Roles of EphA2 in Development and Disease

Abstract
:1. Introduction
2. Eph Receptors and Ephrins
2.1. Domain Configuration
2.2. Signaling
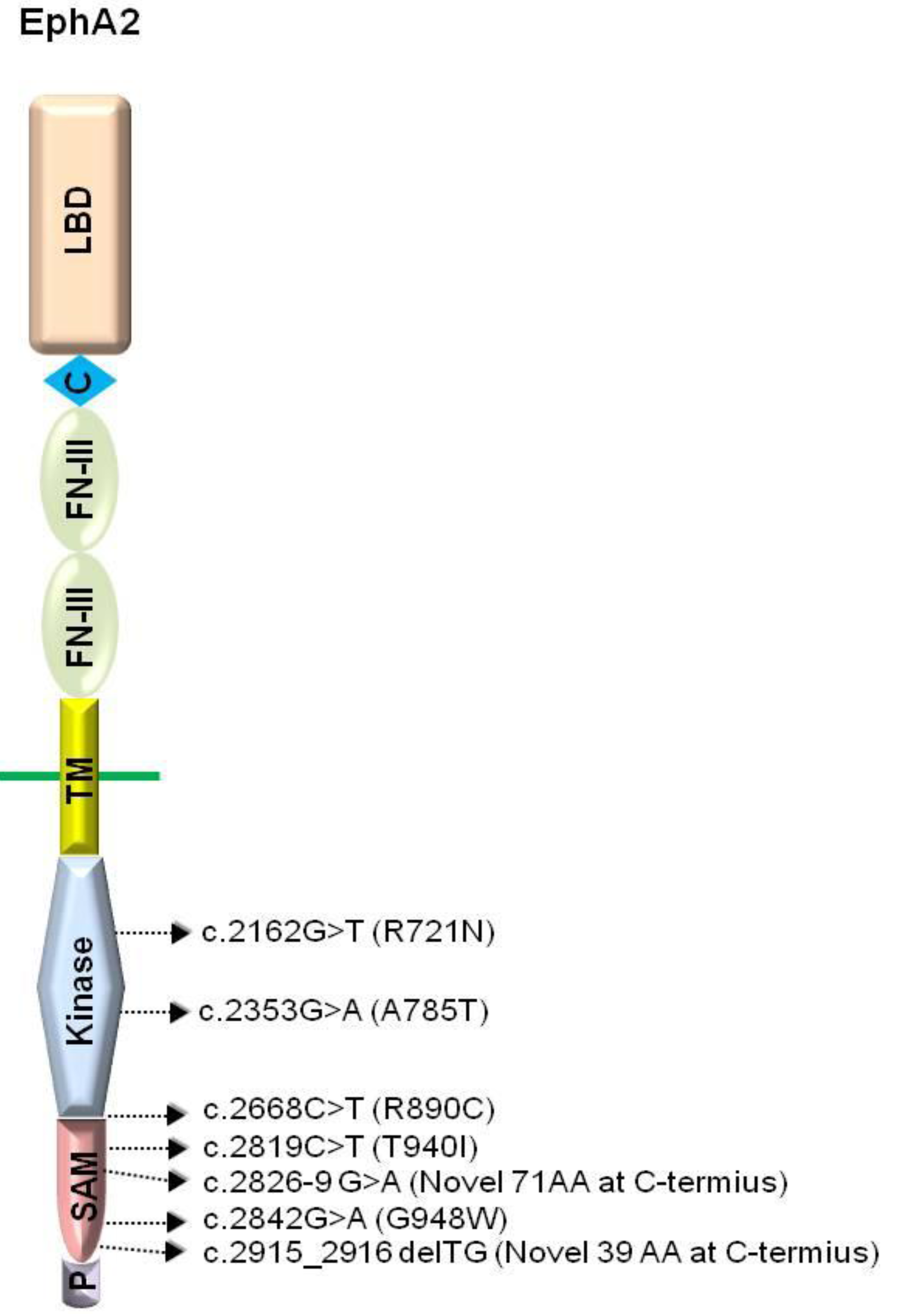
3. EphA2
3.1. Lens Development
3.1.1. EphA2 Expression in the Lens
3.1.2. EphA2 Mutations and Cataractogenesis

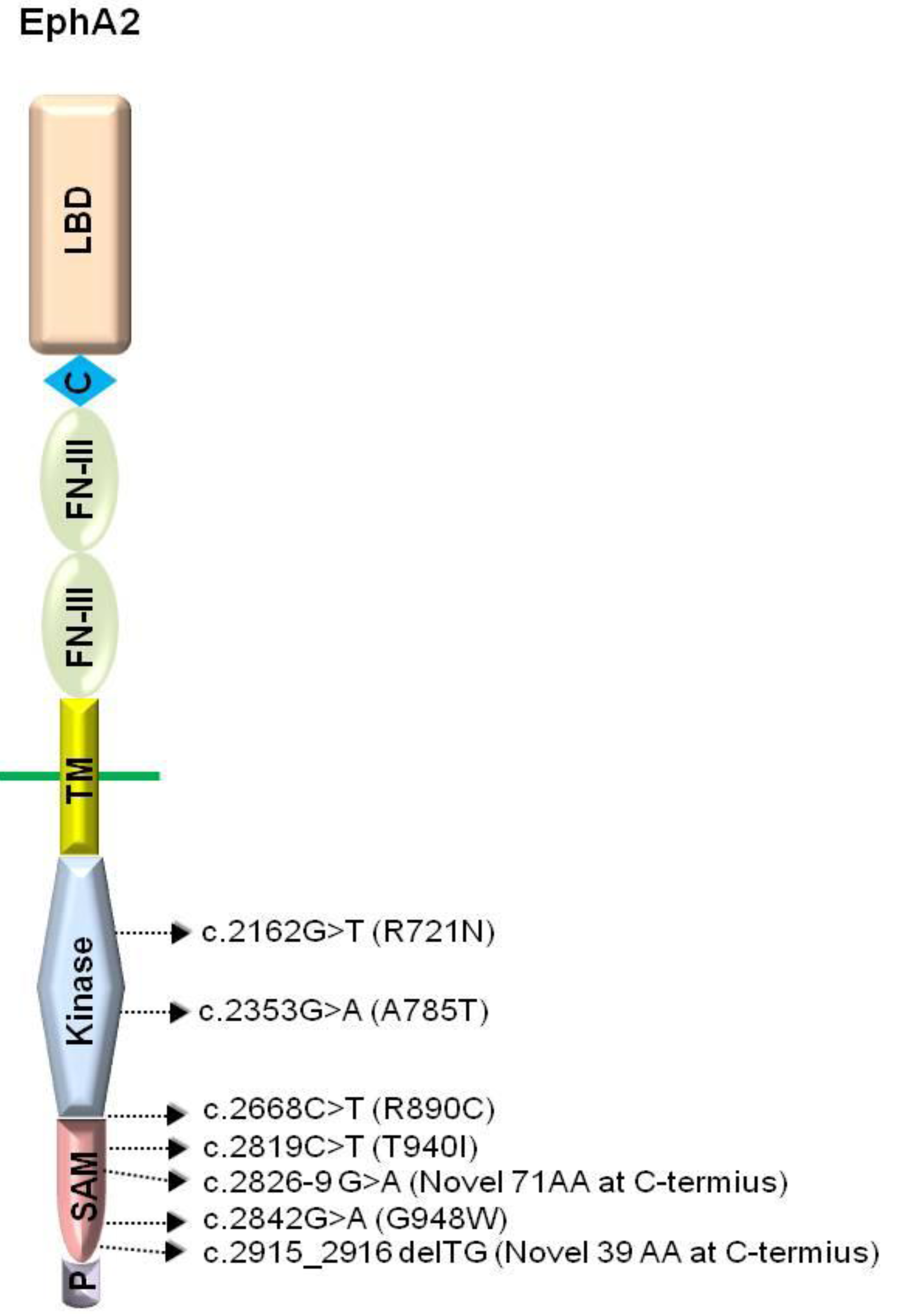{kind=link}
| Mutants | Domain | Mutation | Effect | Phenotype | Ref. |
|---|---|---|---|---|---|
| c.2842G > T | SAM | Missense | G948W | Autosomal dominant posterior polar cataract | [36] |
| c.2819C > T | SAM | Missense | T940I | Autosomal dominant posterior polar cataract | [38] |
| c.2826-9G > A | SAM | Splicing | novel 71 AA | Autosomal dominant total cataract | [38] |
| c.2915_2916delTG | SAM | Frameshift | novel 39 AA | Autosomal dominant posterior polar cataract | [38] |
| c.2162G > T | Kinase | Missense | R721N | Autosomal dominant cortical cataract | [35] |
| c.2353G > A | Kinase | Missense | A785T | Autosomal recessive nuclear cataract | [37] |
| c.2668C > T | Between the kinase and the SAM | Missense | R890C | Autosomal dominant posterior cataract | [41] |
3.1.3. Cataract Mouse Models
3.1.4. Signaling and Molecular Mechanisms
3.2. Retinal Angiogenesis
3.3. Kidney
3.4. Bone
3.5. Mammary Gland Branch Morphogenesis
3.6. Ear
4. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell. Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Fox, G.M.; Holst, P.L.; Chute, H.T.; Lindberg, R.A.; Janssen, A.M.; Basu, R.; Welcher, A.A. cDNA cloning and tissue distribution of five human EPH-like receptor protein-tyrosine kinases. Oncogene 1995, 10, 897–905. [Google Scholar]
- Gurniak, C.B.; Berg, L.J. A new member of the Eph family of receptors that lacks protein tyrosine kinase activity. Oncogene 1996, 13, 777–786. [Google Scholar]
- Pandey, A.; Lindberg, R.A.; Dixit, V.M. Cell signalling. Receptor orphans find a family. Curr. Biol. 1995, 5, 986–989. [Google Scholar] [CrossRef]
- Zhou, R. The Eph family receptors and ligands. Pharmacol. Ther. 1998, 77, 151–181. [Google Scholar] [CrossRef]
- Holder, N.; Klein, R. Eph receptors and ephrins: Effectors of morphogenesis. Development 1999, 126, 2033–2044. [Google Scholar]
- Flenniken, A.M.; Gale, N.W.; Yancopoulos, G.D.; Wilkinson, D.G. Distinct and overlapping expression patterns of ligands for Eph-related receptor tyrosine kinases during mouse embryogenesis. Dev. Biol. 1996, 179, 382–401. [Google Scholar] [CrossRef]
- Rohani, N.; Canty, L.; Luu, O.; Fagotto, F.; Winklbauer, R. EphrinB/EphB signaling controls embryonic germ layer separation by contact-induced cell detachment. PLoS Biol. 2011, 9, e1000597. [Google Scholar]
- Mellitzer, G.; Xu, Q.; Wilkinson, D.G. Eph receptors and ephrins restrict cell intermingling and communication. Nature 1999, 400, 77–81. [Google Scholar] [CrossRef]
- Xu, Q.; Mellitzer, G.; Robinson, V.; Wilkinson, D.G. In vivo cell sorting in complementary segmental domains mediated by Eph receptors and ephrins. Nature 1999, 399, 267–271. [Google Scholar]
- Robinson, V.; Smith, A.; Flenniken, A.M.; Wilkinson, D.G. Roles of Eph receptors and ephrins in neural crest pathfinding. Cell Tissue Res. 1997, 290, 265–274. [Google Scholar] [CrossRef]
- Smith, A.; Robinson, V.; Patel, K.; Wilkinson, D.G. The EphA4 and EphB1 receptor tyrosine kinases and ephrin-B2 ligand regulate targeted migration of branchial neural crest cells. Curr. Biol. 1997, 7, 561–570. [Google Scholar] [CrossRef]
- Flanagan, J.G.; Vanderhaeghen, P. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 1998, 21, 309–345. [Google Scholar] [CrossRef]
- Orioli, D.; Klein, R. The Eph receptor family: Axonal guidance by contact repulsion. Trends Genet. 1997, 13, 354–359. [Google Scholar] [CrossRef]
- Egea, J.; Klein, R. Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell. Biol. 2007, 17, 230–238. [Google Scholar] [CrossRef]
- Wilkinson, D.G. Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci. 2001, 2, 155–164. [Google Scholar] [CrossRef]
- Palmer, A.; Klein, R. Multiple roles of ephrins in morphogenesis, neuronal networking, and brain function. Genes Dev. 2003, 17, 1429–1450. [Google Scholar] [CrossRef]
- Edwards, C.M.; Mundy, G.R. Eph receptors and ephrin signaling pathways: A role in bone homeostasis. Int. J. Med. Sci. 2008, 5, 263–272. [Google Scholar] [CrossRef]
- Matsuo, K.; Otaki, N. Bone cell interactions through Eph/ephrin: Bone modeling, remodeling and associated diseases. Cell Adhes. Migr. 2012, 6, 148–156. [Google Scholar] [CrossRef]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef]
- Irie, N.; Takada, Y.; Watanabe, Y.; Matsuzaki, Y.; Naruse, C.; Asano, M.; Iwakura, Y.; Suda, T.; Matsuo, K. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J. Biol. Chem. 2009, 284, 14637–14644. [Google Scholar] [CrossRef]
- Gale, N.W.; Yancopoulos, G.D. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999, 13, 1055–1066. [Google Scholar] [CrossRef]
- Pandey, A.; Shao, H.; Marks, R.M.; Polverini, P.J.; Dixit, V.M. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNF-alpha-induced angiogenesis. Science 1995, 268, 567–569. [Google Scholar]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef]
- Adams, R.H.; Diella, F.; Hennig, S.; Helmbacher, F.; Deutsch, U.; Klein, R. The cytoplasmic domain of the ligand ephrinB2 is required for vascular morphogenesis but not cranial neural crest migration. Cell 2001, 104, 57–69. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Chen, J. Eph receptor tyrosine kinases in angiogenesis: From development to disease. Angiogenesis 2004, 7, 17–28. [Google Scholar] [CrossRef]
- Shen, J.; Xie, B.; Hatara, C.M.; Hackett, S.F.; Campochiaro, P.A. Vegf or EphA2 antisense polyamide-nucleic acids; vascular localization and suppression of retinal neovascularization. Mol. Ther. 2007, 15, 1924–1930. [Google Scholar] [CrossRef]
- Coulthard, M.G.; Duffy, S.; Down, M.; Evans, B.; Power, M.; Smith, F.; Stylianou, C.; Kleikamp, S.; Oates, A.; Lackmann, M.; et al. The role of the Eph-ephrin signalling system in the regulation of developmental patterning. Int. J. Dev. Biol. 2002, 46, 375–384. [Google Scholar]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef]
- Hafner, C.; Schmitz, G.; Meyer, S.; Bataille, F.; Hau, P.; Langmann, T.; Dietmaier, W.; Landthaler, M.; Vogt, T. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin. Chem. 2004, 50, 490–499. [Google Scholar] [CrossRef]
- Andres, A.C.; Zuercher, G.; Djonov, V.; Flueck, M.; Ziemiecki, A. Protein tyrosine kinase expression during the estrous cycle and carcinogenesis of the mammary gland. Int. J. Cancer 1995, 63, 288–296. [Google Scholar] [CrossRef]
- Andres, A.C.; Reid, H.H.; Zurcher, G.; Blaschke, R.J.; Albrecht, D.; Ziemiecki, A. Expression of two novel eph-related receptor protein tyrosine kinases in mammary gland development and carcinogenesis. Oncogene 1994, 9, 1461–1467. [Google Scholar]
- Jun, G.; Guo, H.; Klein, B.E.; Klein, R.; Wang, J.J.; Mitchell, P.; Miao, H.; Lee, K.E.; Joshi, T.; Buck, M.; et al. EPHA2 is associated with age-related cortical cataract in mice and humans. PLoS Genet. 2009, 5, e1000584. [Google Scholar] [CrossRef]
- Shiels, A.; Bennett, T.M.; Knopf, H.L.; Maraini, G.; Li, A.; Jiao, X.; Hejtmancik, J.F. The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol. Vis. 2008, 14, 2042–2055. [Google Scholar]
- Kaul, H.; Riazuddin, S.A.; Shahid, M.; Kousar, S.; Butt, N.H.; Zafar, A.U.; Khan, S.N.; Husnain, T.; Akram, J.; Hejtmancik, J.F.; et al. Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol. Vis. 2010, 16, 511–517. [Google Scholar]
- Zhang, T.; Hua, R.; Xiao, W.; Burdon, K.P.; Bhattacharya, S.S.; Craig, J.E.; Shang, D.; Zhao, X.; Mackey, D.A.; Moore, A.T.; et al. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum. Mutat. 2009, 30, E603–E611. [Google Scholar] [CrossRef]
- Tan, W.; Hou, S.; Jiang, Z.; Hu, Z.; Yang, P.; Ye, J. Association of EPHA2 polymorphisms and age-related cortical cataract in a Han Chinese population. Mol. Vis. 2011, 17, 1553–1558. [Google Scholar]
- Sundaresan, P.; Ravindran, R.D.; Vashist, P.; Shanker, A.; Nitsch, D.; Talwar, B.; Maraini, G.; Camparini, M.; Nonyane, B.A.; Smeeth, L.; et al. EPHA2 polymorphisms and age-related cataract in India. PLoS One 2012, 7, e33001. [Google Scholar] [CrossRef]
- Shentu, X.C.; Zhao, S.J.; Zhang, L.; Miao, Q. A novel p.R890C mutation in EPHA2 gene associated with progressive childhood posterior cataract in a Chinese family. Int. J. Ophthalmol. 2013, 6, 34–38. [Google Scholar]
- Miao, H.; Nickel, C.H.; Cantley, L.G.; Bruggeman, L.A.; Bennardo, L.N.; Wang, B. EphA kinase activation regulates HGF-induced epithelial branching morphogenesis. J. Cell. Biol. 2003, 162, 1281–1292. [Google Scholar] [CrossRef]
- Xu, H.; Tian, W.; Lindsley, J.N.; Oyama, T.T.; Capasso, J.M.; Rivard, C.J.; Cohen, H.T.; Bagnasco, S.M.; Anderson, S.; Cohen, D.M. EphA2: Expression in the renal medulla and regulation by hypertonicity and urea stress in vitro and in vivo. Am. J. Physiol. Renal Physiol. 2005, 288, F855–F866. [Google Scholar]
- Kouros-Mehr, H.; Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 2006, 235, 3404–3412. [Google Scholar] [CrossRef]
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001, 61, 2301–2306. [Google Scholar]
- Vaught, D.; Chen, J.; Brantley-Sieders, D.M. Regulation of mammary gland branching morphogenesis by EphA2 receptor tyrosine kinase. Mol. Biol. Cell 2009, 20, 2572–2581. [Google Scholar] [CrossRef]
- Park, J.E.; Son, A.I.; Hua, R.; Wang, L.; Zhang, X.; Zhou, R. Human cataract mutations in EPHA2 SAM domain alter receptor stability and function. PLoS One 2012, 7, e36564. [Google Scholar]
- Saeger, B.M.; Suhm, M.; Neubuser, A. Ephrin/ephrin receptor expression during early stages of mouse inner ear development. Dev. Dyn. 2011, 240, 1578–1585. [Google Scholar] [CrossRef]
- Lin, S.; Wang, B.; Getsios, S. Eph/ephrin signaling in epidermal differentiation and disease. Semin. Cell Dev. Biol. 2012, 23, 92–101. [Google Scholar] [CrossRef]
- Arvanitis, D.; Davy, A. Eph/ephrin signaling: Networks. Genes Dev. 2008, 22, 416–429. [Google Scholar] [CrossRef]
- Vaught, D.; Brantley-Sieders, D.M.; Chen, J. Eph receptors in breast cancer: Roles in tumor promotion and tumor suppression. Breast Cancer Res. 2008, 10, 217. [Google Scholar]
- Chen, J. Regulation of tumor initiation and metastatic progression by Eph receptor tyrosine kinases. Adv. Cancer Res. 2012, 114, 1–20. [Google Scholar]
- Janes, P.W.; Nievergall, E.; Lackmann, M. Concepts and consequences of Eph receptor clustering. Semin. Cell Dev. Biol. 2012, 23, 43–50. [Google Scholar] [CrossRef]
- Funk, S.D.; Orr, A.W. Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis. Pharmacol. Res. 2013, 67, 42–52. [Google Scholar] [CrossRef]
- Chen, J.; Zhuang, G.; Frieden, L.; Debinski, W. Eph receptors and Ephrins in cancer: Common themes and controversies. Cancer Res. 2008, 68, 10031–10033. [Google Scholar] [CrossRef]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrinA1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef]
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987, 238, 1717–1720. [Google Scholar]
- Eph Nomenclature Committee. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Cell 1997, 90, 403–404. [CrossRef]
- Bartley, T.D.; Hunt, R.W.; Welcher, A.A.; Boyle, W.J.; Parker, V.P.; Lindberg, R.A.; Lu, H.S.; Colombero, A.M.; Elliott, R.L.; Guthrie, B.A.; et al. B61 is a ligand for the ECK receptor protein-tyrosine kinase. Nature 1994, 368, 558–560. [Google Scholar] [CrossRef]
- Pasquale, E.B. The Eph family of receptors. Curr. Opin. Cell. Biol. 1997, 9, 608–615. [Google Scholar] [CrossRef]
- Gale, N.W.; Holland, S.J.; Valenzuela, D.M.; Flenniken, A.; Pan, L.; Ryan, T.E.; Henkemeyer, M.; Strebhardt, K.; Hirai, H.; Wilkinson, D.G.; et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 1996, 17, 9–19. [Google Scholar] [CrossRef]
- Qin, H.; Noberini, R.; Huan, X.; Shi, J.; Pasquale, E.B.; Song, J. Structural characterization of the EphA4-Ephrin-B2 complex reveals new features enabling Eph-ephrin binding promiscuity. J. Biol. Chem. 2010, 285, 644–654. [Google Scholar]
- Takemoto, M.; Fukuda, T.; Sonoda, R.; Murakami, F.; Tanaka, H.; Yamamoto, N. Ephrin-B3-EphA4 interactions regulate the growth of specific thalamocortical axon populations in vitro. Eur. J. Neurosci. 2002, 16, 1168–1172. [Google Scholar]
- Himanen, J.P.; Chumley, M.J.; Lackmann, M.; Li, C.; Barton, W.A.; Jeffrey, P.D.; Vearing, C.; Geleick, D.; Feldheim, D.A.; Boyd, A.W.; et al. Repelling class discrimination: Ephrin-A5 binds to and activates EphB2 receptor signaling. Nat. Neurosci. 2004, 7, 501–509. [Google Scholar] [CrossRef]
- Himanen, J.P.; Nikolov, D.B. Eph signaling: A structural view. Trends Neurosci. 2003, 26, 46–51. [Google Scholar] [CrossRef]
- Qiao, F.; Bowie, J.U. The many faces of SAM. Sci. STKE 2005, 2005, re7. [Google Scholar] [CrossRef]
- Hui, S.; Xing, X.; Bader, G.D. Predicting PDZ domain mediated protein interactions from structure. BMC Bioinformatics 2013, 14, 27. [Google Scholar] [CrossRef]
- Hock, B.; Bohme, B.; Karn, T.; Yamamoto, T.; Kaibuchi, K.; Holtrich, U.; Holland, S.; Pawson, T.; Rubsamen-Waigmann, H.; Strebhardt, K. PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 9779–9784. [Google Scholar] [CrossRef]
- Smalla, M.; Schmieder, P.; Kelly, M.; Ter Laak, A.; Krause, G.; Ball, L.; Wahl, M.; Bork, P.; Oschkinat, H. Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of two distinct homotypic interaction sites. Protein Sci. 1999, 8, 1954–1961. [Google Scholar] [CrossRef]
- Thanos, C.D.; Goodwill, K.E.; Bowie, J.U. Oligomeric structure of the human EphB2 receptor SAM domain. Science 1999, 283, 833–836. [Google Scholar] [CrossRef]
- Slaughter, B.D.; Huff, J.M.; Wiegraebe, W.; Schwartz, J.W.; Li, R. SAM domain-based protein oligomerization observed by live-cell fluorescence fluctuation spectroscopy. PLoS One 2008, 3, e1931. [Google Scholar] [CrossRef]
- Poliakov, A.; Cotrina, M.; Wilkinson, D.G. Diverse roles of eph receptors and ephrins in the regulation of cell migration and tissue assembly. Dev. Cell. 2004, 7, 465–480. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell. Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef]
- Himanen, J.P.; Yermekbayeva, L.; Janes, P.W.; Walker, J.R.; Xu, K.; Atapattu, L.; Rajashankar, K.R.; Mensinga, A.; Lackmann, M.; Nikolov, D.B.; et al. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. USA 2010, 107, 10860–10865. [Google Scholar] [CrossRef]
- Noren, N.K.; Pasquale, E.B. Eph receptor-ephrin bidirectional signals that target Ras and Rho proteins. Cell. Signal. 2004, 16, 655–666. [Google Scholar] [CrossRef]
- Klein, R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci. 2009, 12, 15–20. [Google Scholar] [CrossRef]
- Murai, K.K.; Pasquale, E.B. 'Eph'ective signaling: Forward, reverse and crosstalk. J. Cell. Sci. 2003, 116, 2823–2832. [Google Scholar] [CrossRef]
- Lim, Y.S.; McLaughlin, T.; Sung, T.C.; Santiago, A.; Lee, K.F.; O’Leary, D.D. p75(NTR) mediates ephrin-A reverse signaling required for axon repulsion and mapping. Neuron 2008, 59, 746–758. [Google Scholar] [CrossRef]
- Yamazaki, T.; Masuda, J.; Omori, T.; Usui, R.; Akiyama, H.; Maru, Y. EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J. Cell. Sci. 2009, 122, 243–255. [Google Scholar] [CrossRef]
- Lee, H.S.; Daar, I.O. EphrinB reverse signaling in cell-cell adhesion: Is it just par for the course? Cell Adhes. Migr. 2009, 3, 250–255. [Google Scholar] [CrossRef]
- Carter, N.; Nakamoto, T.; Hirai, H.; Hunter, T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas). Nat. Cell Biol. 2002, 4, 565–573. [Google Scholar]
- Hattori, M.; Osterfield, M.; Flanagan, J.G. Regulated cleavage of a contact-mediated axon repellent. Science 2000, 289, 1360–1365. [Google Scholar] [CrossRef]
- Sharfe, N.; Freywald, A.; Toro, A.; Roifman, C.M. Ephrin-A1 induces c-Cbl phosphorylation and EphA receptor down-regulation in T cells. J. Immunol. 2003, 170, 6024–6032. [Google Scholar]
- Walker-Daniels, J.; Riese, D.J., 2nd; Kinch, M.S. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res. 2002, 1, 79–87. [Google Scholar]
- Wang, Y.; Ota, S.; Kataoka, H.; Kanamori, M.; Li, Z.; Band, H.; Tanaka, M.; Sugimura, H. Negative regulation of EphA2 receptor by Cbl. Biochem. Biophys. Res. Commun. 2002, 296, 214–220. [Google Scholar] [CrossRef]
- Marmor, M.D.; Yarden, Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004, 23, 2057–2070. [Google Scholar] [CrossRef]
- Palmer, A.; Zimmer, M.; Erdmann, K.S.; Eulenburg, V.; Porthin, A.; Heumann, R.; Deutsch, U.; Klein, R. EphrinB phosphorylation and reverse signaling: Regulation by Src kinases and PTP-BL phosphatase. Mol. Cell 2002, 9, 725–737. [Google Scholar] [CrossRef]
- Baldwin, C.; Chen, Z.W.; Bedirian, A.; Yokota, N.; Nasr, S.H.; Rabb, H.; Lemay, S. Upregulation of EphA2 during in vivo and in vitro renal ischemia-reperfusion injury: Role of Src kinases. Am. J. Physiol. Renal Physiol. 2006, 291, F960–F971. [Google Scholar] [CrossRef]
- Dodelet, V.C.; Pazzagli, C.; Zisch, A.H.; Hauser, C.A.; Pasquale, E.B. A novel signaling intermediate, SHEP1, directly couples Eph receptors to R-Ras and Rap1. J. Biol. Chem. 1999, 274, 31941–31946. [Google Scholar] [CrossRef]
- Marston, D.J.; Dickinson, S.; Nobes, C.D. Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nat. Cell Biol. 2003, 5, 879–888. [Google Scholar] [CrossRef]
- Lu, Q.; Sun, E.E.; Klein, R.S.; Flanagan, J.G. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell 2001, 105, 69–79. [Google Scholar] [CrossRef]
- Feng, Y.X.; Zhao, J.S.; Li, J.J.; Wang, T.; Cheng, S.Q.; Yuan, Y.; Wang, F.; Wang, X.F.; Xie, D. Liver cancer: EphrinA2 promotes tumorigenicity through Rac1/Akt/NF-kappaB signaling pathway 120. Hepatology 2010, 51, 535–544. [Google Scholar] [CrossRef]
- Zhuang, G.; Hunter, S.; Hwang, Y.; Chen, J. Regulation of EphA2 receptor endocytosis by SHIP2 lipid phosphatase via phosphatidylinositol 3-Kinase-dependent Rac1 activation. J. Biol. Chem. 2007, 282, 2683–2694. [Google Scholar] [CrossRef]
- Fang, W.B.; Ireton, R.C.; Zhuang, G.; Takahashi, T.; Reynolds, A.; Chen, J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J. Cell Sci. 2008, 121, 358–368. [Google Scholar] [CrossRef]
- Margolis, S.S.; Salogiannis, J.; Lipton, D.M.; Mandel-Brehm, C.; Wills, Z.P.; Mardinly, A.R.; Hu, L.; Greer, P.L.; Bikoff, J.B.; Ho, H.Y.; et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 2010, 143, 442–455. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Zhuang, G.; Hicks, D.; Fang, W.B.; Hwang, Y.; Cates, J.M.; Coffman, K.; Jackson, D.; Bruckheimer, E.; Muraoka-Cook, R.S.; et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Invest. 2008, 118, 64–78. [Google Scholar] [CrossRef]
- Cowan, C.A.; Henkemeyer, M. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature 2001, 413, 174–179. [Google Scholar] [CrossRef]
- Bruckner, K.; Pablo Labrador, J.; Scheiffele, P.; Herb, A.; Seeburg, P.H.; Klein, R. EphrinB ligands recruit GRIP family PDZ adaptor proteins into raft membrane microdomains. Neuron 1999, 22, 511–524. [Google Scholar] [CrossRef]
- Ellis, C.; Kasmi, F.; Ganju, P.; Walls, E.; Panayotou, G.; Reith, A.D. A juxtamembrane autophosphorylation site in the Eph family receptor tyrosine kinase, Sek, mediates high affinity interaction with p59fyn. Oncogene 1996, 12, 1727–1736. [Google Scholar]
- Kalo, M.S.; Pasquale, E.B. Multiple in vivo tyrosine phosphorylation sites in EphB receptors. Biochemistry 1999, 38, 14396–14408. [Google Scholar] [CrossRef]
- Stein, E.; Huynh-Do, U.; Lane, A.A.; Cerretti, D.P.; Daniel, T.O. Nck recruitment to Eph receptor, EphB1/ELK, couples ligand activation to c-Jun kinase. J. Biol. Chem. 1998, 273, 1303–1308. [Google Scholar]
- Han, D.C.; Shen, T.L.; Miao, H.; Wang, B.; Guan, J.L. EphB1 associates with Grb7 and regulates cell migration. J. Biol. Chem. 2002, 277, 45655–45661. [Google Scholar]
- Holland, S.J.; Gale, N.W.; Gish, G.D.; Roth, R.A.; Songyang, Z.; Cantley, L.C.; Henkemeyer, M.; Yancopoulos, G.D.; Pawson, T. Juxtamembrane tyrosine residues couple the Eph family receptor EphB2/Nuk to specific SH2 domain proteins in neuronal cells. EMBO J. 1997, 16, 3877–3888. [Google Scholar] [CrossRef]
- Hu, Q.; Milfay, D.; Williams, L.T. Binding of NCK to SOS and activation of ras-dependent gene expression. Mol. Cell. Biol. 1995, 15, 1169–1174. [Google Scholar]
- Han, D.C.; Shen, T.L.; Guan, J.L. The Grb7 family proteins: Structure, interactions with other signaling molecules and potential cellular functions. Oncogene 2001, 20, 6315–6321. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Chen, J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002, 13, 75–85. [Google Scholar] [CrossRef]
- Singh, A.; Winterbottom, E.; Daar, I.O. Eph/ephrin signaling in cell-cell and cell-substrate adhesion. Front. Biosci. 2012, 17, 473–497. [Google Scholar] [CrossRef]
- Lindberg, R.A.; Hunter, T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol. Cell Biol. 1990, 10, 6316–6324. [Google Scholar]
- Sulman, E.P.; Tang, X.X.; Allen, C.; Biegel, J.A.; Pleasure, D.E.; Brodeur, G.M.; Ikegaki, N. ECK, a human EPH-related gene, maps to 1p36.1, a common region of alteration in human cancers. Genomics 1997, 40, 371–374. [Google Scholar] [CrossRef]
- Ruiz, J.C.; Robertson, E.J. The expression of the receptor-protein tyrosine kinase gene, eck, is highly restricted during early mouse development. Mech. Dev. 1994, 46, 87–100. [Google Scholar] [CrossRef]
- Cooper, M.A.; Son, A.I.; Komlos, D.; Sun, Y.; Kleiman, N.J.; Zhou, R. Loss of ephrin-A5 function disrupts lens fiber cell packing and leads to cataract. Proc. Natl. Acad. Sci. USA 2008, 105, 16620–16625. [Google Scholar]
- Noberini, R.; Koolpe, M.; Peddibhotla, S.; Dahl, R.; Su, Y.; Cosford, N.D.; Roth, G.P.; Pasquale, E.B. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J. Biol. Chem. 2008, 283, 29461–29472. [Google Scholar] [CrossRef]
- Walker-Daniels, J.; Hess, A.R.; Hendrix, M.J.; Kinch, M.S. Differential regulation of EphA2 in normal and malignant cells. Am. J. Pathol. 2003, 162, 1037–1042. [Google Scholar] [CrossRef]
- Davis, S.; Gale, N.W.; Aldrich, T.H.; Maisonpierre, P.C.; Lhotak, V.; Pawson, T.; Goldfarb, M.; Yancopoulos, G.D. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 1994, 266, 816–819. [Google Scholar]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar]
- Foster, A.; Resnikoff, S. The impact of Vision 2020 on global blindness. Eye (Lond.) 2005, 19, 1133–1135. [Google Scholar] [CrossRef]
- Rahi, J.S.; Dezateux, C. Measuring and interpreting the incidence of congenital ocular anomalies: Lessons from a national study of congenital cataract in the UK. Invest. Ophthalmol. Vis. Sci. 2001, 42, 1444–1448. [Google Scholar]
- Shiels, A.; Mackay, D.; Ionides, A.; Berry, V.; Moore, A.; Bhattacharya, S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am. J. Hum. Genet. 1998, 62, 526–532. [Google Scholar] [CrossRef]
- Mackay, D.; Ionides, A.; Kibar, Z.; Rouleau, G.; Berry, V.; Moore, A.; Shiels, A.; Bhattacharya, S. Connexin46 mutations in autosomal dominant congenital cataract. Am. J. Hum. Genet. 1999, 64, 1357–1364. [Google Scholar] [CrossRef]
- Litt, M.; Kramer, P.; LaMorticella, D.M.; Murphey, W.; Lovrien, E.W.; Weleber, R.G. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum. Mol. Genet. 1998, 7, 471–474. [Google Scholar] [CrossRef]
- Berry, V.; Francis, P.; Reddy, M.A.; Collyer, D.; Vithana, E.; MacKay, I.; Dawson, G.; Carey, A.H.; Moore, A.; Bhattacharya, S.S.; et al. Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am. J. Hum. Genet. 2001, 69, 1141–1145. [Google Scholar] [CrossRef]
- Mackay, D.S.; Boskovska, O.B.; Knopf, H.L.; Lampi, K.J.; Shiels, A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am. J. Hum. Genet. 2002, 71, 1216–1221. [Google Scholar] [CrossRef]
- Litt, M.; Carrero-Valenzuela, R.; LaMorticella, D.M.; Schultz, D.W.; Mitchell, T.N.; Kramer, P.; Maumenee, I.H. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum. Mol. Genet. 1997, 6, 665–668. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Yasmeen, A.; Yao, W.; Sergeev, Y.V.; Zhang, Q.; Zulfiqar, F.; Riaz, A.; Riazuddin, S.; Hejtmancik, J.F. Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest. Ophthalmol. Vis. Sci. 2005, 46, 2100–2106. [Google Scholar] [CrossRef]
- Kannabiran, C.; Rogan, P.K.; Olmos, L.; Basti, S.; Rao, G.N.; Kaiser-Kupfer, M.; Hejtmancik, J.F. Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol. Vis. 1998, 4, 21. [Google Scholar]
- Billingsley, G.; Santhiya, S.T.; Paterson, A.D.; Ogata, K.; Wodak, S.; Hosseini, S.M.; Manisastry, S.M.; Vijayalakshmi, P.; Gopinath, P.M.; Graw, J.; Heon, E. CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am. J. Hum. Genet. 2006, 79, 702–709. [Google Scholar] [CrossRef]
- Sun, H.; Ma, Z.; Li, Y.; Liu, B.; Li, Z.; Ding, X.; Gao, Y.; Ma, W.; Tang, X.; Li, X.; et al. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J. Med. Genet. 2005, 42, 706–710. [Google Scholar] [CrossRef]
- Heon, E.; Priston, M.; Schorderet, D.F.; Billingsley, G.D.; Girard, P.O.; Lubsen, N.; Munier, F.L. The gamma-crystallins and human cataracts: A puzzle made clearer. Am. J. Hum. Genet. 1999, 65, 1261–1267. [Google Scholar] [CrossRef]
- Muller, M.; Bhattacharya, S.S.; Moore, T.; Prescott, Q.; Wedig, T.; Herrmann, H.; Magin, T.M. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum. Mol. Genet. 2009, 18, 1052–1057. [Google Scholar] [CrossRef]
- Shi, Y.; de Maria, A.; Bennett, T.; Shiels, A.; Bassnett, S. A role for epha2 in cell migration and refractive organization of the ocular lens. Invest. Ophthalmol. Vis. Sci. 2012, 53, 551–559. [Google Scholar] [CrossRef]
- Cheng, C.; Gong, X. Diverse roles of Eph/ephrin signaling in the mouse lens. PLoS One 2012, 6, e28147. [Google Scholar] [CrossRef]
- Son, A.I.; Cooper, M.A.; Sheleg, M.; Sun, Y.; Kleiman, N.J.; Zhou, R. Further analysis of the lens of ephrin-A5(-/-) mice: Development of postnatal defects. Mol. Vis. 2013, 19, 254–266. [Google Scholar]
- Guo, H.; Miao, H.; Gerber, L.; Singh, J.; Denning, M.F.; Gilliam, A.C.; Wang, B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006, 66, 7050–7058. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Pinson, K.I.; Kelly, O.G.; Brennan, J.; Zupicich, J.; Scherz, P.; Leighton, P.A.; Goodrich, L.V.; Lu, X.; Avery, B.J.; et al. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat. Genet. 2001, 28, 241–249. [Google Scholar] [CrossRef]
- Naruse-Nakajima, C.; Asano, M.; Iwakura, Y. Involvement of EphA2 in the formation of the tail notochord via interaction with ephrinA1. Mech. Dev. 2001, 102, 95–105. [Google Scholar] [CrossRef]
- Fang, W.B.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.J.; Chen, J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J. Biol. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef]
- Dufour, A.; Egea, J.; Kullander, K.; Klein, R.; Vanderhaeghen, P. Genetic analysis of EphA-dependent signaling mechanisms controlling topographic mapping in vivo. Development 2006, 133, 4415–4420. [Google Scholar] [CrossRef]
- Park, E.K.; Warner, N.; Bong, Y.S.; Stapleton, D.; Maeda, R.; Pawson, T.; Daar, I.O. Ectopic EphA4 receptor induces posterior protrusions via FGF signaling in Xenopus embryos. Mol. Biol. Cell 2004, 15, 1647–1655. [Google Scholar] [CrossRef]
- Kikawa, K.D.; Vidale, D.R.; van Etten, R.L.; Kinch, M.S. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J. Biol. Chem. 2002, 277, 39274–39279. [Google Scholar]
- Parri, M.; Buricchi, F.; Taddei, M.L.; Giannoni, E.; Raugei, G.; Ramponi, G.; Chiarugi, P. EphrinA1 repulsive response is regulated by an EphA2 tyrosine phosphatase. J. Biol. Chem. 2005, 280, 34008–34018. [Google Scholar]
- Saint-Geniez, M.; D’Amore, P.A. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 2004, 48, 1045–1058. [Google Scholar] [CrossRef]
- Gariano, R.F.; Kalina, R.E.; Hendrickson, A.E. Normal and pathological mechanisms in retinal vascular development. Surv. Ophthalmol. 1996, 40, 481–490. [Google Scholar] [CrossRef]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef]
- Stahl, A.; Connor, K.M.; Sapieha, P.; Chen, J.; Dennison, R.J.; Krah, N.M.; Seaward, M.R.; Willett, K.L.; Aderman, C.M.; Guerin, K.I.; et al. The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 2010, 51, 2813–2826. [Google Scholar] [CrossRef]
- Miller, J.W.; Le Couter, J.; Strauss, E.C.; Ferrara, N. Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 2013, 120, 106–114. [Google Scholar] [CrossRef]
- Kempen, J.H.; O’Colmain, B.J.; Leske, M.C.; Haffner, S.M.; Klein, R.; Moss, S.E.; Taylor, H.R.; Hamman, R.F. The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol. 2004, 122, 552–563. [Google Scholar]
- Klein, B.E. Overview of epidemiologic studies of diabetic retinopathy. Ophthalmic Epidemiol. 2007, 14, 179–183. [Google Scholar] [CrossRef]
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar] [CrossRef]
- Friedman, D.S.; O’Colmain, B.J.; Munoz, B.; Tomany, S.C.; McCarty, C.; de Jong, P.T.; Nemesure, B.; Mitchell, P.; Kempen, J. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar] [CrossRef]
- Mechoulam, H.; Pierce, E.A. Retinopathy of prematurity: Molecular pathology and therapeutic strategies. Am. J. Pharmacogenomics 2003, 3, 261–277. [Google Scholar] [CrossRef]
- Hess, A.R.; Seftor, E.A.; Gardner, L.M.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.; Kinch, M.S.; Hendrix, M.J. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001, 61, 3250–3255. [Google Scholar]
- Ogawa, K.; Pasqualini, R.; Lindberg, R.A.; Kain, R.; Freeman, A.L.; Pasquale, E.B. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000, 19, 6043–6052. [Google Scholar] [CrossRef]
- Brantley, D.M.; Cheng, N.; Thompson, E.J.; Lin, Q.; Brekken, R.A.; Thorpe, P.E.; Muraoka, R.S.; Cerretti, D.P.; Pozzi, A.; Jackson, D.; et al. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo . Oncogene 2002, 21, 7011–7026. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Liu, H.; Lin, Q.; Enriquez, M.; Gale, N.; Yancopoulos, G.; Cerretti, D.P.; Daniel, T.O.; Chen, J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol. Cancer Res. 2002, 1, 2–11. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.; Fang, W.B.; Liu, H.; Fanslow, W.; Cerretti, D.P.; Bussell, K.N.; Reith, A.; Jackson, D.; Chen, J. Inhibition of VEGF-dependent multistage carcinogenesis by soluble EphA receptors. Neoplasia 2003, 5, 445–456. [Google Scholar]
- Dobrzanski, P.; Hunter, K.; Jones-Bolin, S.; Chang, H.; Robinson, C.; Pritchard, S.; Zhao, H.; Ruggeri, B. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004, 64, 910–919. [Google Scholar] [CrossRef]
- Recchia, F.M.; Xu, L.; Penn, J.S.; Boone, B.; Dexheimer, P.J. Identification of genes and pathways involved in retinal neovascularization by microarray analysis of two animal models of retinal angiogenesis. Invest. Ophthalmol. Vis. Sci. 2010, 51, 1098–1105. [Google Scholar] [CrossRef]
- Chen, J.; Hicks, D.; Brantley-Sieders, D.; Cheng, N.; McCollum, G.W.; Qi-Werdich, X.; Penn, J. Inhibition of retinal neovascularization by soluble EphA2 receptor. Exp. Eye Res. 2006, 82, 664–673. [Google Scholar] [CrossRef]
- Wang, J.L.; Liu, Y.L.; Li, Y.; Dai, W.B.; Guo, Z.M.; Wang, Z.H.; Zhang, Q. EphA2 targeted doxorubicin stealth liposomes as a therapy system for choroidal neovascularization in rats. Invest. Ophthalmol. Vis. Sci. 2012, 53, 7348–7357. [Google Scholar]
- Ojima, T.; Takagi, H.; Suzuma, K.; Oh, H.; Suzuma, I.; Ohashi, H.; Watanabe, D.; Suganami, E.; Murakami, T.; Kurimoto, M.; et al. EphrinA1 inhibits vascular endothelial growth factor-induced intracellular signaling and suppresses retinal neovascularization and blood-retinal barrier breakdown. Am. J. Pathol. 2006, 168, 331–339. [Google Scholar] [CrossRef]
- Miao, H.; Wang, B. Eph/ephrin signaling in epithelial development and homeostasis. Int. J. Biochem. Cell Biol. 2009, 41, 762–770. [Google Scholar] [CrossRef]
- Costantini, F.; Kopan, R. Patterning a complex organ: Branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 2010, 18, 698–712. [Google Scholar] [CrossRef]
- Wakayama, Y.; Miura, K.; Sabe, H.; Mochizuki, N. EphrinA1-EphA2 signal induces compaction and polarization of Madin-Darby canine kidney cells by inactivating Ezrin through negative regulation of RhoA. J. Biol. Chem. 2011, 286, 44243–44253. [Google Scholar] [CrossRef]
- Kellerman, P.S.; Norenberg, S.L.; Jones, G.M. Early recovery of the actin cytoskeleton during renal ischemic injury in vivo. Am. J. Kidney Dis. 1996, 27, 709–714. [Google Scholar] [CrossRef]
- Molitoris, B.A. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004, 66, 871–883. [Google Scholar] [CrossRef]
- Molitoris, B.A. Ischemia-induced loss of epithelial polarity: Potential role of the actin cytoskeleton. Am. J. Physiol. 1991, 260, F769–F778. [Google Scholar]
- Molitoris, B.A.; Dahl, R.; Geerdes, A. Cytoskeleton disruption and apical redistribution of proximal tubule Na(+)-K(+)-ATPase during ischemia. Am. J. Physiol. 1992, 263, F488–F495. [Google Scholar]
- Murai, K.K.; Pasquale, E.B. New exchanges in eph-dependent growth cone dynamics. Neuron 2005, 46, 161–163. [Google Scholar] [CrossRef]
- Nahm, O.; Woo, S.K.; Handler, J.S.; Kwon, H.M. Involvement of multiple kinase pathways in stimulation of gene transcription by hypertonicity. Am. J. Physiol. Cell Physiol. 2002, 282, C49–C58. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Steiner, A.A.; Scheck, A.C.; Romanovsky, A.A. Expression of Eph receptors and their ligands, ephrins, during lipopolysaccharide fever in rats. Physiol. Genomics 2005, 21, 152–160. [Google Scholar] [CrossRef]
- Li, Z.; Tanaka, M.; Kataoka, H.; Nakamura, R.; Sanjar, R.; Shinmura, K.; Sugimura, H. EphA2 up-regulation induced by deoxycholic acid in human colon carcinoma cells, an involvement of extracellular signal-regulated kinase and p53-independence. J. Cancer Res. Clin. Oncol. 2003, 129, 703–708. [Google Scholar] [CrossRef]
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef]
- Matsuo, K.; Ray, N. Osteoclasts, mononuclear phagocytes, and c-Fos: New insight into osteoimmunology. Keio J. Med. 2004, 53, 78–84. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef]
- Hennighausen, L.; Robinson, G.W. Information networks in the mammary gland. Nat. Rev. Mol. Cell. Biol. 2005, 6, 715–725. [Google Scholar] [CrossRef]
- Watson, C.J.; Khaled, W.T. Mammary development in the embryo and adult: A journey of morphogenesis and commitment. Development 2008, 135, 995–1003. [Google Scholar] [CrossRef]
- Andres, A.-C.; Ziemiecki, A. Eph and ephrin signaling in mammary gland morphogenesis and cancer. J. Mammary Gland Biol. Neoplasia 2003, 8, 475–485. [Google Scholar] [CrossRef]
- Munarini, N.; Jager, R.; Abderhalden, S.; Zuercher, G.; Rohrbach, V.; Loercher, S.; Pfanner-Meyer, B.; Andres, A.C.; Ziemiecki, A. Altered mammary epithelial development, pattern formation and involution in transgenic mice expressing the EphB4 receptor tyrosine kinase. J. Cell. Sci. 2002, 115, 25–37. [Google Scholar]
- Haldimann, M.; Custer, D.; Munarini, N.; Stirnimann, C.; Zurcher, G.; Rohrbach, V.; Djonov, V.; Ziemiecki, A.; Andres, A.C. Deregulated ephrin-B2 expression in the mammary gland interferes with the development of both the glandular epithelium and vasculature and promotes metastasis formation. Int. J. Oncol. 2009, 35, 525–536. [Google Scholar]
- Sternlicht, M.D.; Kouros-Mehr, H.; Lu, P.; Werb, Z. Hormonal and local control of mammary branching morphogenesis. Differentiation 2006, 74, 365–381. [Google Scholar] [CrossRef]
- Nikolova, Z.; Djonov, V.; Zuercher, G.; Andres, A.C.; Ziemiecki, A. Cell-type specific and estrogen dependent expression of the receptor tyrosine kinase EphB4 and its ligand ephrin-B2 during mammary gland morphogenesis. J. Cell. Sci. 1998, 111, 2741–2751. [Google Scholar]
- Zelinski, D.P.; Zantek, N.D.; Walker-Daniels, J.; Peters, M.A.; Taparowsky, E.J.; Kinch, M.S. Estrogen and Myc negatively regulate expression of the EphA2 tyrosine kinase. J. Cell. Biochem. 2002, 85, 714–720. [Google Scholar] [CrossRef]
- Martin, K.J.; Patrick, D.R.; Bissell, M.J.; Fournier, M.V. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS One 2008, 3, e2994. [Google Scholar] [CrossRef]
- Fournier, M.V.; Martin, K.J.; Kenny, P.A.; Xhaja, K.; Bosch, I.; Yaswen, P.; Bissell, M.J. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res. 2006, 66, 7095–7102. [Google Scholar] [CrossRef]
- Ireton, R.C.; Chen, J. EphA2 receptor tyrosine kinase as a promising target for cancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 149–157. [Google Scholar] [CrossRef]
- Kamat, A.A.; Coffey, D.; Merritt, W.M.; Nugent, E.; Urbauer, D.; Lin, Y.G.; Edwards, C.; Broaddus, R.; Coleman, R.L.; Sood, A.K. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer 2009, 115, 2684–2692. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-stromal interactions. Transforming growth factor-beta isoforms and hepatocyte growth factor/scatter factor in mammary gland ductal morphogenesis. Breast Cancer Res. 2001, 3, 230–237. [Google Scholar] [CrossRef]
- Bianchi, L.M.; Liu, H. Comparison of ephrin-A ligand and EphA receptor distribution in the developing inner ear. Anat. Rec. 1999, 254, 127–134. [Google Scholar] [CrossRef]
- Pickles, J.O.; Claxton, C.; van Heumen, W.R. Complementary and layered expression of Ephs and ephrins in developing mouse inner ear. J. Comp. Neurol. 2002, 449, 207–216. [Google Scholar] [CrossRef]
- Van Heumen, W.R.; Claxton, C.; Pickles, J.O. Expression of EphA4 in developing inner ears of the mouse and guinea pig. Hear. Res. 2000, 139, 42–50. [Google Scholar] [CrossRef]
- Howard, M.A.; Rodenas-Ruano, A.; Henkemeyer, M.; Martin, G.K.; Lonsbury-Martin, B.L.; Liebl, D.J. Eph receptor deficiencies lead to altered cochlear function. Hear. Res. 2003, 178, 118–130. [Google Scholar] [CrossRef]
- Torres, M.; Giraldez, F. The development of the vertebrate inner ear. Mech. Dev. 1998, 71, 5–21. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, J.E.; Son, A.I.; Zhou, R. Roles of EphA2 in Development and Disease. Genes 2013, 4, 334-357. https://doi.org/10.3390/genes4030334
Park JE, Son AI, Zhou R. Roles of EphA2 in Development and Disease. Genes. 2013; 4(3):334-357. https://doi.org/10.3390/genes4030334
Chicago/Turabian StylePark, Jeong Eun, Alexander I. Son, and Renping Zhou. 2013. "Roles of EphA2 in Development and Disease" Genes 4, no. 3: 334-357. https://doi.org/10.3390/genes4030334
APA StylePark, J. E., Son, A. I., & Zhou, R. (2013). Roles of EphA2 in Development and Disease. Genes, 4(3), 334-357. https://doi.org/10.3390/genes4030334