as at the time it provided the greatest density and most even genomic coverage of any CNV arrays. All data was analysed through ChAS which uses a HMM-based algorithm and was specifically developed to use with the Cyto2.7M array data.
To improve our predictive accuracy we did consider merging other CNV data sets that were at our disposal [
82], but this would result in a significant loss of information since the our original data set was created using a SNP based array which did not have even coverage across the genome whereas the one used in this report was an oligo-based array with uniform coverage. Any merged dataset would lose a significant amount of information.
Finally, a recent report suggesting the use of a minimum of three different algorithms when conducting CNV association analysis, this was not possible due to limitations in the data generated by the array used in this study [
87].
4. Conclusions
We were unable to identify any DNA mismatch repair genes targeted by CNVs that may contribute to a significant proportion of HNPCC patients recruited into this study. We did identify several genomic regions that were altered in multiple unrelated HNPCC patients that could potentially be associated with disease risk. The genomic regions encompassed by these CNVs warrant further study to define precisely their role in disease development. We could not rule out the existence of CNVs, smaller than the limits of detection provided by this array, from involvement in the aetiology of HNPCC.
Pathway analysis was thus utilized to identify possible common pathways associated with the heterogeneous outcomes of the analysis. We identified a total of 317 genes impacted by CNVs uniquely identified in patients (compared to controls). Results from KEGG pathway analysis identified the enrichment of pathways involved in metabolism, and these are known to be required for cancer development. It is likely that these loci may contribute to CRC disease risk in the affected individuals. miR enrichment analysis has further highlighted a series of miRs which are suggested to contribute to carcinogenesis. It was found that over 40% of these miRs had been previously reported to play a role in CRC development. As such we have shown that CNV altered genes are over represented in pathways leading to carcinogenesis, tumour growth and disease susceptibility, including CRC. The genes driving pathway enrichments require further investigation to elucidate their precise role in disease development.
The annotation of 114 miRs (reported in the pathway analysis) identified significant functional miR categories associated with cancer, including specifically adenocarcinomas and colorectal neoplasms. Placental tissue was identified to be among the tissues most significantly enriched with the miR looked at in this investigation. We speculate that processes of angiogenesis and vascularisation necessary for placental development are also present during tumour formation including those observed in CRC. As such the genes associated with CNVs we have identified are targeted by miRs which are implicated in various processes leading to malignancy. We conclude that while we have not shown direct consequences of miRs interacting with our CNV altered genes, the separate effects of aberrant miR expression and CNVs impacting on genes that such miRs target may have similar consequences.
Overall the results of this study provide some evidence of CNV involvement in the aetiology of HNPCC and furthermore reinforce that CNV probe arrays compared to SNP arrays appear to be of limited utility for CNV detection.
Acknowledgements
This work has been supported by the following funding bodies and institutions: Australian Rotary Health/Rotary District 9650, the Commonwealth Scientific and Industrial Research Organization (CSIRO), the University of Newcastle and the Hunter Medical Research Institute. Samples were provided by the Hunter Area Pathology Service and the Hunter Community Study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International collaborative group on hereditary non-polyposis colorectal cancer (icg-hnpcc). Dis. Colon. Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef]
- Kemp, Z.; Thirlwell, C.; Sieber, O.; Silver, A.; Tomlinson, I. An update on the genetics of colorectal cancer. Hum. Mol. Genet. 2004, 13, R177–R185. [Google Scholar] [CrossRef]
- Peltomaki, P. Deficient DNA mismatch repair: A common etiologic factor for colon cancer. Hum. Mol. Genet. 2001, 10, 735–740. [Google Scholar] [CrossRef]
- Thompson, E.; Meldrum, C.J.; Crooks, R.; McPhillips, M.; Thomas, L.; Spigelman, A.D.; Scott, R.J. Hereditary non-polyposis colorectal cancer and the role of hPMS2 and hEXO1 mutations. Clin. Genet. 2004, 65, 215–225. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Vissers, L.E.; Venkatachalam, R.; Bodmer, D.; Hoenselaar, E.; Goossens, M.; Haufe, A.; Kamping, E.; Niessen, R.C.; Hogervorst, F.B.; et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011, 32, 407–414. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef]
- McPhillips, M.; Meldrum, C.J.; Creegan, R.; Edkins, E.; Scott, R.J. Deletion Mutations in an Australian Series of HNPCC Patients. Hered. Cancer Clin. Pract. 2005, 3, 43–47. [Google Scholar] [CrossRef]
- Bonis, P.A.; Trikalinos, T.A.; Chung, M.; Chew, P.; Ip, S.; DeVine, D.A.; Lau, J. Hereditary nonpolyposis colorectal cancer: Diagnostic strategies and their implications. Evid. Rep. Technol. Assess. 2007, 150, 1–180. [Google Scholar]
- Obermair, A.; Youlden, D.R.; Young, J.P.; Lindor, N.M.; Baron, J.A.; Newcomb, P.A.; Parry, S.; Hopper, J.L.; Haile, R.; Jenkins, M.A. Risk of Endometrial Cancer for women diagnosed with HNPCC-related colorectal cancer. Int. J. Cancer 2010, 127, 7. [Google Scholar]
- Ionita-Laza, I.; Rogers, A.J.; Lange, C.; Raby, B.A.; Lee, C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 2009, 93, 22–26. [Google Scholar] [CrossRef]
- Almal, S.H.; Padh, H. Implications of gene copy-number variation in health and diseases. J. Hum. Genet. 2012, 57, 6–13. [Google Scholar] [CrossRef]
- Bronstad, I.; Wolff, A.S.; Lovas, K.; Knappskog, P.M.; Husebye, E.S. Genome-wide copy number variation (CNV) in patients with autoimmune Addison’s disease. BMC Med. Genet. 2011, 12, 111. [Google Scholar] [CrossRef]
- Grozeva, D.; Kirov, G.; Ivanov, D.; Jones, I.R.; Jones, L.; Green, E.K.; St Clair, D.M.; Young, A.H.; Ferrier, N.; Farmer, A.E.; et al. Rare copy number variants: A point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch. Gen. Psychiatry 2010, 67, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Hai, R.; Pei, Y.F.; Shen, H.; Zhang, L.; Liu, X.G.; Lin, Y.; Ran, S.; Pan, F.; Tan, L.J.; Lei, S.F.; et al. Genome-wide association study of copy number variation identified gremlin1 as a candidate gene for lean body mass. J. Hum. Genet. 2012, 57, 33–37. [Google Scholar] [CrossRef]
- Jiang, Q.; Ho, Y.Y.; Hao, L.; Nichols Berrios, C.; Chakravarti, A. Copy number variants in candidate genes are genetic modifiers of Hirschsprung disease. PLoS One 2011, 6, e21219. [Google Scholar]
- Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010, 464, 713–720. [Google Scholar] [CrossRef]
- Delnatte, C.; Sanlaville, D.; Mougenot, J.F.; Vermeesch, J.R.; Houdayer, C.; Blois, M.C.; Genevieve, D.; Goulet, O.; Fryns, J.P.; Jaubert, F.; et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am. J. Hum. Genet. 2006, 78, 1066–1074. [Google Scholar] [CrossRef]
- Van Hattem, W.A.; Brosens, L.A.; de Leng, W.W.; Morsink, F.H.; Lens, S.; Carvalho, R.; Giardiello, F.M.; Offerhaus, G.J. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut 2008, 57, 623–627. [Google Scholar] [CrossRef]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Nagasaka, T.; Rhees, J.; Kloor, M.; Gebert, J.; Naomoto, Y.; Boland, C.R.; Goel, A. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010, 70, 3098–3108. [Google Scholar] [CrossRef]
- McEvoy, M.; Smith, W.; D’Este, C.; Duke, J.; Peel, R.; Schofield, P.; Scott, R.; Byles, J.; Henry, D.; Ewald, B.; et al. Cohort profile: The hunter community study. Int. J. Epidemiol. 2010, 39, 1452–1463. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A national cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: Meeting highlights and bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef]
- GraphPad Software. Available online: http://www.graphpad.com/quickcalcs/ttest1/ (accessed on 27 February 2013).
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Lu, M.; Shi, B.; Wang, J.; Cao, Q.; Cui, Q. TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinf. 2010, 11, 419. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Maner, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef]
- Krepischi, A.C.; Achatz, M.I.; Santos, E.M.; Costa, S.S.; Lisboa, B.C.; Brentani, H.; Santos, T.M.; Goncalves, A.; Nobrega, A.F.; Pearson, P.L.; et al. Germline DNA copy number variation in familial and early-onset breast cancer. Breast Cancer Res. 2012, 14, R24. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.J.; Farrington, S.M.; Dunlop, M.G.; Campbell, H. Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: A HuGE review. Am. J. Epidemiol. 2002, 156, 885–902. [Google Scholar] [CrossRef]
- Foran, E.; McWilliam, P.; Kelleher, D.; Croke, D.T.; Long, A. The leukocyte protein L-plastin induces proliferation, invasion and loss of E-cadherin expression in colon cancer cells. Int. J. Cancer 2006, 118, 2098–2104. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, M.; Yoshikawa, T.; Chen, H.; Brown, E.J.; Masuho, Y.; Omata, M.; Seki, N. Differential expression of the L-plastin gene in human colorectal cancer progression and metastasis. Biochem. Biophys. Res. Commun. 2001, 289, 876–881. [Google Scholar] [CrossRef]
- Park, T.; Chen, Z.P.; Leavitt, J. Activation of the leukocyte plastin gene occurs in most human cancer cells. Cancer Res. 1994, 54, 1775–1781. [Google Scholar]
- Sygut, A.; Przybylowska, K.; Ferenc, T.; Dziki, L.; Spychalski, M.; Mik, M.; Dziki, A. Genetic Variations of the CTNNA1 And The CTNNB1 Genes in Sporadic Colorectal Cancer in Polish Population. Pol. Przegl. Chir. 2012, 84, 560–564. [Google Scholar]
- Giannini, A.L.; Vivanco, M.; Kypta, R.M. Alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J. Biol. Chem. 2000, 275, 21883–21888. [Google Scholar] [CrossRef]
- Patel, S.G.; Ahnen, D.J. Familial colon cancer syndromes: An update of a rapidly evolving field. Curr. Gastroenterol. Rep. 2012, 14, 428–438. [Google Scholar] [CrossRef]
- Albuquerque, C.; Baltazar, C.; Filipe, B.; Penha, F.; Pereira, T.; Smits, R.; Cravo, M.; Lage, P.; Fidalgo, P.; Claro, I.; et al. Colorectal cancers show distinct mutation spectra in members of the canonical WNT signaling pathway according to their anatomical location and type of genetic instability. Genes Chromosomes Cancer 2010, 49, 746–759. [Google Scholar]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta-catenin signalling thresholds for tumourigenesis. Oncogene 2012. [Google Scholar] [CrossRef]
- Watanabe, T.; Suda, T.; Tsunoda, T.; Uchida, N.; Ura, K.; Kato, T.; Hasegawa, S.; Satoh, S.; Ohgi, S.; Tahara, H.; et al. Identification of immunoglobulin superfamily 11 (IGSF11) as a novel target for cancer immunotherapy of gastrointestinal and hepatocellular carcinomas. Cancer Sci. 2005, 96, 498–506. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Munoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef]
- Joo, Y.E. Increased expression of brain-derived neurotrophic factor in irritable bowel syndrome and its correlation with abdominal pain (Gut 2012;61:685–694). J. Neurogastroenterol. Motil. 2013, 19, 109–111. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability, and autoimmunity: Celiac disease and type 1 diabetes paradigms. Ann. N. Y. Acad. Sci. 2009, 1165, 195–205. [Google Scholar] [CrossRef]
- Akil, H.; Perraud, A.; Melin, C.; Jauberteau, M.O.; Mathonnet, M. Fine-tuning roles of endogenous brain-derived neurotrophic factor, TrkB and sortilin in colorectal cancer cell survival. PLoS One 2011, 6, e25097. [Google Scholar]
- Enam, S.; Gan, D.D.; White, M.K.; del Valle, L.; Khalili, K. Regulation of human neurotropic JCV in colon cancer cells. Anticancer Res. 2006, 26, 833–841. [Google Scholar]
- Wang, X.; Tully, O.; Ngo, B.; Zitin, M.; Mullin, J.M. Epithelial tight junctional changes in colorectal cancer tissues. Sci. World J. 2011, 11, 826–841. [Google Scholar] [CrossRef]
- Soler, A.P.; Miller, R.D.; Laughlin, K.V.; Carp, N.Z.; Klurfeld, D.M.; Mullin, J.M. Increased tight junctional permeability is associated with the development of colon cancer. Carcinogenesis 1999, 20, 1425–1431. [Google Scholar] [CrossRef]
- Grindedal, E.M.; Moller, P.; Eeles, R.; Stormorken, A.T.; Bowitz-Lothe, I.M.; Landro, S.M.; Clark, N.; Kvale, R.; Shanley, S.; Maehle, L. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2460–2467. [Google Scholar] [CrossRef]
- Desai, M.D.; Saroya, B.S.; Lockhart, A.C. Investigational therapies targeting the ErbB (EGFR, HER2, HER3, HER4) family in GI cancers. Expert Opin. Invest. Drugs 2013, 22, 341–356. [Google Scholar] [CrossRef]
- Khelwatty, S.A.; Essapen, S.; Seddon, A.M.; Modjtahedi, H. Prognostic significance and targeting of HER family in colorectal cancer. Front. Biosci. 2013, 18, 394–421. [Google Scholar] [CrossRef]
- Baraniskin, A.; Birkenkamp-Demtroder, K.; Maghnouj, A.; Zollner, H.; Munding, J.; Klein-Scory, S.; Reinacher-Schick, A.; Schwarte-Waldhoff, I.; Schmiegel, W.; Hahn, S.A. MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis 2012, 33, 732–739. [Google Scholar] [CrossRef]
- Bauer, K.M.; Hummon, A.B. Effects of the miR-143/-145 microRNA cluster on the colon cancer proteome and transcriptome. J. Proteome Res. 2012, 11, 4744–4754. [Google Scholar] [CrossRef]
- Cekaite, L.; Rantala, J.K.; Bruun, J.; Guriby, M.; Agesen, T.H.; Danielsen, S.A.; Lind, G.E.; Nesbakken, A.; Kallioniemi, O.; Lothe, R.A.; et al. MiR-9, -31, and -182 deregulation promote proliferation and tumor cell survival in colon cancer. Neoplasia 2012, 14, 868–879. [Google Scholar]
- Cheng, H.; Zhang, L.; Cogdell, D.E.; Zheng, H.; Schetter, A.J.; Nykter, M.; Harris, C.C.; Chen, K.; Hamilton, S.R.; Zhang, W. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One 2011, 6, e17745. [Google Scholar] [CrossRef]
- Dai, L.; Wang, W.; Zhang, S.; Jiang, Q.; Wang, R.; Dai, L.; Cheng, L.; Yang, Y.; Wei, Y.Q.; Deng, H.X. Vector-based miR-15a/16-1 plasmid inhibits colon cancer growth in vivo. Cell. Biol. Int. 2012, 36, 765–770. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- He, X.; Dong, Y.; Wu, C.W.; Zhao, Z.; Ng, S.S.; Chan, F.K.; Sung, J.J.; Yu, J. MicroRNA-218 inhibits cell cycle progression and promotes apoptosis in colon cancer by downregulating oncogene BMI-1. Mol. Med. 2012, 18, 1491–1498. [Google Scholar]
- Kenny, E.E.; Pe’er, I.; Karban, A.; Ozelius, L.; Mitchell, A.A.; Ng, S.M.; Erazo, M.; Ostrer, H.; Abraham, C.; Abreu, M.T.; et al. A genome-wide scan of Ashkenazi Jewish Crohn’s disease suggests novel susceptibility loci. PLoS Genet. 2012, 8, e1002559. [Google Scholar] [CrossRef] [Green Version]
- Migliore, C.; Martin, V.; Leoni, V.P.; Restivo, A.; Atzori, L.; Petrelli, A.; Isella, C.; Zorcolo, L.; Sarotto, I.; Casula, G.; et al. MiR-1 downregulation cooperates with MACC1 in promoting MET overexpression in human colon cancer. Clin. Cancer Res. 2012, 18, 737–747. [Google Scholar] [CrossRef]
- Nie, J.; Liu, L.; Zheng, W.; Chen, L.; Wu, X.; Xu, Y.; Du, X.; Han, W. MicroRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis 2012, 33, 220–225. [Google Scholar] [CrossRef]
- Okamoto, K.; Ishiguro, T.; Midorikawa, Y.; Ohata, H.; Izumiya, M.; Tsuchiya, N.; Sato, A.; Sakai, H.; Nakagama, H. MiR-493 induction during carcinogenesis blocks metastatic settlement of colon cancer cells in liver. EMBO J. 2012, 31, 1752–1763. [Google Scholar] [CrossRef]
- Okayama, H.; Schetter, A.J.; Harris, C.C. MicroRNAs and inflammation in the pathogenesis and progression of colon cancer. Dig. Dis. 2012, 30, 9–15. [Google Scholar] [CrossRef]
- Qased, A.B.; Yi, H.; Liang, N.; Ma, S.; Qiao, S.; Liu, X. MicroRNA-18a upregulates autophagy and ataxia telangiectasia mutated gene expression in HCT116 colon cancer cells. Mol. Med. Rep. 2013, 7, 559–564. [Google Scholar]
- Roy, S.; Levi, E.; Majumdar, A.P.; Sarkar, F.H. Expression of miR-34 is lost in colon cancer which can be re-expressed by a novel agent CDF. J. Hematol. Oncol. 2012, 5, 58. [Google Scholar] [CrossRef]
- Slaby, O.; Svoboda, M.; Michalek, J.; Vyzula, R. MicroRNAs in colorectal cancer: Translation of molecular biology into clinical application. Mol. Cancer 2009, 8, 102. [Google Scholar] [CrossRef]
- Strillacci, A.; Valerii, M.C.; Sansone, P.; Caggiano, C.; Sgromo, A.; Vittori, L.; Fiorentino, M.; Poggioli, G.; Rizzello, F.; Campieri, M.; et al. Loss of miR-101 expression promotes Wnt/beta-catenin signalling pathway activation and malignancy in colon cancer cells. J. Pathol. 2013, 229, 379–389. [Google Scholar] [CrossRef]
- Sun, J.Y.; Huang, Y.; Li, J.P.; Zhang, X.; Wang, L.; Meng, Y.L.; Yan, B.; Bian, Y.Q.; Zhao, J.; Wang, W.Z.; et al. MicroRNA-320a suppresses human colon cancer cell proliferation by directly targeting beta-catenin. Biochem. Biophys. Res. Commun. 2012, 420, 787–792. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Yang, Z.; Du, H.; Wu, Z.; Gong, J.; Yan, J.; Zheng, Q. MiR-145 regulates PAK4 via the MAPK pathway and exhibits an antitumor effect in human colon cells. Biochem. Biophys. Res. Commun. 2012, 427, 444–449. [Google Scholar] [CrossRef]
- Weissmann-Brenner, A.; Kushnir, M.; Lithwick Yanai, G.; Aharonov, R.; Gibori, H.; Purim, O.; Kundel, Y.; Morgenstern, S.; Halperin, M.; Niv, Y.; et al. Tumor microRNA-29a expression and the risk of recurrence in stage II colon cancer. Int. J. Oncol. 2012, 40, 2097–2103. [Google Scholar]
- Wu, J.; Ji, X.; Zhu, L.; Jiang, Q.; Wen, Z.; Xu, S.; Shao, W.; Cai, J.; Du, Q.; Zhu, Y.; et al. Up-regulation of microRNA-1290 impairs cytokinesis and affects the reprogramming of colon cancer cells. Cancer Lett. 2013, 329, 155–163. [Google Scholar] [CrossRef]
- Wu, J.; Wu, G.; Lv, L.; Ren, Y.F.; Zhang, X.J.; Xue, Y.F.; Li, G.; Lu, X.; Sun, Z.; Tang, K.F. MicroRNA-34a inhibits migration and invasion of colon cancer cells via targeting to Fra-1. Carcinogenesis 2012, 33, 519–528. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, Z.; Lai, D.; Sun, J.; He, C.; Chu, Z.; Ye, H.; Chen, S.; Wang, J. MiR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br. J. Cancer 2012, 107, 352–359. [Google Scholar] [CrossRef]
- Zhu, R.; Yang, Y.; Tian, Y.; Bai, J.; Zhang, X.; Li, X.; Peng, Z.; He, Y.; Chen, L.; Pan, Q.; et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS One 2012, 7, e32170. [Google Scholar]
- Harada, O.; Suga, T.; Suzuki, T.; Nakamoto, K.; Kobayashi, M.; Nomiyama, T.; Nadano, D.; Ohyama, C.; Fukuda, M.N.; Nakayama, J. The role of trophinin, an adhesion molecule unique to human trophoblasts, in progression of colorectal cancer. Int. J. Cancer 2007, 121, 1072–1078. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Fukuda, Y.; Ohshima, K.; Terashima, M.; Yamaguchi, K.; Mochizuki, T. Placenta—Specific novel splice variants of Rho GDP dissociation inhibitor beta are highly expressed in cancerous cells. BMC Res. Notes 2012, 5, 666. [Google Scholar] [CrossRef]
- Wei, S.C.; Tsao, P.N.; Yu, S.C.; Shun, C.T.; Tsai-Wu, J.J.; Wu, C.H.; Su, Y.N.; Hsieh, F.J.; Wong, J.M. Placenta growth factor expression is correlated with survival of patients with colorectal cancer. Gut 2005, 54, 666–672. [Google Scholar] [CrossRef]
- Hrasovec, S.; Glavac, D. MicroRNAs as Novel Biomarkers in Colorectal Cancer. Front. Genet. 2012, 3, 180. [Google Scholar]
- Girirajan, S.; Eichler, E.E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010, 19, R176–R187. [Google Scholar] [CrossRef]
- Talseth-Palmer, B.A.; Holliday, E.G.; Evans, T.J.; McEvoy, M.; Attia, J.; Grice, D.M.; Masson, A.L.; Meldrum, C.; Spigelman, A.; Scott, R.J. Continuing difficulties in interpreting CNV data: lessons from a genome-wide CNV association study of Australian HNPCC/lynch syndrome patients. BMC Med. Genomics 2013, 6, 10. [Google Scholar] [CrossRef]
- Dellinger, A.E.; Saw, S.M.; Goh, L.K.; Seielstad, M.; Young, T.L.; Li, Y.J. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38, e105. [Google Scholar] [CrossRef]
- Tsuang, D.W.; Millard, S.P.; Ely, B.; Chi, P.; Wang, K.; Raskind, W.H.; Kim, S.; Brkanac, Z.; Yu, C.E. The effect of algorithms on copy number variant detection. PLoS One 2010, 5, e14456. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, Y.; Akula, N.; Alliey-Rodriguez, N.; Tang, J.; Bipolar Genome, S.; Gershon, E.S.; Liu, C. Accuracy of CNV detection from GWAS data. PLoS One 2011, 6, e14511. [Google Scholar] [CrossRef]
- Pinto, D.; Darvishi, K.; Shi, X.; Rajan, D.; Rigler, D.; Fitzgerald, T.; Lionel, A.C.; Thiruvahindrapuram, B.; Macdonald, J.R.; Mills, R.; et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011, 29, 512–520. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, J.H.; Chung, Y.J. Effect of combining multiple CNV defining algorithms on the reliability of CNV calls from snp genotyping data. Genomics Inform. 2012, 10, 194–199. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).

 ,
, 

{kind=link}