Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
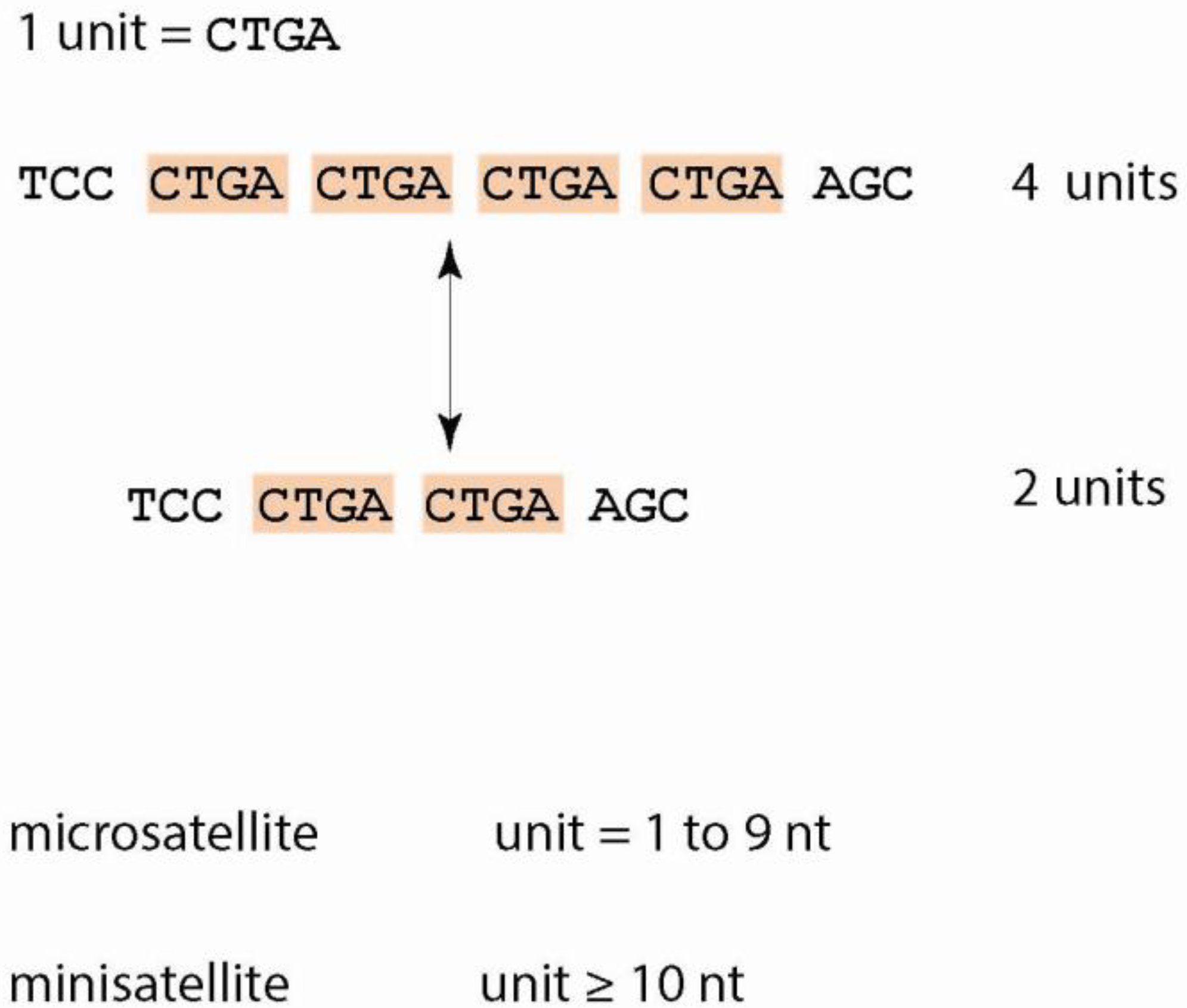
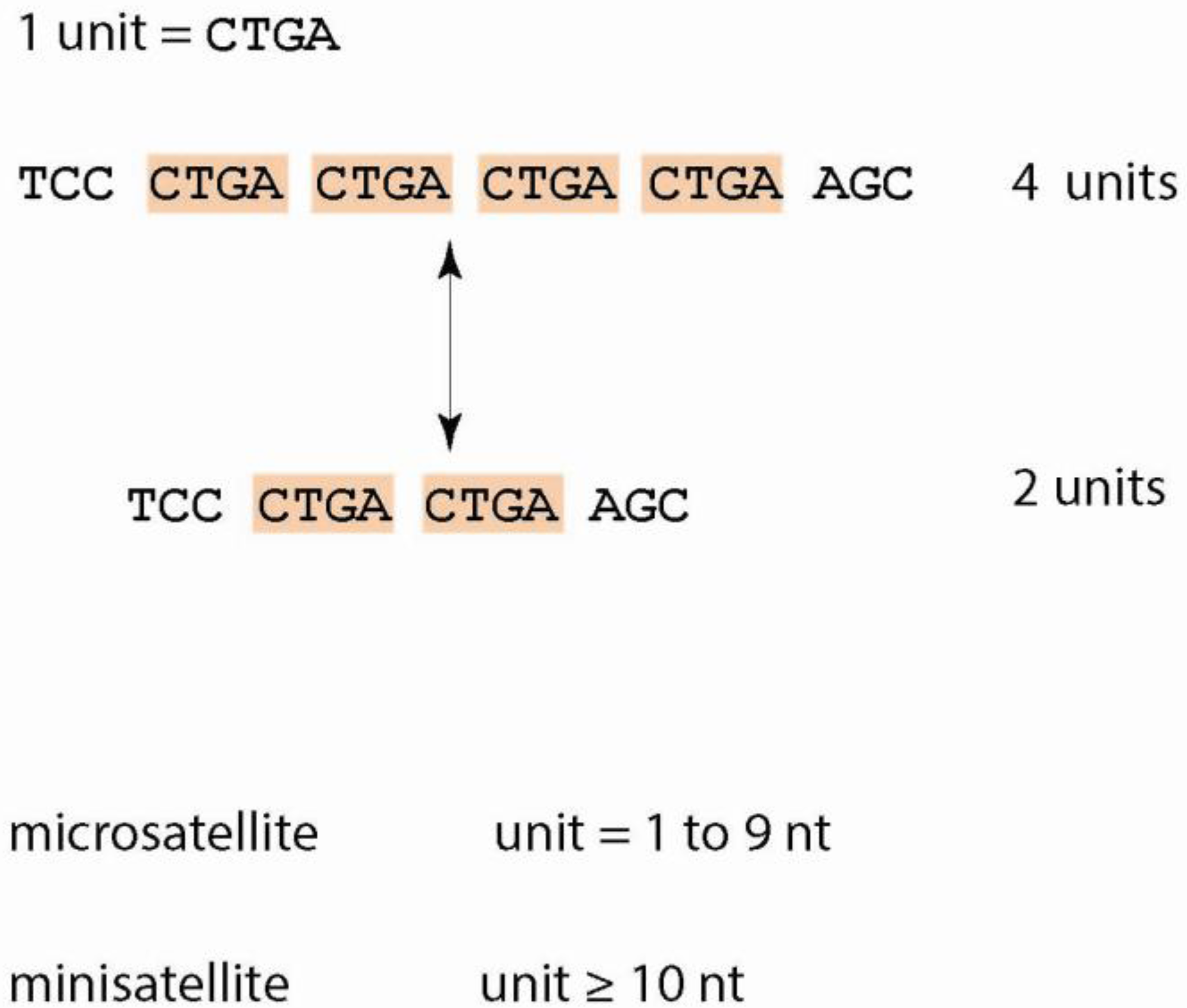
1.1. Tandem Repeats—Definitions and Characteristics
1.2. Instability of Repeats
1.3. Factors Influencing Repeat Instability
1.4. In Search of Repeats
1.5. Location of Repeats in Genomes: Coding and Regulatory Regions
2. Repeats in Non-Coding Regions
3. Promoter Evolution Through Tandem Repeat Variation
3.1. Tandemly Repeated Transcription Factor Binding Sites
3.2. Variable Repeats Induce Variable Spacing Between Functional Promoter Elements

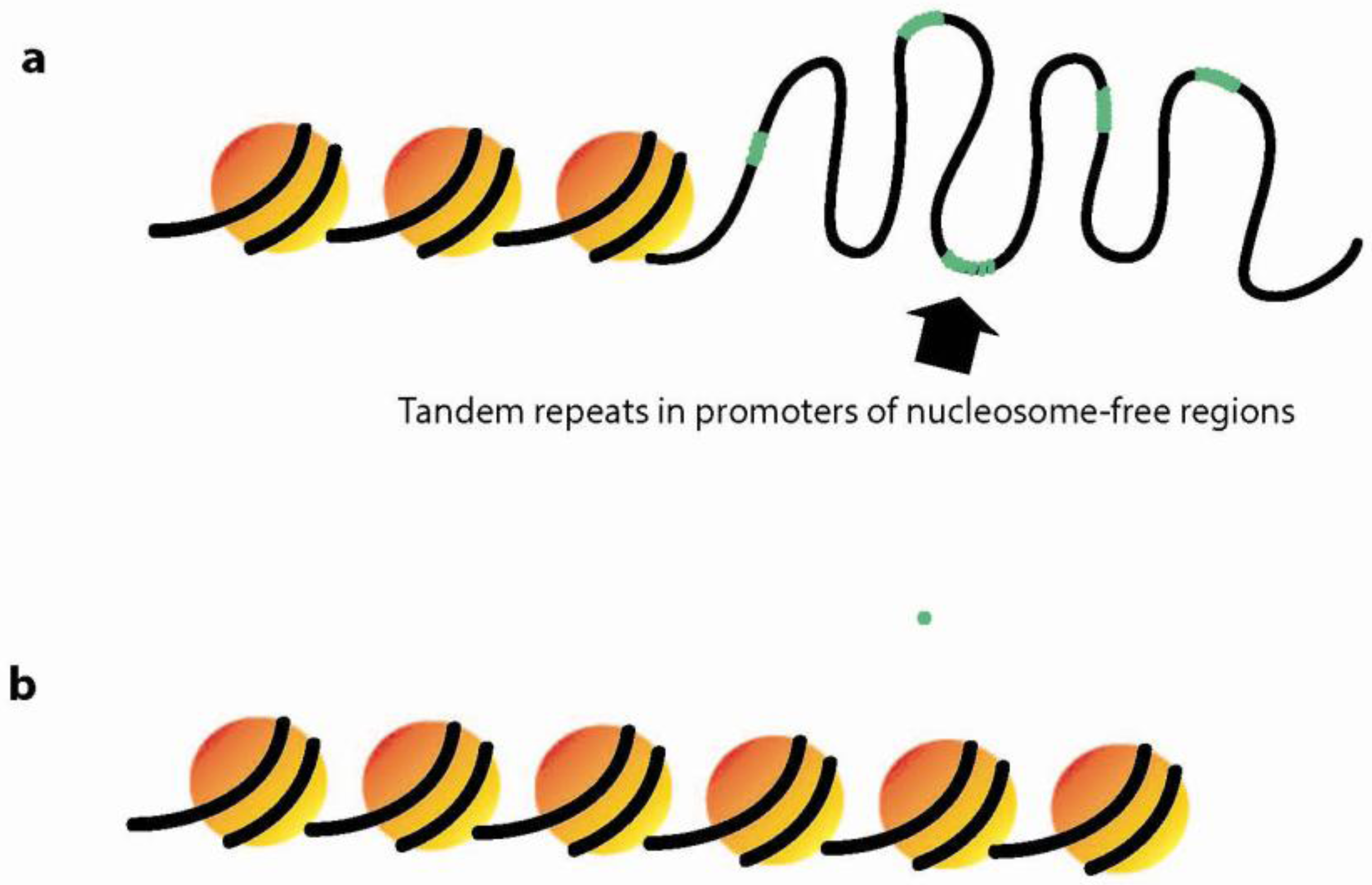
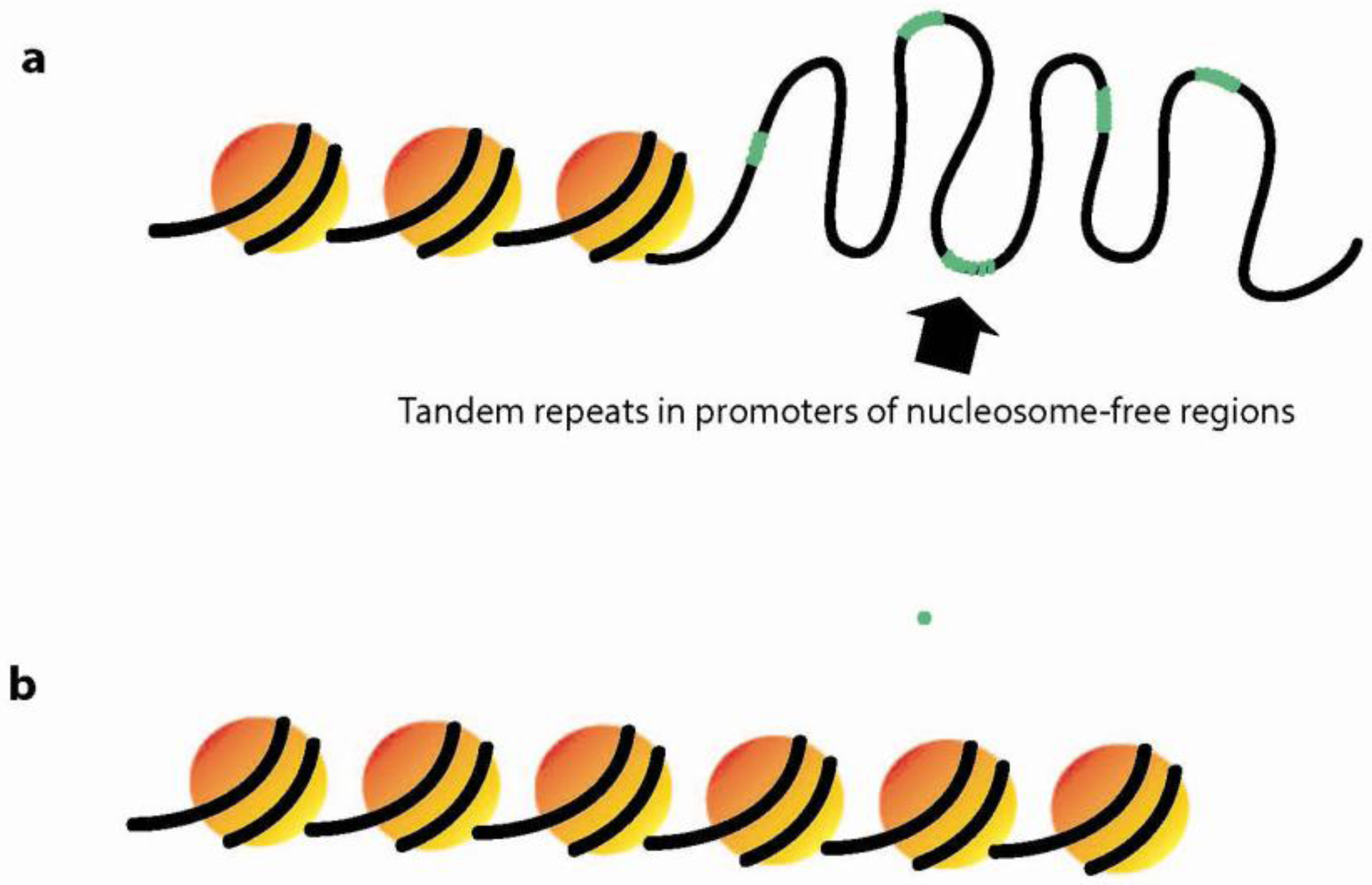
3.3. Dynamic Chromatin Structure and Nucleosome Positioning
3.4. Intrinsic Properties of Tandem Repeats Affect DNA Structure
4. Repeats in Introns and Untranslated Regions
4.1. Recruitment of RNA Binding Proteins
4.2. Variable mRNA Splicing Patterns

4.3. How Can Variable Tandem Repeats in Regulatory Regions Promote Evolvability?
5. Repeats in Coding Regions
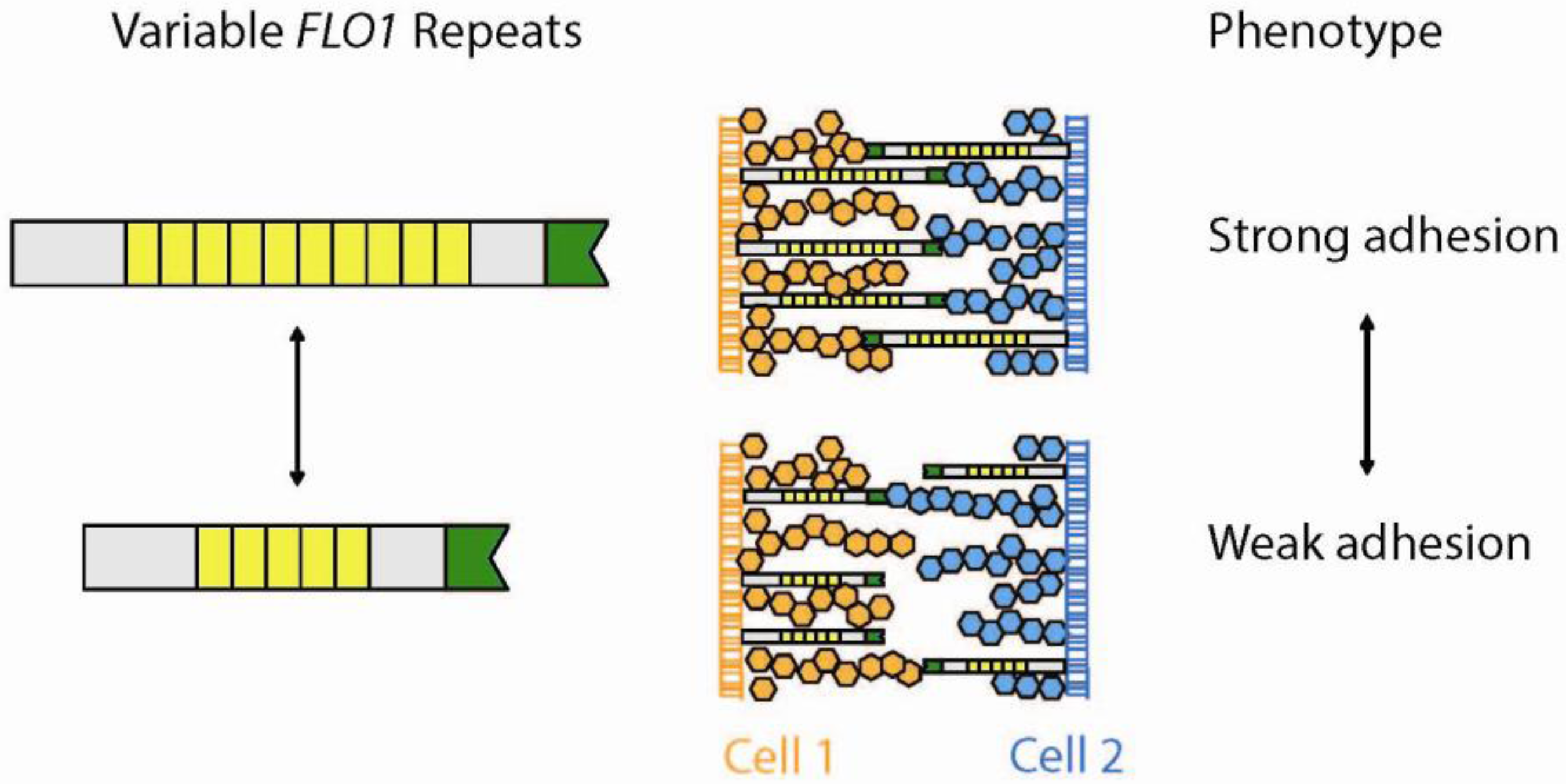
5.1. Minisatellites—Functional Variability in Cell Surface Proteins

5.2. Microsatellites—Beyond Diseases: A Greater Purpose for Coding Repeats
5.3. Repeats as Molecular Switches in Bacteria
5.4. Significance of Polyglutamine Repeats
5.4.1. Polyglutamine Repeats Regulate Circadian Clocks
5.4.2. Polyglutamine Repeats and Morphological Variation
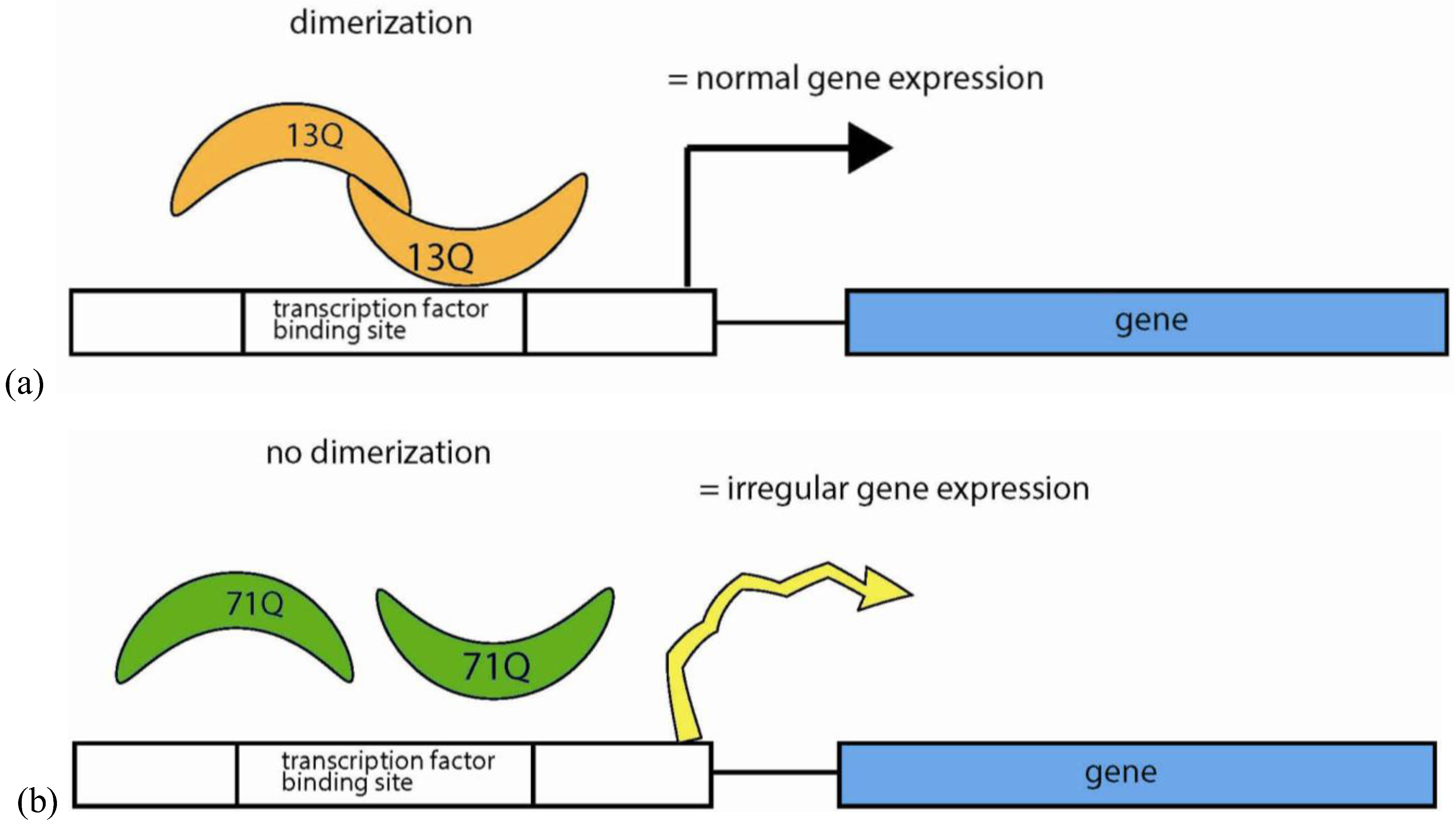
5.4.3. Polyglutamine Repeats and Gene Transcription
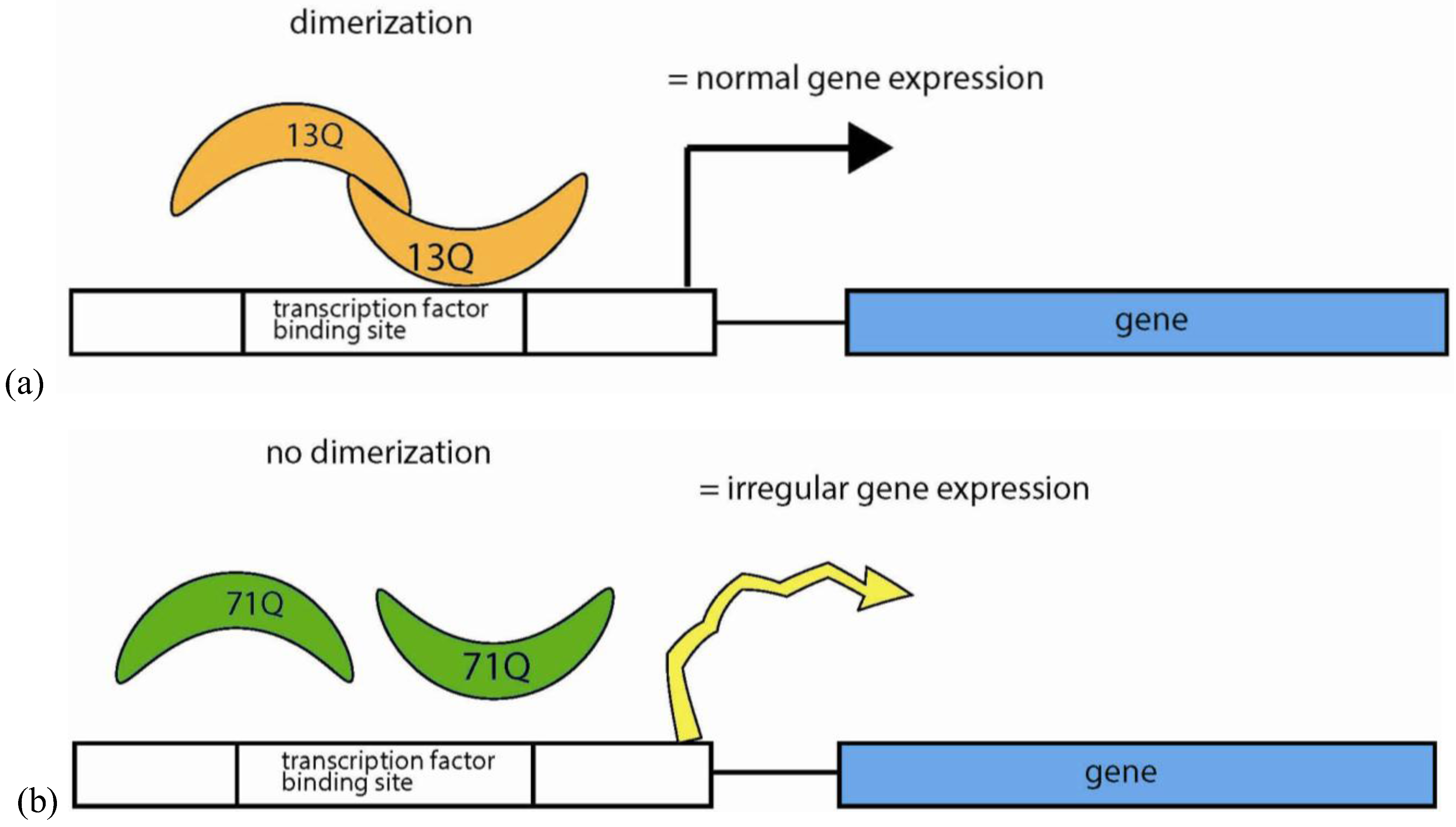
5.4.4. Polyglutamine Repeats in Protein Interactions
6. Conclusions
Acknowledgements
References
- Hartl, D.L. Molecular melodies in high and low C. Nat. Rev. Genet. 2000, 1, 145–149. [Google Scholar] [CrossRef]
- van Belkum, A.; Scherer, S.; van Alphen, L.; Verbrugh, H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. 1998, 62, 275–293. [Google Scholar]
- Feschotte, C.; Keswani, U.; Ranganathan, N.; Guibotsy, M.L.; Levine, D. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol. Evol. 2009, 1, 205–220. [Google Scholar]
- Kit, S. Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 1961, 3, 711–716. [Google Scholar] [CrossRef]
- Verstrepen, K.J.; Jansen, A.; Lewitter, F.; Fink, G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005, 37, 986–990. [Google Scholar]
- Paques, F.; Leung, W.Y.; Haber, J.E. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell. Biol. 1998, 18, 2045–2054. [Google Scholar]
- Kokoska, R.J.; Stefanovic, L.; Tran, H.T.; Resnick, M.A.; Gordenin, D.A.; Petes, T.D. Destabilization of yeast micro- and minisatellite DNA sequences by mutations affecting a nuclease involved in Okazaki fragment processing (rad27) and DNA polymerase delta (pol3-t). Mol. Cell. Biol. 1998, 18, 2779–2788. [Google Scholar]
- Richard, G.F.; Paques, F. Mini- and microsatellite expansions: The recombination connection. EMBO Rep. 2000, 1, 122–126. [Google Scholar] [CrossRef]
- Legendre, M.; Pochet, N.; Pak, T.; Verstrepen, K.J. Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res. 2007, 17, 1787–1796. [Google Scholar]
- Petes, T.D.; Greenwell, P.W.; Dominska, M. Stabilization of microsatellite sequences by variant repeats in the yeast Saccharomyces cerevisiae. Genetics 1997, 146, 491–498. [Google Scholar]
- Gragg, H.; Harfe, B.D.; Jinks-Robertson, S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 8756–8762. [Google Scholar]
- Wierdl, M.; Greene, C.N.; Datta, A.; Jinks-Robertson, S.; Petes, T.D. Destabilization of simple repetitive DNA sequences by transcription in yeast. Genetics 1996, 143, 713–721. [Google Scholar]
- Schmidt, A.L.; Mitter, V. Microsatellite mutation directed by an external stimulus. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 568, 233–243. [Google Scholar] [CrossRef]
- Rosenberg, S.M. Evolving responsively: Adaptive mutation. Nat. Rev. Genet. 2001, 2, 504–515. [Google Scholar] [CrossRef]
- Mittelman, D.; Sykoudis, K.; Hersh, M.; Lin, Y.; Wilson, J.H. Hsp90 modulates CAG repeat instability in human cells. Cell Stress Chaperones 2010, 15, 753–759. [Google Scholar] [CrossRef]
- Merkel, A.; Gemmell, N. Detecting short tandem repeats from genome data: Opening the software black box. Brief Bioinform. 2008, 9, 355–366. [Google Scholar] [CrossRef]
- Gemayel, R.; Vinces, M.D.; Legendre, M.; Verstrepen, K.J. Variable Tandem Repeats Accelerate Evolution of Coding and Regulatory Sequences. Annu. Rev. Genet. 2010, 44, 445–477. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.; Gemayel, R.; Verstrepen, K.J. Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn. 2012, 7, 108–125. [Google Scholar] [CrossRef]
- Young, E.T.; Sloan, J.S.; van Riper, K. Trinucleotide repeats are clustered in regulatory genes in Saccharomyces cerevisiae. Genetics 2000, 154, 1053–1068. [Google Scholar]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar]
- O'Dushlaine, C.T.; Edwards, R.J.; Park, S.D.; Shields, D.C. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 2005, 6, 12. [Google Scholar]
- Mularoni, L.; Ledda, A.; Toll-Riera, M.; Alba, M.M. Natural selection drives the accumulation of amino acid tandem repeats in human proteins. Genome Res. 2010, 20, 745–754. [Google Scholar] [CrossRef]
- Legendre, M.; Verstrepen, K.J. Harvard University: Cambridge, MA, USA, 2007; Unpublished work.
- Faux, N.G.; Bottomley, S.P.; Lesk, A.M.; Irving, J.A.; Morrison, J.R.; de la Banda, M.C.; Whisstock, J.C. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 2005, 15, 537–551. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Sapienza, C. Selfish genes, the phenotype paradigm and genome evolution. Nature 1980, 284, 601–603. [Google Scholar] [CrossRef]
- Ohno, S. So much "junk" DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Orgel, L.E.; Crick, F.H. Selfish DNA: the ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- King, M.C.; Wilson, A.C. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar]
- Wray, G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef]
- Rockman, M.V.; Wray, G.A. Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol. 2002, 19, 1991–2004. [Google Scholar] [CrossRef]
- Vinces, M.D.; Legendre, M.; Caldara, M.; Hagihara, M.; Verstrepen, K.J. Unstable tandem repeats in promoters confer transcriptional evolvability. Science 2009, 324, 1213–1216. [Google Scholar]
- Martin, P.; Makepeace, K.; Hill, S.A.; Hood, D.W.; Moxon, E.R. Microsatellite instability regulates transcription factor binding and gene expression. Proc. Natl. Acad. Sci. USA 2005, 102, 3800–3804. [Google Scholar]
- Johnson, A.C.; Jinno, Y.; Merlino, G.T. Modulation of epidermal growth factor receptor proto-oncogene transcription by a promoter site sensitive to S1 nuclease. Mol. Cell. Biol. 1988, 8, 4174–4184. [Google Scholar]
- Whetstine, J.R.; Witt, T.L.; Matherly, L.H. The human reduced folate carrier gene is regulated by the AP2 and Sp1 transcription factor families and a functional 61-base pair polymorphism. J. Biol. Chem. 2002, 277, 43873–43880. [Google Scholar]
- Iglesias, A.R.; Kindlund, E.; Tammi, M.; Wadelius, C. Some microsatellites may act as novel polymorphic cis-regulatory elements through transcription factor binding. Gene 2004, 341, 149–165. [Google Scholar] [CrossRef]
- Metruccio, M.M.E.; Pigozzi, E.; Roncarati, D.; Scorza, F.B.; Norais, N.; Hill, S.A.; Scarlato, V.; Delany, I. A Novel Phase Variation Mechanism in the Meningococcus Driven by a Ligand-Responsive Repressor and Differential Spacing of Distal Promoter Elements. PLoS Pathog. 2009, 5, e1000710. [Google Scholar]
- van der Ende, A.; Hopman, C.T.; Zaat, S.; Essink, B.B.; Berkhout, B.; Dankert, J. Variable expression of class 1 outer membrane protein in Neisseria meningitidis is caused by variation in the spacing between the -10 and -35 regions of the promoter. J. Bacteriol. 1995, 177, 2475–2480. [Google Scholar]
- van Ham, S.M.; van Alphen, L.; Mooi, F.R.; van Putten, J.P. Phase variation of H. influenzae fimbriae: Transcriptional control of two divergent genes through a variable combined promoter region. Cell 1993, 73, 1187–1196. [Google Scholar] [CrossRef]
- Willems, R.; Paul, A.; van der Heide, H.G.; ter Avest, A.R.; Mooi, F.R. Fimbrial phase variation in Bordetella pertussis: A novel mechanism for transcriptional regulation. EMBO J. 1990, 9, 2803–2809. [Google Scholar]
- Yogev, D.; Rosengarten, R.; Watson-McKown, R.; Wise, K.S. Molecular basis of Mycoplasma surface antigenic variation: A novel set of divergent genes undergo spontaneous mutation of periodic coding regions and 5' regulatory sequences. EMBO J. 1991, 10, 4069–4079. [Google Scholar]
- Saunders, N.J.; Jeffries, A.C.; Peden, P.F.; Hood, D.W.; Tettelin, H.; Rappuoli, R.; Moxon, E.R. Repeat-associated phase variable genes in the complete genome sequence of Neisseria meningitidis strain MC58. Mol. Microbiol. 2000, 37, 207–215. [Google Scholar] [CrossRef]
- Iyer, V.; Struhl, K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995, 14, 2570–2579. [Google Scholar]
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366. [Google Scholar]
- Yuan, G.C.; Liu, Y.J.; Dion, M.F.; Slack, M.D.; Wu, L.F.; Altschuler, S.J.; Rando, O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 2005, 309, 626–630. [Google Scholar]
- Naylor, L.H.; Clark, E.M. d(CA)n sequences upstream of the rat prolactin gene form Z-DNA and inhibit gene transcription. Nucleic Acids Res. 1990, 18, 1595–1601. [Google Scholar] [CrossRef]
- Oh, D.B.; Kim, Y.G.; Rich, A. Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 16666–16671. [Google Scholar]
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755. [Google Scholar] [CrossRef]
- Shang, E.; Cui, Q.; Wang, X.; Beseler, C.; Greenberg, D.A.; Wolgemuth, D.J. The Bromodomain-Containing Gene BRD2 Is Regulated at Transcription, Splicing, and Translation Levels. J. Cell. Biochem. 2011, 112, 2784–2793. [Google Scholar]
- Pagani, F.; Buratti, E.; Stuani, C.; Romano, M.; Zuccato, E.; Niksic, M.; Giglio, L.; Faraguna, D.; Baralle, F.E. Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J. Biol. Chem. 2000, 275, 21041–21047. [Google Scholar]
- Rockman, M.V.; Hahn, M.W.; Soranzo, N.; Loisel, D.A.; Goldstein, D.B.; Wray, G.A. Positive selection on MMP3 regulation has shaped heart disease risk. Curr. Biol. 2004, 14, 1531–1539. [Google Scholar]
- Fidalgo, M.; Barrales, R.R.; Ibeas, J.I.; Jimenez, J. Adaptive evolution by mutations in the FLO11 gene. Proc. Natl. Acad. Sci. USA 2006, 103, 11228–11233. [Google Scholar]
- Hoyer, L.L. The ALS gene family of Candida albicans. Trends Microbiol. 2001, 9, 176–180. [Google Scholar] [CrossRef]
- Verstrepen, K.J.; Reynolds, T.B.; Fink, G.R. Origins of variation in the fungal cell surface. Nat. Rev. Microbiol. 2004, 2, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Kita, E.; Katsui, N.; Emoto, M.; Sawaki, M.; Oku, D.; Nishikawa, F.; Hamuro, A.; Kashiba, S. Virulence of transparent and opaque colony types of Neisseria gonorrhoeae for the genital tract of mice. J. Med. Microbiol. 1991, 34, 355–362. [Google Scholar] [CrossRef]
- Stern, A.; Brown, M.; Nickel, P.; Meyer, T.F. Opacity genes in Neisseria gonorrhoeae: Control of phase and antigenic variation. Cell 1986, 47, 61–71. [Google Scholar] [CrossRef]
- Weiser, J.N.; Love, J.M.; Moxon, E.R. The molecular mechanism of phase variation of H. influenzae lipopolysaccharide. Cell 1989, 59, 657–665. [Google Scholar] [CrossRef]
- de Bolle, X.; Bayliss, C.D.; Field, D.; van de Ven, T.; Saunders, N.J.; Hood, D.W.; Moxon, E.R. The length of a tetranucleotide repeat tract in Haemophilus influenzae determines the phase variation rate of a gene with homology to type III DNA methyltransferases. Mol. Microbiol. 2000, 35, 211–222. [Google Scholar] [CrossRef]
- Srikhanta, Y.N.; Dowideit, S.J.; Edwards, J.L.; Falsetta, M.L.; Wu, H.J.; Harrison, O.B.; Fox, K.L.; Seib, K.L.; Maguire, T.L.; Wang, A.H.J.; et al. Phasevarions Mediate Random Switching of Gene Expression in Pathogenic Neisseria. PLoS Pathog. 2009, 5, e1000400. [Google Scholar]
- Mrazek, J.; Guo, X.X.; Shah, A. Simple sequence repeats in prokaryotic genomes. Proc. Natl. Acad. Sci. USA 2007, 104, 8472–8477. [Google Scholar]
- Lin, W.H.; Kussell, E. Evolutionary pressures on simple sequence repeats in prokaryotic coding regions. Nucleic Acids Res. 2012, 40, 2399–2413. [Google Scholar] [CrossRef]
- Orr, H.; Zoghbi, H. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Karlin, S.; Burge, C. Trinucleotide repeats and long homopeptides in genes and proteins associated with nervous system disease and development. Proc. Natl. Acad. Sci. USA 1996, 93, 1560–1565. [Google Scholar] [CrossRef]
- Froehlich, A.C.; Liu, Y.; Loros, J.J.; Dunlap, J.C. White collar-1, a circadian blue light photoreceptor, binding to the frequency promoter. Science 2002, 297, 815–819. [Google Scholar] [CrossRef]
- Michael, T.P.; Park, S.; Kim, T.S.; Booth, J.; Byer, A.; Sun, Q.; Chory, J.; Lee, K. Simple Sequence Repeats Provide a Substrate for Phenotypic Variation in the Neurospora crassa Circadian Clock. PLoS One 2007, 2, 10. [Google Scholar]
- Johnsen, A.; Fidler, A.E.; Kuhn, S.; Carter, K.L.; Hoffmann, A.; Barr, I.R.; Biard, C.; Charmantier, A.; Eens, M.; Korsten, P.; et al. Avian Clock gene polymorphism: evidence for a latitudinal cline in allele frequencies. Mol. Ecol. 2007, 16, 4867–4880. [Google Scholar] [CrossRef]
- Fondon, J.W.; Garner, H.R. Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 18058–18063. [Google Scholar]
- Sears, K.E.; Goswami, A.; Flynn, J.J.; Niswander, L.A. The correlated evolution of Runx2 tandem repeats, transcriptional activity, and facial length in Carnivora. Evol. Dev. 2007, 9, 555–565. [Google Scholar] [CrossRef]
- Gerber, H.P.; Seipel, K.; Georgiev, O.; Hofferer, M.; Hug, M.; Rusconi, S.; Schaffner, W. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science 1994, 263, 808–811. [Google Scholar]
- Friedman, M.J.; Shah, A.G.; Fang, Z.-H.; Ward, E.G.; Warren, S.T.; Li, S.; Li, X.-J. Polyglutamine domain modulates the TBP-TFIIB interaction: Implications for its normal function and neurodegeneration. Nat. Neurosci. 2007, 10, 1519–1528. [Google Scholar] [CrossRef]
- Jackson-Fisher, A.J.; Chitikila, C.; Mitra, M.; Pugh, B.F. A role for TBP dimerization in preventing unregulated gene expression. Mol. Cell 1999, 3, 717–727. [Google Scholar] [CrossRef]
- Bachtrog, D.; Weiss, S.; Zangerl, B.; Brem, G.; Schlotterer, C. Distribution of dinucleotide microsatellites in the Drosophila melanogaster genome. Mol. Biol. Evol. 1999, 16, 602–610. [Google Scholar] [CrossRef]
- Schroth, G.P.; Chou, P.J.; Ho, P.S. Mapping Z-DNA in the human genome. Computer-aided mapping reveals a nonrandom distribution of potential Z-DNA-forming sequences in human genes. J. Biol. Chem. 1992, 267, 11846–11855. [Google Scholar]
- Riley, D.E.; Krieger, J.N. UTR dinucleotide simple sequence repeat evolution exhibits recurring patterns including regulatory sequence motif replacements. Gene 2009, 429, 80–86. [Google Scholar] [CrossRef]
- Riley, D.E.; Krieger, J.N. Embryonic nervous system genes predominate in searches for dinucleotide simple sequence repeats flanked by conserved sequences. Gene 2009, 429, 74–79. [Google Scholar] [CrossRef]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Chen, M.; Tan, Z.; Jiang, J.; Li, M.; Chen, H.; Shen, G.; Yu, R. Similar distribution of simple sequence repeats in diverse completed Human Immunodeficiency Virus Type 1 genomes. FEBS Lett. 2009, 583, 2959–2963. [Google Scholar] [CrossRef]
- King, D.G.; Soller, M.; Kashi, Y. Evolutionary tuning knobs. Endeavour 1997, 21, 36–40. [Google Scholar] [CrossRef]
- Lo Sardo, V.; Zuccato, C.; Gaudenzi, G.; Vitali, B.; Ramos, C.; Tartari, M.; Myre, M.A.; Walker, J.A.; Pistocchi, A.; Conti, L.; et al. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat. Neurosci. 2012, 15, 713–721. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gemayel, R.; Cho, J.; Boeynaems, S.; Verstrepen, K.J. Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences. Genes 2012, 3, 461-480. https://doi.org/10.3390/genes3030461
Gemayel R, Cho J, Boeynaems S, Verstrepen KJ. Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences. Genes. 2012; 3(3):461-480. https://doi.org/10.3390/genes3030461
Chicago/Turabian StyleGemayel, Rita, Janice Cho, Steven Boeynaems, and Kevin J. Verstrepen. 2012. "Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences" Genes 3, no. 3: 461-480. https://doi.org/10.3390/genes3030461
APA StyleGemayel, R., Cho, J., Boeynaems, S., & Verstrepen, K. J. (2012). Beyond Junk-Variable Tandem Repeats as Facilitators of Rapid Evolution of Regulatory and Coding Sequences. Genes, 3(3), 461-480. https://doi.org/10.3390/genes3030461