The Genetics of Symbiotic Nitrogen Fixation: Comparative Genomics of 14 Rhizobia Strains by Resolution of Protein Clusters

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Key Characteristics of the Fourteen Strains of the Order Rhizobiales
2.1.1. Azorhizobium
2.1.2. Bradyrhizobium

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rhizobium species | Chr | Plasmid1 | Plasmid2 | Plasmid3 | Plasmid4 | Plasmid5 | Plasmid6 |
|---|---|---|---|---|---|---|---|
| Azorhizobium caulinodans ORS 571 | NC_009937 | - | - | - | - | - | - |
| Bradyrhizobium japonicum USDA 110 | NC_004463 | - | - | - | - | - | - |
| Bradyrhizobium sp. BTAi1 | NC_009485 | NC_009475 | |||||
| Bradyrhizobium sp. ORS 278 | NC_009445 | - | - | - | - | - | - |
| Mesorhizobium loti MAFF303099 | NC_002678 | NC_002679 | NC_002682 | ||||
| Mesorhizobium sp. BNC1 (Chelativorans sp. BNC1) | NC_008254 | NC_008242 | NC_008243 | NC_008244 | |||
| Rhizobium etli CFN 42 | NC_007761 | NC_007762 | NC_007763 | NC_007764 | NC_004041 | NC_007765 | NC_007766 |
| Rhizobium etli CIAT 652 | NC_010994 | NC_010998 | NC_010996 | NC_010997 | |||
| Rhizobium leguminosarum bv. trifolii WSM1325 | NC_012850 | NC_012848 | NC_012858 | NC_012853 | NC_012852 | NC_012854 | |
| Rhizobium leguminosarum bv. trifolii WSM2304 | NC_011369 | NC_011368 | NC_011366 | NC_011370 | NC_011371 | ||
| Rhizobium leguminosarum bv. viciae 3841 | NC_008380 | NC_008382 | NC_008383 | NC_008379 | NC_008381 | NC_008384 | NC_008378 |
| Sinorhizobium fredii sp. NGR 234 | NC_012587 | NC_000914 | NC_012586 | ||||
| Sinorhizobium medicae WSM419 | NC_009636 | NC_009620 | NC_009621 | NC_009622 | |||
| Sinorhizobium meliloti 1021 | NC_003047 | NC_003037 | NC_003078 |
| Rhizobiales species | Species Code | No. of Plasmids | Total Genome Length (nucleotides) | Protein Coding Genes | tRNA genes | Pseudo genes | GC Content (%) | Proportion of Genome that is Gene Coding (%) | Host (Scientific/common name) |
|---|---|---|---|---|---|---|---|---|---|
| Azorhizobium caulinodans ORS 571 | azc | 0 | 5,369,772 | 4717 | 63 | - | 67 | 89 | Sesbania rostrata (sesbania) |
| Bradyrhizobium japonicum USDA 110 | bja | 0 | 9,105,828 | 8317 | 56 | - | 64 | 86 | Glycine max (soybean) |
| Bradyrhizobium sp. BTAi1 | bbt | 1 | 8,493,513 | 7621 | 70 | 90 | 64 | 85 | Aeschynomene indica (Indian joint-vetch) |
| Bradyrhizobium sp. ORS 278 | bra | 0 | 7,456,587 | 6717 | 66 | 35 | 65 | 85 | Aeschynomene sensitiva (sensitive joint-vetch) |
| Mesorhizobium loti MAFF303099 | mlo | 2 | 7,596,297 | 7272 | 57 | - | 62 | 86 | Lotus sp., including Lotus japonicas (trifoils, vetches) |
| Mesorhizobium sp. BNC1 | mes | 3 | 4,935,185 | 4543 | 68 | 40 | 61 | 89 | non-symbiotic |
| Rhizobium etli CFN 42 | ret | 6 | 6,530,228 | 5963 | 59 | 32 | 61 | 86 | Phaseolus vulgaris (common bean) |
| Rhizobium etli CIAT 652 | rec | 3 | 6,448,048 | 6056 | 60 | 15 | 61 | 86 | Phaseolus vulgaris (common bean) |
| Rhizobium leguminosarum bv. trifolii WSM1325 | rlg | 5 | 7,418,122 | 7001 | 63 | 75 | 61 | 86 | Trifolli pratense and other Mediterraneum Trifollium (clovers), |
| Rhizobium leguminosarum bv. trifolii WSM2304 | rlt | 4 | 6,872,702 | 6415 | 65 | 45 | 61 | 86 | Trifolium polymorphum from Uruguay (clover) |
| Rhizobium leguminosarum bv. viciae 3841 | rle | 6 | 7,751,309 | 7143 | 61 | 37 | 61 | 86 | Tribe Viciae –Vicia, Pisum, Lathyrus, Lens (vetchs, peas, lathyrus, lentils) |
| Sinorhizobium fredii NGR 234 | rhi | 2 | 6,891,900 | 6363 | 70 | - | 63 | 87 | 112 legume species and the non-legume Parasponia (family Ulmaceae) |
| Sinorhizobium medicae WSM419 | smd | 3 | 6,817,576 | 6213 | 63 | 43 | 61 | 87 | Medicago spp. |
| Sinorhizobium meliloti 1021 | sme | 2 | 6,691,694 | 6218 | 66 | 4 | 62 | 86 | Medicago, Melilotus, Trigonella (alfalfa) |
2.1.3. Mesorhizobium
2.1.4. Rhizobium
2.1.5. Sinorhizobium
2.2. Phylogeny and Taxonomy of Rhizobiales
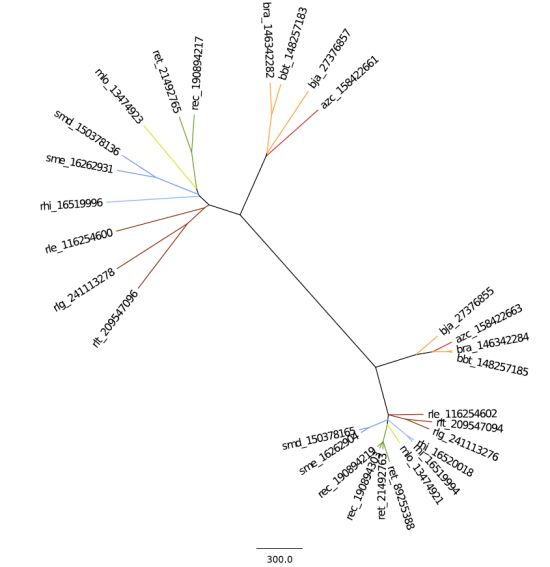
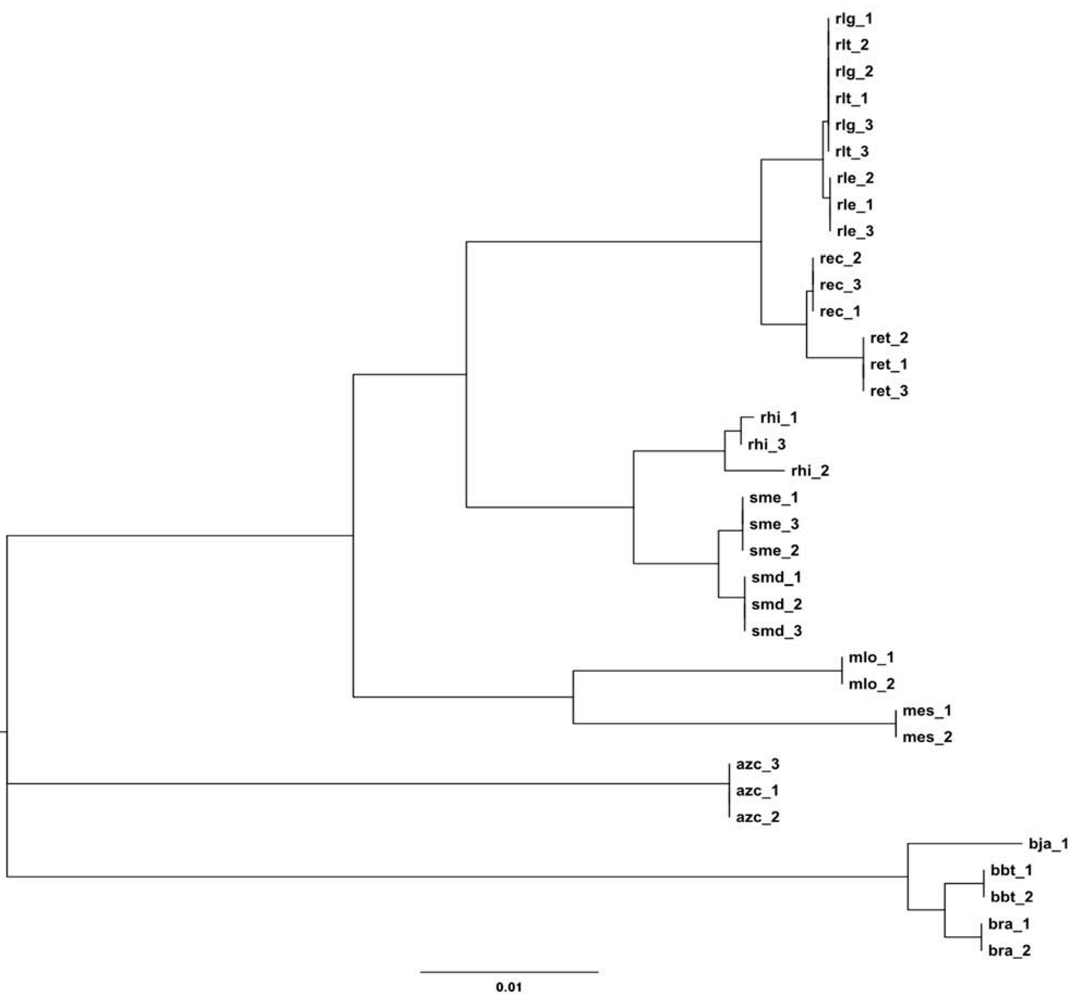
2.2.1. 16S rRNA Taxonomy

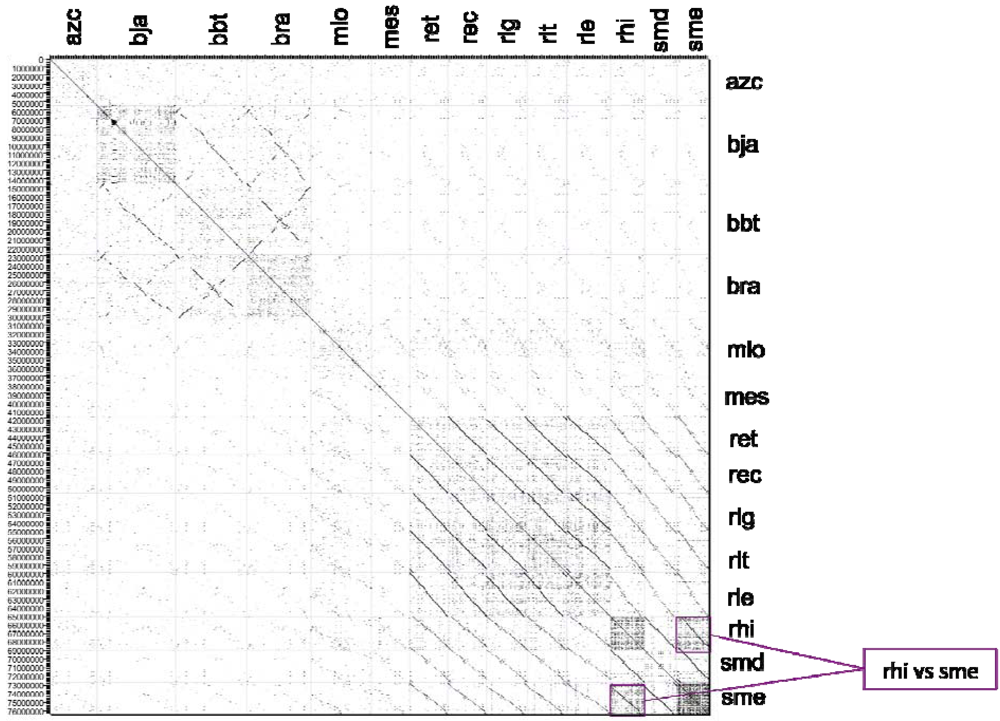
2.2.2. Dotplot Analysis

2.3. KEGG Orthology and Protein Clustering
2.3.1. KEGG Pathway Analysis
2.3.2. Protein Cluster Analysis
| Relevant KEGG Pathways | Number of KEGG Protein Orthologs | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| azc | bja | bbt | bra | mlo | mes | ret | rec | rlg | rlt | rle | rhi | smd | sme | |
| 1.1 Carbohydrate Metabolism | 240 | 295 | 295 | 296 | 274 | 245 | 249 | 279 | 257 | 256 | 299 | 262 | 267 | 273 |
| 1.2 Energy Metabolism | 114 | 147 | 146 | 145 | 113 | 114 | 112 | 111 | 99 | 99 | 118 | 111 | 124 | 138 |
| 1.3 Lipid Metabolism | 52 | 63 | 72 | 72 | 72 | 53 | 61 | 76 | 58 | 57 | 81 | 56 | 58 | 71 |
| 1.4 Nucleotide Metabolism | 102 | 96 | 100 | 99 | 111 | 105 | 100 | 108 | 97 | 99 | 112 | 97 | 98 | 114 |
| 1.5 Amino Acid Metabolism | 221 | 249 | 268 | 258 | 275 | 224 | 235 | 261 | 227 | 241 | 272 | 239 | 236 | 253 |
| 1.6 Metabolism of Other Amino Acids | 50 | 58 | 62 | 57 | 60 | 48 | 54 | 58 | 54 | 57 | 58 | 56 | 54 | 62 |
| 1.7 Glycan Biosynthesis and Metabolism | 30 | 29 | 31 | 33 | 32 | 25 | 33 | 35 | 32 | 34 | 36 | 31 | 22 | 32 |
| 1.8 Metabolism of Cofactors and Vitamins | 117 | 132 | 143 | 137 | 131 | 103 | 122 | 126 | 114 | 117 | 130 | 119 | 117 | 124 |
| 1.9 Biosynthesis of Polyketides and Terpenoids | 29 | 29 | 42 | 43 | 29 | 27 | 28 | 36 | 28 | 27 | 34 | 27 | 27 | 34 |
| 1.10 Biosynthesis of Other Secondary Metabolites | 8 | 21 | 32 | 35 | 31 | 20 | 27 | 34 | 26 | 26 | 37 | 26 | 23 | 28 |
| 1.11 Xenobiotics Biodegradation and Metabolism | 85 | 134 | 166 | 163 | 97 | 72 | 58 | 129 | 60 | 59 | 138 | 55 | 53 | 101 |
| 2.1 Transcription | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| 2.2 Translation | 134 | 131 | 128 | 129 | 130 | 123 | 128 | 128 | 129 | 129 | 127 | 124 | 129 | 132 |
| 2.3 Folding, Sorting and Degradation | 40 | 40 | 38 | 38 | 36 | 34 | 36 | 37 | 38 | 38 | 37 | 36 | 38 | 38 |
| 2.4 Replication and Repair | 69 | 74 | 71 | 71 | 70 | 70 | 71 | 71 | 73 | 71 | 73 | 70 | 71 | 71 |
| 3.1 Membrane Transport | 119 | 141 | 121 | 114 | 172 | 135 | 159 | 163 | 138 | 131 | 162 | 165 | 148 | 170 |
| 3.2 Signal Transduction | 57 | 61 | 55 | 57 | 50 | 39 | 50 | 55 | 48 | 48 | 53 | 49 | 46 | 49 |
| 4.2 Cell Motility | 38 | 41 | 43 | 43 | 34 | 38 | 40 | 41 | 40 | 40 | 41 | 40 | 40 | 40 |
| Total KEGG Protein Orthologs | 1509 | 1745 | 1817 | 1794 | 1721 | 1479 | 1567 | 1752 | 1522 | 1533 | 1812 | 1567 | 1555 | 1734 |
| Cluster Family | Clusters | Proteins | Proteins in Chromosomes | Proteins in Plasmids | Percent in Chromosomes | Percent in Plasmids |
|---|---|---|---|---|---|---|
| Pan-genome (all 14) | 1126 | 28110 | 23686 | 4424 | 84.26 | 15.74 |
| 13 Fix+ genomes | 105 | 1126 | 619 | 507 | 54.97 | 45.03 |
| 11 NodABC+ genomes | 9 | 113 | 60 | 53 | 53.10 | 46.90 |
| azc+bbt+bja+bra | 206 | 1150 | 1141 | 9 | 99.22 | 0.78 |
| bbt+bja+bra | 857 | 2981 | 2976 | 5 | 99.83 | 0.17 |
| bbt+bra | 577 | 1224 | 1224 | 0 | 100.00 | 0.00 |
| mlo+mes | 86 | 192 | 167 | 25 | 86.98 | 13.02 |
| mes+mlo+rhi+ret+rec+rlg+rlt+rle+smd+sme | 214 | 2424 | 2081 | 343 | 85.85 | 14.15 |
| mlo+rhi+ret+rec+rlg+rlt+rle+smd+sme | 161 | 1661 | 1140 | 521 | 68.63 | 31.37 |
| rhi+ret+rec+rlg+rlt+rle+smd+sme | 155 | 1347 | 1122 | 225 | 83.30 | 16.70 |
| rhi+ret+rec+rlg+rlt+rle | 51 | 347 | 191 | 156 | 55.04 | 44.96 |
| rlg+rlt+rle | 92 | 286 | 197 | 89 | 68.88 | 31.12 |
| smd+sme | 253 | 555 | 182 | 373 | 32.79 | 67.21 |
| rhi+ret+rec | 6 | 21 | 5 | 16 | 23.81 | 76.19 |
| rhi+smd+sme | 242 | 767 | 476 | 291 | 62.06 | 37.94 |
| ret+rec | 123 | 262 | 71 | 191 | 27.10 | 72.90 |
| ret+rec+rlg+rlt+rle | 352 | 1866 | 1436 | 430 | 76.96 | 23.04 |
| Singletons | ||||||
| azc | 956 | 986 | 986 | 0 | 100.00 | 0.00 |
| bja | 1760 | 1839 | 1839 | 0 | 100.00 | 0.00 |
| bbt | 1051 | 1107 | 1005 | 102 | 90.79 | 9.21 |
| bra | 959 | 987 | 987 | 0 | 100.00 | 0.00 |
| mlo | 1706 | 1809 | 1613 | 196 | 89.17 | 10.83 |
| mes | 857 | 899 | 761 | 138 | 84.65 | 15.35 |
| ret | 333 | 344 | 196 | 148 | 56.98 | 43.02 |
| rec | 563 | 563 | 374 | 189 | 66.43 | 33.57 |
| rlg | 576 | 602 | 260 | 342 | 43.19 | 56.81 |
| rlt | 490 | 492 | 286 | 206 | 58.13 | 41.87 |
| rle | 542 | 550 | 276 | 274 | 50.18 | 49.82 |
| rhi | 758 | 797 | 356 | 441 | 44.67 | 55.33 |
| smd | 598 | 639 | 317 | 322 | 49.61 | 50.39 |
| sme | 472 | 498 | 170 | 328 | 34.14 | 65.86 |
2.4. The “Symbiome”- Nodulation, Secretion, Exopolysaccharide Production, Oxygen Transport and Nitrogen Fixation
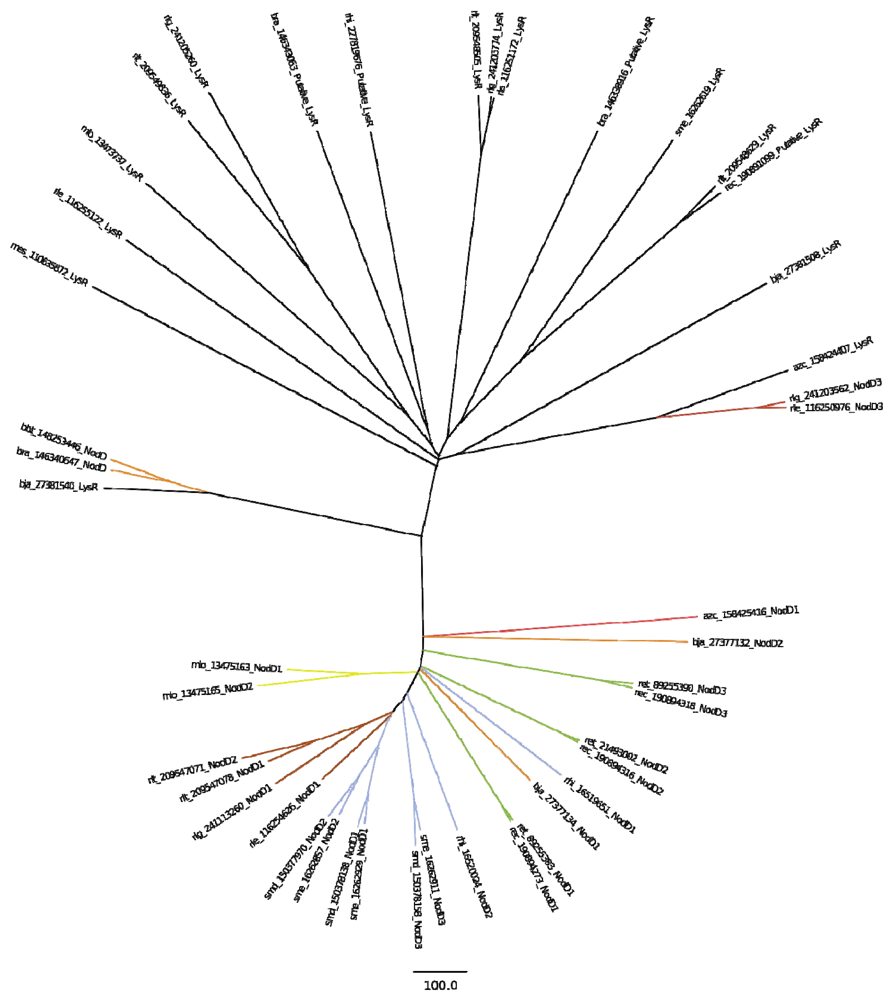
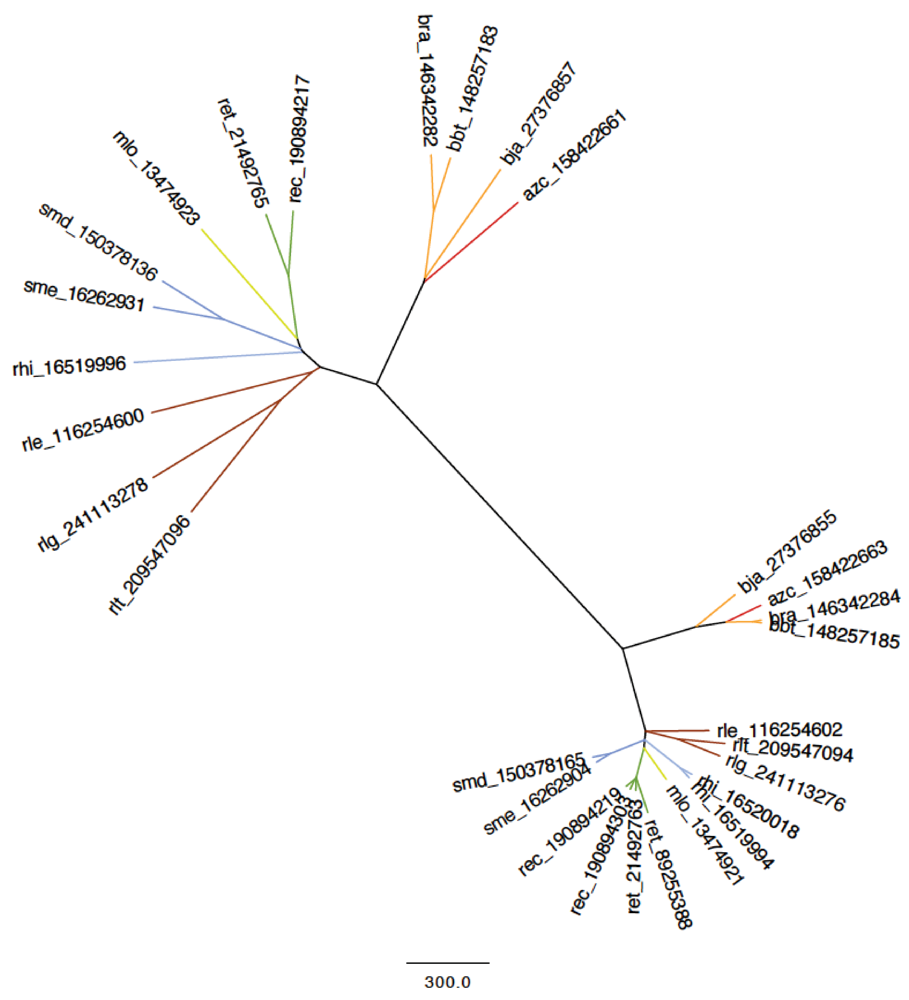
2.4.1. Nod Genes

2.4.2. Other Nodulation Genes
2.4.3. Bacterial Secretion Systems
2.4.3.1. Tat, Type I and II Secretion Systems
2.4.3.2. Tat, Type III and IV Secretion Systems
2.4.3.3. Type V and Type VI Secretion Systems
2.4.4. Exopolysaccharide Production
2.4.5. Nif and Fix Genes
2.5. Is it Possible to Define a Symbiome?

3. Experimental Section
3.1. Genomes
3.2. Bioinformatics Analysis
3.3. Protein Clustering
4. Concluding Remarks
Acknowledgements
References
- Cheng, Q. Perspectives in biological nitrogen fixation research. J. Integr. Plant Biol. 2008, 50, 786–798. [Google Scholar] [CrossRef]
- Hungria, M.; Vargas, M.A. Environmental factors affecting N2 fixation in gratn legumes in the tropics, with an emphasis on Brazil. Field Crops Res. 2000, 65, 151–164. [Google Scholar] [CrossRef]
- Hungria, M.; Loureiro, M.F.; Mendes, I.C.; Campo, R.J.; Graham, P.H. Inoculant Preparation, Production and Application. In Nitrogen Fixation in Agriculture, Forestry, Ecology and the Environment; Werner, W., Newton, W.E., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp. 223–254. [Google Scholar]
- Guerrero, G.; Peralta, H.; Aguilar, A.; Diaz, R.; Villalobos, M.A.; Medrano-Soto, A.; Mora, J. Evolutionary, structural and functional relationships revealed by comparative analysis of syntenic genes in Rhizobiales. BMC Evol. Biol. 2005, 5, 55–73. [Google Scholar] [CrossRef]
- Carvalho, F.M.; Souza, R.C.; Barcellos, F.G.; Hungria, M.; Vasconcelos, A.T.R. Genomic and evolutionary comparisons of diazotrophic and pathogenic bacteria of the order Rhizobiales. BMC Microbiol. 2010, 10. [Google Scholar] [CrossRef]
- Vavilov, N.I. Centers of origin of cultivated plants. Trends Pract. Bot. Genet. Sel. 1926, 16, 3–248. (in Russian). [Google Scholar]
- Lee, K.B.; de Backer, P.; Aono, T.; Liu, C.T.; Suzuki, S.; Suzuki, T.; Kaneko, T.; Yamada, M.; Tabata, S.; Kupfer, D.M.; et al. The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 2008, 9. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, S.; Aono, T.; Akiba, N.; Lee, K.B.; Liu, C.T.; Toyazaki, H.; Oyaizu, H. Comparative genome-wide transcriptional profiling of Azorhizobium caulinodans ORS571 grown under free-living and symbiotic conditions. Appl. Environ. Microbiol. 2009, 75, 5037–5046. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002, 9, 189–197. [Google Scholar] [CrossRef]
- Bonaldi, K.; Gourion, B.; Fardoux, J.; Hannibal, L.; Cartieaux, F.; Boursot, M.; Vallenet, D.; Chaintreuil, C.; Prin, Y.; Nouwen, N.; et al. Large-scale transposon mutagenesis of photosynthetic Bradyrhizobium sp. strain ORS278 reveals new genetic loci putatively important for nod-independent symbiosis with Aeschynomene indica. Mol. Plant Microbe Interact. 2010, 23, 760–770. [Google Scholar] [CrossRef]
- Giraud, E.; Moulin, L.; Vallenet, D.; Barbe, V.; Cytryn, E.; Avarre, J.C.; Jaubert, M.; Simon, D.; Cartieaux, F.; Prin, Y.; et al. Legumes symbioses: Absence of nod genes in photosynthetic bradyrhizobia. Science 2007, 316, 1307–1312. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Ishikawa, A.; Kawashima, K.; et al. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000, 7, 331–338. [Google Scholar] [CrossRef]
- Bohuslavek, J.; Payne, J.W.; Liu, Y.; Bolton, H.; Xun, L.Y. Cloning, sequencing, and characterization of a gene cluster involved in EDTA degradation from the bacterium BNC1. Appl. Environ. Microbiol. 2001, 67, 688–695. [Google Scholar] [CrossRef]
- Baldwin, I.L.; Fred, E.B. Nomenclature of the root-nodule bacteria of the Leguminosae. J. Bacteriol. 1929, 17, 141–150. [Google Scholar]
- Gonzalez, V.; Bustos, P.; Ramirez-Romero, M.A.; Medrano-Soto, A.; Salgado, H.; Hernandez-Gonzalez, I.; Hernandez-Celis, J.C.; Quintero, V.; Moreno-Hagelsieb, G.; Girard, L.; et al. The mosaic structure of the symbiotic plasmid of Rhizobium etli CFN42 and its relation to other symbiotic genome compartments. Genome Biol. 2003, 4. [Google Scholar] [CrossRef] [Green Version]
- González, V.; Santamaría, R.I.; Bustos, P.; Hernández-González, I.; Medrano-Soto, A.; Moreno-Hagelsieb, G.; Janga, S.C.; Ramírez, M.A.; Jiménez-Jacinto, V.; Collado-Vides, J.; et al. The partitioned Rhizobium etli genome: Genetic and metabolic redundancy in seven interacting replicons. Proc. Natl. Acad. Sci. USA 2006, 103, 3834–3839. [Google Scholar]
- González, V.; Acosta, J.L.; Santamaría, R.I.; Bustos, P.; Fernández, J.L.; Hernández González, I.L.; Díaz, R.; Flores, M.; Palacios, R.; Mora, J.; et al. Conserved symbiotic plasmid DNA sequences in the multireplicon pangenomic structure of Rhizobium etli. Appl. Environ. Microbiol. 2010, 76, 1604–1614. [Google Scholar]
- Lozano, L.; Hernández-González, I.; Bustos, P.; Santamaría, R.I.; Souza, V.; Young, J.P.W.; Dávila, G.; González, V. Evolutionary dynamics of insertion sequences in relation to the evolutionary histories of the chromosome and symbiotic plasmid genes of Rhizobium etli populations. Appl. Environ. Microbiol. 2010, 76, 6504–6513. [Google Scholar]
- Howieson, J.G.; Yates, R.J.; Ryder, M.; Real, D. The interactions of Rhizobium leguminosarum biovar trifolii in nodulation of annual and perennial Trifolium spp. from diverse centres of origin. Aust. J. Exp. Agric. 2005, 45, 199–207. [Google Scholar] [CrossRef]
- Yates, R.; Howieson, J.; Reeve, W.; O’Hara, G. A re-appraisal of the biology and terminology describing rhizobial strain success in nodule occupancy of legumes in agriculture. Plant Soil 2011, 348, 255–267. [Google Scholar] [CrossRef]
- Young, J.P.; Crossman, L.; Johnston, A.; Thomson, N.; Ghazoui, Z.; Hull, K.; Wexler, M.; Curson, A.; Todd, J.; Poole, P.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7. [Google Scholar] [CrossRef]
- Chen, W.X.; Yan, G.H.; Li, J.L. Numerical taxonomic study of fast-growing soybean rhizobia and a proposal that Rhizobium fredii be assigned to Sinorhizobium gen. nov. Int. J. Syst. Bacteriol. 1988, 38, 392–397. [Google Scholar] [CrossRef]
- Young, J.M. The genus name Ensifer Casida 1982 takes priority over Sinorhizobium Chen et al. 1988, and Sinorhizobium morelense Wang et al. 2002 is a later synonym of Ensfer adhaerens Casida 1982. Is the combination ‘Sinorhizobium adhaerens’ (Casida 1982) Willems et al. 2003 legitimate? Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2003, 53, 2107–2110. [Google Scholar] [CrossRef]
- Sawada, H.; Kuykendall, L.D.; Young, J.M. Changing concepts in the systematics of bacterial nitrogen-fixing legume symbionts. J. Gen. Appl. Microbiol. 2003, 49, 155–179. [Google Scholar] [CrossRef]
- Graham, P.H. Ecology of the Root-Nodule Bacteria of Legumes. In Nitrogen-Fixing Leguminous Symbioses; Dilworth, M.J., James, E.K., Sprent, J.I., Newton, W.E., Eds.; Springer: Dordrecht, The Netherlands, 2007; Volume 7, pp. 23–58. [Google Scholar]
- Capela, D.; Barloy-Hubler, F.; Gouzy, J.; Bothe, G.; Ampe, F.; Batut, J.; Boistard, P.; Becker, A.; Boutry, M.; Cadieu, E.; et al. Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc. Natl. Acad. Sci. USA 2001, 98, 9877–9882. [Google Scholar]
- Barnett, M.J.; Fisher, R.F.; Jones, T.; Komp, C.; Abola, A.P.; Gurjal, M.; Hong, A.; Huizar, L.; Bowser, L.; Capela, D.; et al. Nucleotide sequence and predicted functions of the entire Sinorhizobium meliloti pSymA megaplasmid. Proc. Natl. Acad. Sci. USA 2001, 98, 9883–9888. [Google Scholar]
- Finan, T.M.; Weidner, S.; Wong, K.; Buhrmester, J.; Chain, P.; Vorholter, F.J.; Hernandez-lucas, I.; Becker, A.; Cowie, A.; Gouzy, J.; et al. The complete sequence of the 1,683-kb pSymB megaplasmid from the N2-fixing endosymbiont Sinorhizobium meliloti. Proc. Natl. Acad. Sci. USA 2001, 98, 9889–9894. [Google Scholar]
- Terpolilli, J.J.; Tiwari, R.P.; Dilworth, M.J.; O'Hara, G.W.; Howieson, J.G. Investigating nitrogen fixation in the Medicago-Sinorhizobium symbiosis. Biol. Nitrogen Fixat. 2008, 42, 145–146. [Google Scholar]
- Terpolilli, J.J.; O'Hara, G.W.; Tiwari, R.P.; Dilworth, M.J.; Howieson, J.G. The model legume Medicago truncatula A17 is poorly matched for N2 fixation with the sequenced microsymbiont Sinorhizobium meliloti 1021. New Phytol. 2008, 179, 62–66. [Google Scholar] [CrossRef]
- Garau, G.; Reeve, W.G.; Brau, L.; Deiana, P.; Yates, R.J.; James, D.; Tiwari, R.; O’Hara, G.W.; Howieson, J.G. The symbiotic requirements of different Medicago spp. suggest the evolution of Sinorhizobium meliloti and S-Medicae with hosts differentially adapted to soil pH. Plant Soil 2005, 276, 263–277. [Google Scholar] [CrossRef]
- Pueppke, S.G.; Broughton, W.J. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant Microbe Interact. 1999, 12, 293–318. [Google Scholar] [CrossRef]
- Viprey, V.; Rosenthal, A.; Broughton, W.J.; Perret, X. Genetic snapshots of the Rhizobium species NGR234 genome. Genome Biol. 2000, 1, 1–17. [Google Scholar]
- Schmeisser, C.; Liesegang, H.; Krysciak, D.; Bakkou, N.; le Quéré, A.; Wollherr, A.; Heinemeyer, I.; Morgenstern, B.; Pommerening-Röser, A.; Flores, M.; et al. Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systems. Appl. Environ. Microbiol. 2009, 75, 4035–4045. [Google Scholar]
- Turner, S.L.; Zhang, X.X.; Li, F.D.; Young, J.P. What does a bacterial genome sequence represent? Mis-assignment of MAFF 303099 to the genospecies Mesorhizobium loti. Microbiology 2002, 148, 3330–3331. [Google Scholar]
- Willems, A. The taxonomy of rhizobia: An overview. Plant and Soil 2006, 287, 3–14. [Google Scholar] [CrossRef]
- Landeta, C.; Davalos, A.; Cevallos, M.A.; Geiger, O.; Brom, S.; Romero, D. Plasmids with a chromosome-like role in Rhizobium. J. Bacteriol. 2011, 193, 1317–1326. [Google Scholar]
- Harrison, P.W.; Lower, R.P.J.; Kim, N.K.D.; Young, J.P.W. Introducing the bacterial ‘chromid’: Not a chromosome, not a plasmid. Trends Microbiol. 2010, 18, 141–148. [Google Scholar] [CrossRef]
- Gevers, D.; Cohan, F.M.; Lawrence, J.G.; Spratt, B.G.; Coenye, T.; Feil, E.J.; Stackebrandt, E.; Peer, Y.V.D.; Vandamme, P.; THOMPSON, F.L.; et al. Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 2005, 3, 733–739. [Google Scholar] [CrossRef]
- Gogarten, J.P.; Townsend, J.P. Horizontal gene transfer, genomes innovation and evolution. Nat. Rev. Microbiol. 2005, 3, 679–687. [Google Scholar] [CrossRef]
- Gómez-Hernández, N.; Reyes-González, A.; Sánchez, C.; Mora, Y.; Delgado, M.J.; Girard, L. Regulation and symbiotic role of nirK and norC expression in Rhizobium etli. Mol. Plant Microbe Interact. 2011, 24, 233–235. [Google Scholar] [CrossRef]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Nicolás, M.F.; Barcellos, M.F.; Hess, P.N.; Hungria, M. ABC transporters in Mycoplasma hyopneumoniae and Mycoplasma synoviae: Insights into evolution and pathogenicity. Genet. Mol. Biol. 2007, 30, 202–211. [Google Scholar] [CrossRef]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Ishikawa, A.; Kawashima, K.; et al. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti (supplement). DNA Res. 2000, 7, 381–406. [Google Scholar] [CrossRef]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar]
- Jones, K.M.; Kobayashi, H.; Davies, B.W.; Taga, M.E.; Walker, G.C. How rhizobial symbionts invade plants: The Sinorhizobium–Medicago model. Nat. Rev. Microbiol. 2007, 5, 619–633. [Google Scholar] [CrossRef]
- Oliveira, L.R.; Marcelino, F.C.; Barcellos, F.G.; Rodrigues, E.P.; Megías, M.; Hungria, M. The nodC, nodG, and glgX genes of Rhizobium tropici strain PRF 81. Funct. Integr. Genomics 2010, 10, 425–431. [Google Scholar] [CrossRef]
- Downie, J.A. The roles of extracellular proteins, polysaccharides and signals in the interactions of rhizobia with legume roots. FEMS Microbiol. Rev. 2010, 34, 150–170. [Google Scholar] [CrossRef]
- Walker, S.A.; Viprey, V.; Downie, J.A. Dissection of nodulation signaling using pea mutants defective for calcium spiking induced by nod factors and chitin oligomers. Proc. Natl. Acad. Sci. USA 2000, 97, 13413–13418. [Google Scholar]
- Vlassak, K.M.; de Wilde, P.; Snoeck, C.; Luyten, E.; van Rhijn, P.; Vanderleyden, J. The Rhizobium sp. BR816 nodD3 gene is regulated by a transcriptional regulator of the AraC/XylS family. Mol. Gen. Genet. 1998, 258, 558–561. [Google Scholar] [CrossRef]
- Cooper, J.E. Multiple responses of rhizobia to flavonoids during legume root infection. Adv. Bot. Res. Inc. Adv. Plant Pathol. 2004, 41, 1–62. [Google Scholar] [CrossRef]
- Cooper, J.E. Early interactions between legumes and rhizobia: Disclosing complexity in a molecular dialogue. J. Appl. Microbiol. 2007, 103, 1355–1365. [Google Scholar] [CrossRef]
- Fauvart, M.; Michiels, J. Rhizobial secreted proteins as determinants of host specificity in the rhizobium-legume symbiosis. FEMS Microbiol. Lett. 2008, 285, 1–9. [Google Scholar] [CrossRef]
- Deakin, W.J.; Broughton, W.J. Symbiotic use of pathogenic strategies: Rhizobial protein secretion systems. Nat. Rev. Microbiol. 2009, 7, 312–320. [Google Scholar]
- Pickering, B.S.; Oresnik, I.J. The twin arginine transport system appears to be essential for viability in Sinorhizobium meliloti. J. Bacteriol. 2010, 192, 5173–5180. [Google Scholar] [CrossRef]
- Alvarez-Martinez, C.E.; Christie, P.J. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 2009, 73, 775–808. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Lorio, J.; Kim, W.S.; Jiang, G.Q.; Kim, K.Y.; DeBoer, M.; Pueppke, S.G. Extracellular proteins involved in soybean cultivar-specific nodulation are associated with pilus-like surface appendages and exported by a type III protein secretion system in Sinorhizobium fredii USDA257. Mol. Plant Microbe Interact. 2003, 16, 617–625. [Google Scholar] [CrossRef]
- Desvaux, M.; Parham, N.J.; Henderson, I.R. Type V protein secretion: Simplicity gone awry? Curr. Issues Mol. Biol. 2004, 6, 111–124. [Google Scholar]
- Schwarz, S.; Hood, R.D.; Mougous, J.D. What is type VI secretion doing in all those bugs? Trends Microbiol. 2010, 18, 531–537. [Google Scholar] [CrossRef]
- Bonemann, G.; Pietrosiuk, A.; Mogk, A. Tubules and donuts: A type VI secretion story. Mol. Microbiol. 2010, 76, 815–821. [Google Scholar] [CrossRef]
- Skorupska, A.; Janczarek, M.; Marczak, M.; Mazur, A.; Król, J. Rhizobial exopolysaccharides: Genetic control and symbiotic functions. Microbial Cell Factories 2006, 5. [Google Scholar] [CrossRef]
- Cytryn, E.J.; Sangurdekar, D.P.; Streeter, J.G.; Franck, W.L.; Chang, W.-S.; Stacey, G.; Emerich, D.W.; Joshi, T.; Xu, D.; Sadowsky, M.J. Transcriptional and physiological responses of Bradyrhizobium japonicum to desiccation-induced stress. J. Bacteriol. 2007, 189, 6751–6762. [Google Scholar]
- Batista, J.S.S.; Torres, A.R.; Hungria, M. Towards a two-dimensional proteomic reference map of Bradyrhizobium japonicum CPAC 15: Spotlighting on “hypothetical proteins”. Proteomics 2010, 10, 3176–3189. [Google Scholar] [CrossRef]
- Raymond, J.; Siefert, J.L.; Staples, C.R.; Blankenship, R.E. The natural history of nitrogen fixation. Mol. Biol. Evol. 2004, 21, 541–554. [Google Scholar]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef]
- Rubio, L.M.; Ludden, P.W. Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 2008, 62, 93–111. [Google Scholar] [CrossRef]
- Halary, S.; Leigh, J.W.; Cheaib, B.; Lopez, P.; Bapteste, E. Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. USA 2010, 107, 127–132. [Google Scholar]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Bellgard, M.I.; Wanchanthuek, P.; La, T.; Ryan, K.; Moolhuijzen, P.; Albertyn, Z.; Shaban, B.; Motro, Y.; Dunn, D.S.; Schibeci, D.; et al. Genome sequence of the pathogenic intestinal spirochete brachyspira hyodysenteriae reveals adaptations to its lifestyle in the porcine large intestine. Plos One 2009, 4. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Garau, G.; Yates, R.; Deiana, P.; Howieson, J. Novel strains of nodulating Burkholderia have a role in nitrogen fixation with papilionoid herbaceous legumes adapted to acid, infertile soils. Soil Biol. Biochem. 2009, 41, 125–134. [Google Scholar] [CrossRef]
- Amadou, C.; Pascal, G.; Mangenot, S.; Glew, M.; Bontemps, C.; Capela, D.; Carrère, S.; Cruveiller, S.; Dossat, C.; Lajus, A.; et al. Genome sequence of the beta-rhizobium Cupriavidus taiwanensis and comparative genomics of rhizobia. Genome Res. 2008, 18, 1472–1483. [Google Scholar] [CrossRef]
- Pokharel, A.; Mirza, B.S.; Dawson, J.O.; Hahn, D. Frankia populations in soil and root nodules of sympatrically grown Alnus taxa. Microb. Ecol. 2011, 61, 92–100. [Google Scholar] [CrossRef]
- Pini, F.; Galardini, M.; Bazzicalupo, M.; Mengoni, A. Plant-bacteria association and symbiosis: Are there common genomic traits in alphaproteobacteria? Genes 2011, 2, 1017–1032. [Google Scholar] [CrossRef]
- Rodriguez-llorente, I.; Caviedes, M.A.; Dary, M.; Palomares, A.J. The symbiosis interactome: A computational approach reveals novel components, functional interactions and modules in Sinorhizobium meliloti. BMC Syst. Biol. 2009, 3, 1–18. [Google Scholar] [CrossRef]
- Delmotte, N.; Ahrens, C.; Knief, C.; Qeli, E.; Koch, M. An integrated proteomics and transcriptomics reference data set provides new insights into the Bradyrhizobium japonicum bacteroid metabolism in soybean root nodules. Proteomics 2010, 10, 1391–1400. [Google Scholar] [CrossRef]
- Ballesteros-Almanza, L.; Altamirano-Hernandez, J.; Pena-Cabriales, J.J.; Santoyo, G.; Sanchez-Yanez, J.M.; Valencia-Cantero, E.; Macias-Rodriguez, L.; Lopez-Bucio, J.; Cardenas-Navarro, R.; Farias-Rodriguez, R. Effect of co-inoculation with mycorrhiza and rhizobia on the nodule trehalose content of different bean genotypes. Open Microbiol. J. 2010, 4, 83–92. [Google Scholar] [CrossRef]
- Batut, J.; Mergaert, P.; Masson-Boivin, C. Peptide signalling in the rhizobium-legume symbiosis. Current Opinion in Microbiology 2011, 14, 181–187. [Google Scholar]
- Prell, J.; Bourdès, A.; Kumar, S.; Lodwig, E.; Hosie, A.; Kinghorn, S.; White, J.; Poole, P. Role of symbiotic auxotrophy in the Rhizobium-legume symbioses. Plos One 2010, 5. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Black, M.; Moolhuijzen, P.; Chapman, B.; Barrero, R.; Howieson, J.; Hungria, M.; Bellgard, M. The Genetics of Symbiotic Nitrogen Fixation: Comparative Genomics of 14 Rhizobia Strains by Resolution of Protein Clusters. Genes 2012, 3, 138-166. https://doi.org/10.3390/genes3010138
Black M, Moolhuijzen P, Chapman B, Barrero R, Howieson J, Hungria M, Bellgard M. The Genetics of Symbiotic Nitrogen Fixation: Comparative Genomics of 14 Rhizobia Strains by Resolution of Protein Clusters. Genes. 2012; 3(1):138-166. https://doi.org/10.3390/genes3010138
Chicago/Turabian StyleBlack, Michael, Paula Moolhuijzen, Brett Chapman, Roberto Barrero, John Howieson, Mariangela Hungria, and Matthew Bellgard. 2012. "The Genetics of Symbiotic Nitrogen Fixation: Comparative Genomics of 14 Rhizobia Strains by Resolution of Protein Clusters" Genes 3, no. 1: 138-166. https://doi.org/10.3390/genes3010138