Plant-Bacteria Association and Symbiosis: Are There Common Genomic Traits in Alphaproteobacteria?



Abstract
: Alphaproteobacteria show a great versatility in adapting to a broad range of environments and lifestyles, with the association between bacteria and plants as one of the most intriguing, spanning from relatively unspecific nonsymbiotic association (as rhizospheric or endophytic strains) to the highly species-specific interaction of rhizobia. To shed some light on possible common genetic features in such a heterogeneous set of plant associations, the genomes of 92 Alphaproteobacteria strains were analyzed with a fuzzy orthologs-species detection approach. This showed that the different habitats and lifestyles of plant-associated bacteria (soil, plant colonizers, symbiont) are partially reflected by the trend to have larger genomes with respect to nonplant-associated species. A relatively large set of genes specific to symbiotic bacteria (73 orthologous groups) was found, with a remarkable presence of regulators, sugar transporters, metabolic enzymes, nodulation genes and several genes with unknown function that could be good candidates for further characterization. Interestingly, 15 orthologous groupspresent in all plant-associated bacteria (symbiotic and nonsymbiotic), but absent in nonplant-associated bacteria, were also found, whose functions were mainly related to regulation of gene expression and electron transport. Two of these orthologous groups were also detected in fully sequenced plant-associated Betaproteobacteria and Gammaproteobacteria. Overall these results lead us to hypothesize that plant-bacteria associations, though quite variable, are partially supported by a conserved set of unsuspected gene functions.1. Introduction
The phylum Proteobacteria is the most numerous group currently recognized in the domain Bacteria [1]. Within this group, the class of Alphaproteobacteria harbors a miscellaneous set of metabolisms, cellular phenotypes and a wide range of habitats, including phototrophic genera (Rhodobacter), symbionts of plants (Rhizobium, Sinorhizobium, Mesorhizobium and Azorhizobium [2]), animal and plant pathogens (Rickettsia, Brucella, Agrobacterium) and also genera able to metabolize C1 compounds (Methylobacterium). In addition, mitochondria have a common origin with SAR11 clade, as a sister group of the order Rickettsiales [3]. Habitats that are colonized by Alphaproteobacteria, range from the ocean floor volcanic environments, to soil, in which they may interact with plant roots, to surface waters of oceans [1].
Alphaproteobacteria, with nearly 600 completely sequenced genomes, is one of the most studied bacterial classes [1], showing a large heterogeneity in genome size, from 1.1 to 9.2 Mbp [4] and genome architecture, with the presence of additional replicons, such as chromids [5], and plasmids [6]. Because of these genomic traits, and also thanks to their versatility in adapting to different habitats, Alphaproteobacteria constitute an excellent model system to study how bacterial genomes evolve and how genomic features are related to environmental adaptation [1,4].
Particularly intriguing is the alphaproteobacterial ability to interact with plants, as pathogens and as nonpathogenic mutualist/commensals (symbionts/nonsymbionts) (e.g., Rhizobium, Azospirillum). Plant-associated bacteria sensu lato can be found in, and around roots, in the vasculature, and on aerial tissues or in specifically developed organs (e.g., root nodules) [7], allowing to categorize strains as phyllospheric, rhizospheric and endophytic.
Phyllospheric bacteria inhabits the aerial parts of the plant (leaves, stems, buds, flowers and fruits), possibly affecting plant fitness and productivity of agricultural crops [8]. The rhizosphere is the part of soil around plant roots populated by microbes (bacteria and fungi); microorganisms from the rhizosphere interact with roots in several process such as the decomposition of organic matter, the maintenance of soil structure and water relationships, as a consequence rhizosphere is a fundamental niche of the soil ecosystem [9]. Endophytic bacteria can be defined as those bacteria that colonize the internal tissue of the plants (endosphere) with no external sign of infection or negative effect on their host [10]; they can be classified as ‘obligate’ or ‘facultative’ endophytes in accordance with their life strategies. Obligate endophytes are strictly dependent on the host plant for their growth and survival and transmission to other plants could occur only by seeds or via vectors, while facultative endophytes could grow outside host plants [11]. Finally, a noteworthy endophytic example within Alphaproteobacteria, is the nitrogen-fixing symbiosis established with leguminous plants by rhizobia, which is coupled with the development of a new plant structure, the nodule, in the root or in the stem of the plant [12]. All these heterogenous phenotypes suggest that it could be difficult to find common genetic traits for Plant-associated bacteria.
An additional degree of complexity is given by the fact that single species or even single strains inhabit both soil and plant tissues and can show multiple types of plant association. For example, Azospirillum strains are known as model plant-growth promoting rhizosphere (PGPR) bacteria, but they have also been shown within plant tissue, as endophytes of cereals [13]; on the other hand, the specific alfalfa symbiont Sinorhizobium meliloti is also able to grow as rhizospheric of nontarget host plants and it behaves as endophytes with cereals like rice [14], besides free-living in bulk soil. Such observations led to doubt whether a genetic common background is present within all plant-associated Alphaproteobacteria. In fact, concerning symbiotic species, it is fairly accepted that the symbiotic lifestyle needs some specific genetic functions (e.g., nod genes), which are not present in nonsymbiotic nitrogen fixers [15]. However, stem-nodulating bradyrhizobia have shown that a nod-independent symbiosis can be established [15,16]. Two questions therefore arise: (i) Is the symbiotic lifestyle in α-rhizobia characterized by the presence of a common gene set? (ii) Do all plant-associated species (both symbiotic and nonsymbiotic) share some common genes conferring the ability to associate with plants? One way to begin to answer these questions is to apply a comparative genomics approach. Previous investigations on the comparison of α- and β-rhizobia have been performed [15,17] as well as the comparison of some Plant-associated endophytes in Gammaproteobacteria [18], however no systematic analyses have been attempted in Alphaproteobacteria.
Here we report a bioinformatic analysis aimed at the scanning of all the alphaproteobacterial sequenced genomes trying to sort out the possible exclusive or distinctive genes which enable some of the Alphaproteobacteria to be associated with plants, evaluating if plant-bacteria association needs a specific assortment of gene functions or if, as suggested by its phenotypic heterogeneity, it is rather unrelated to the presence of a dedicated set of genes.
2. Results and Discussion
2.1. Plant-Associated Bacteria Have Larger Genomes than Nonplant-Associated
First a dataset of the relevant Alphaproteobacteria (“alphas” for short) was constructed by downloading all the alphaproteobacterial genomes available in NCBI genome database. All animal obligate pathogens were excluded, since they show extensive genome reductions, linked with intracellular lifestyle [4,19], as well as the SAR11 clade due to the extensive gene loss described for this group [1]. A total of 92 genomes were then analyzed (Figure 1 and Supplementary Material S1), and divided into three groups: (i) solely free-living, (ii) plant-associated and (iii) symbiont (that is a sub-set of plant-associated) combining the information available on GOLD database [20,21], Bergey's manual of systematic bacteriology [22] and bibliographic search on Pubmed. Plant-associated bacteria include 27 genomes (2 pathogens, 7 associated and 18 symbionts), all but two (25/27) grouped within the order Rhizobiales (Figure 1), the only exceptions are the species Gluconacetobacter diazotrophicus and Azospirillum B510 which fall in the order Rhodospirillales; of course we cannot exclude that among the 65 nonplant-associated bacteria some could have also experienced the plant environment, even if those putative events have not been reported.
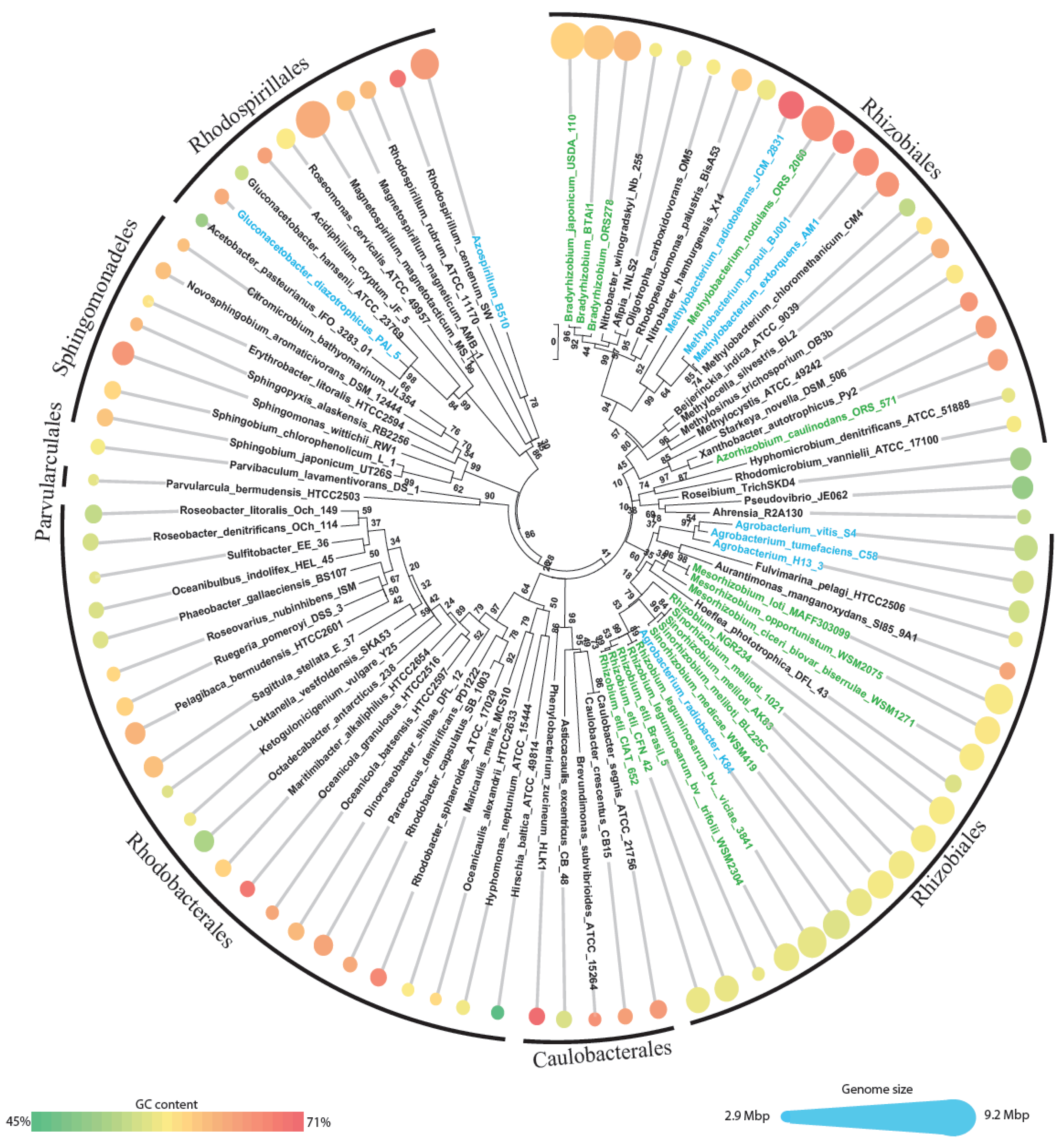
A quick look at these genomes shows a wide range of genome sizes, spanning from 2.9 Mbp (Parvularcula bermudensis HTCC2503) to 9.2 Mbp (the magnetotactic bacterium Magnetospirillum magnetotacticum MS1). The average genome size (±standard deviation) of the dataset is 5.04 ± 1.53 Mbp. Plant-associated bacteria have significant (P < 0.0001, one-way ANOVA) larger genomes (6.73 ± 1.26 Mbp) than nonplant-associated ones (4.34 ± 0.99 Mbp), as previously noticed [19]. The same trend was observed considering only the order Rhizobiales, which accounts for near half of the entire dataset (42 out of 92 genomes with average length of 5.83 ± 1.53 Mbp), with plant-associated Rhizobiales having genomes larger than those of nonplant-associated Rhizobiales (6.81 Mbp and 4.47 Mbp, respectively, P < 0.0001). Average GC content is 63.1 ± 4.4% and is similar for plant-associated and nonplant-associated genomes (63.03% and 62.71%, respectively, P < 0.8) and ranges from 45.2% (Hirschia baltica ATCC49814) to 71.1% (Phenylobacterium zucineum HLK1); within the Rhizobiales we observed the same trend: an average of 63.1% with 63.0% for plant-associated and 63.1% for nonplant-associated).
2.2. Are There Life-Style Specific Genes in Alphaproteobacteria?
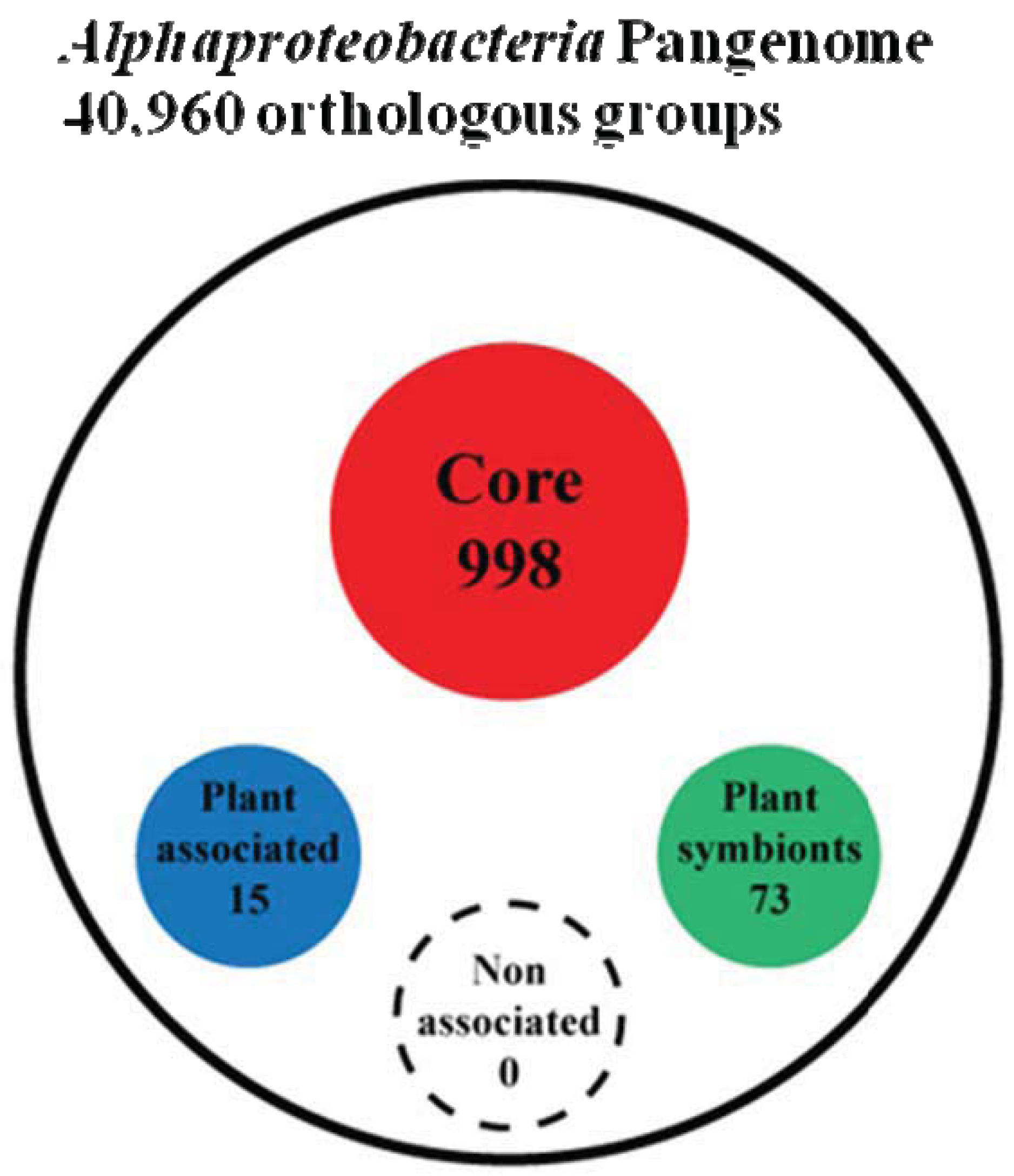
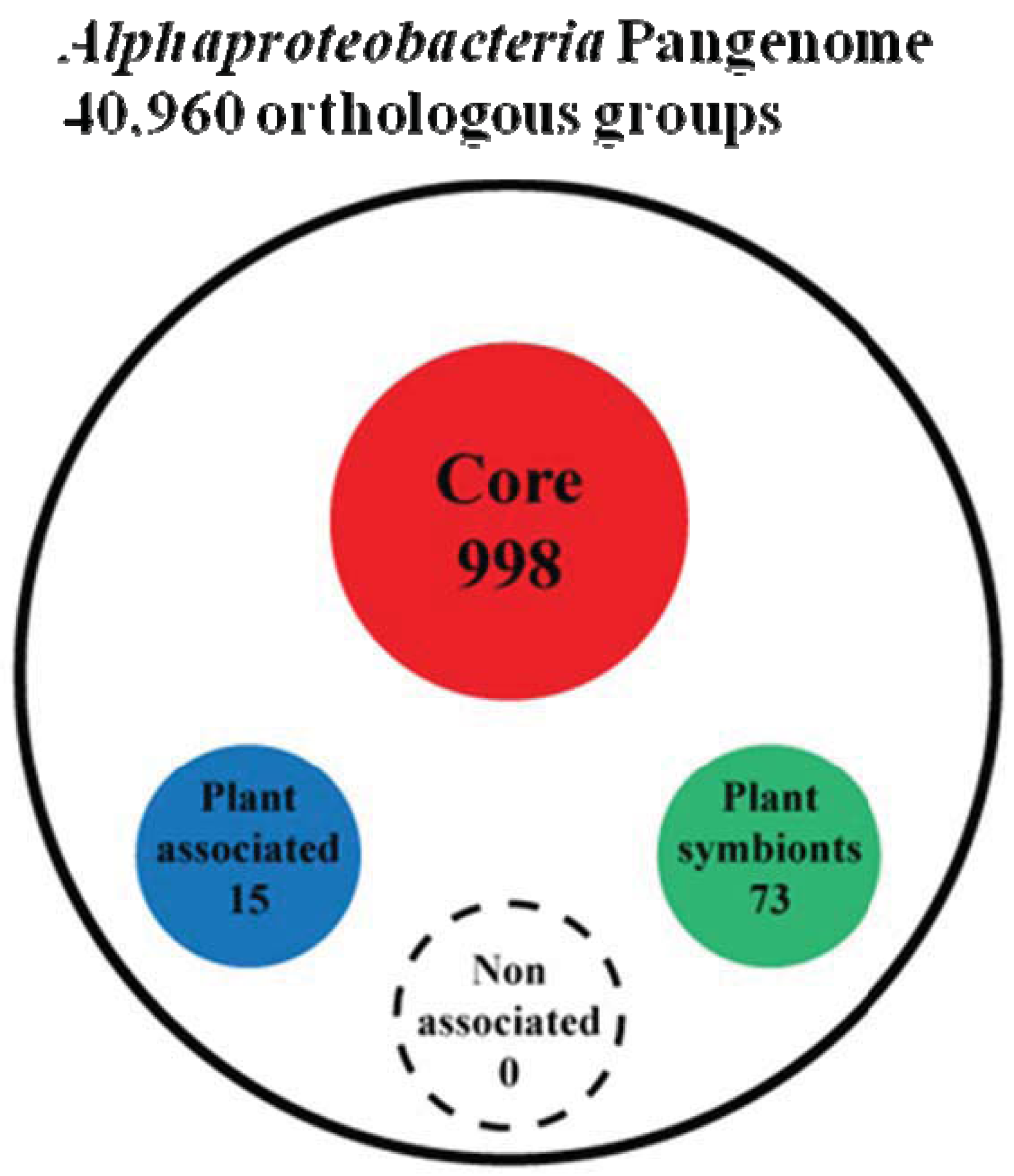
To answer this question we first peformed a genome clusterization of all protein coding genes present in the 92 genomes, obtaining 40,960 groups of orthologs (out of a total number of 434,411 proteins analyzed). Next, starting from these groups of orthologs, we proceeded trying to extract four groups named as: (i) Alpha Core (common to all the analyzed organisms), (ii) Plant-Associated (common to and exclusive of plant associated bacteria), (iii) Plant-Symbionts (common to and exclusive of plant symbionts), and (iv) NonPlant-Associated (common to and exclusive of nonplant-associated bacteria). Since genetic elements inside bacteria are prone to horizontal gene transfer and therefore genes that may be specific for a certain life-style may be found also in other bacterial species, by chance or because they might carry out a different function, we developed an orthologs-species clustering approach capable of taking into account this dynamical behavior. This “Fuzzy orthologs-species clusterization” analysis (see Materials and Methods) sorted out 998 orthologous groups for the Alpha Core subset, while life-style specific subset of NonPlant-Associated, Plant-Associated and Plant-Symbionts accounted for 88, 15 and 73 orthologous groups, respectively (Figure 2 and Supplementary Material S2). As expected, the Non-Plant subset of orthologous groups was found to be inconsistent, since a series of random subsets having the same number of species (see materials and methods) showed a similar number of orthologous groups. This finding is not surprising, since the NonPlant subset consists of species with no unique distinctive habitat, varying from soil to marine and freshwater organisms. On the contrary, for the other subsets, the number of orthologous groups generated from random species list was zero; the same result was observed when sampling only inside the order Rhizobiales (where most of the Plant-associated species are), retrieving 1.7 orthologous groups on average (data not shown). Notably, when we did not apply fuzziness to the orthologs-species clusterization, we did not find anyorthologous groups in any subset, but only in the Alpha Coreone. This discrepancy suggests that the plant association behavior may not be dependent on a strongly conserved defined set of genes strictly common to all the species of the subset. However, interestingly, by applying the “Fuzzy orthologs-species clusterization”, the larger Plant-Associated subset was found to contain only 15 orthologous groups, while in contrast the smaller Plant Symbionts subset contains 73 orthologous groups. This finding again confirms that the generic plant association behavior does not require a large repertoire of specific genes, while the symbiotic interaction in α-rhizobia is more dependent on the presence of specific gene traits [23].
2.3. Which Biological Functions Are Encoded by Life-Style Associated Orthologous Groups?
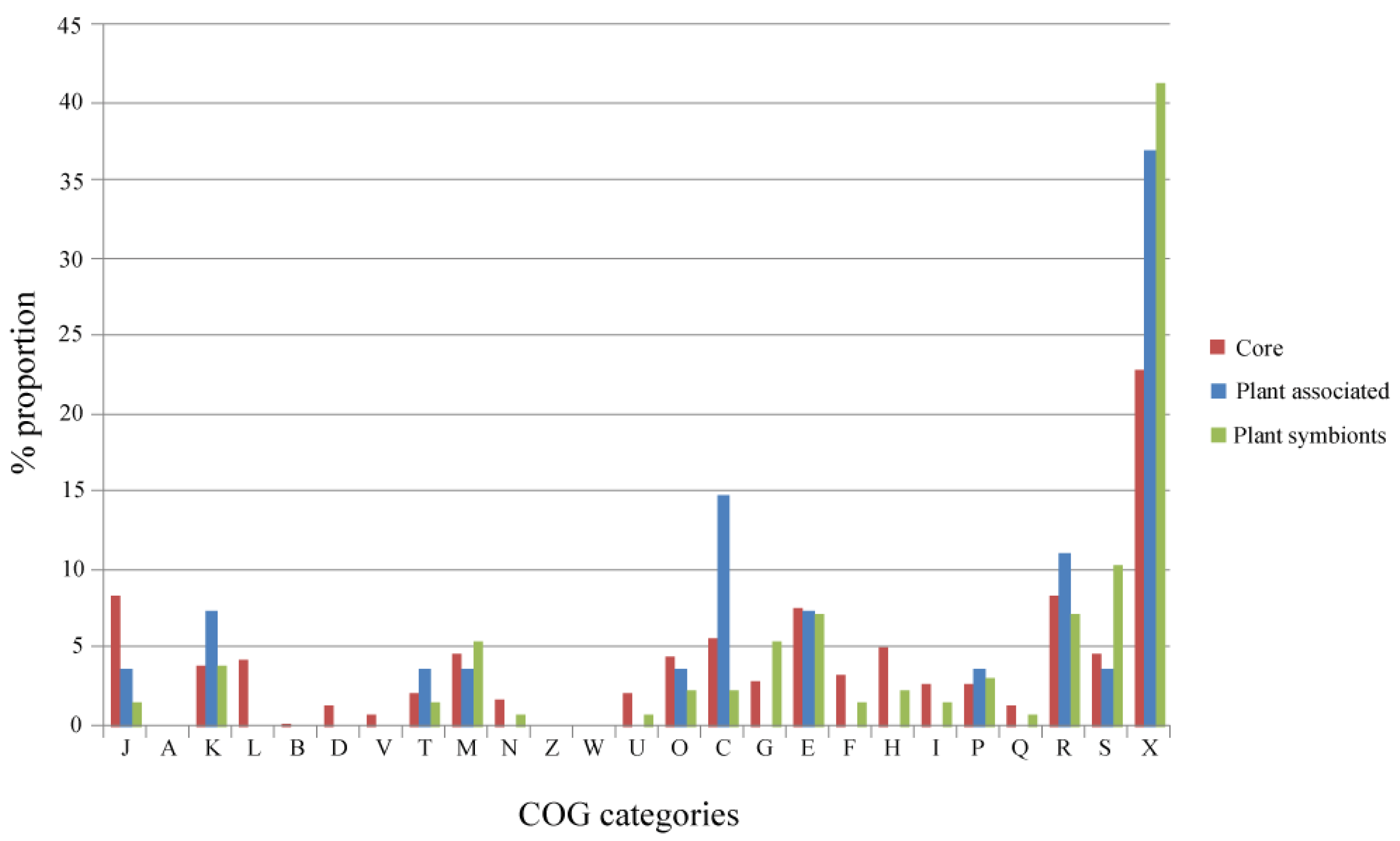
An overview of the distribution among the COGs (Cluster of Orthologous Groups) categories of the genes present in the Alpha Core, Plant-Associated and Plant Symbionts subsets is reported in Figure 3. Regarding the Plant-Associated and the Plant Symbionts subsets, COG categories related to basic cell functions (L, Replication, recombination and repair; B, Chromatin structure and dynamics D, Cell cycle control, cell division, chromosome partitioning; V, Defense mechanisms) are not represented, since they are mostly present in the Alpha Core subset, as expected; on the other hand, COG categories poorly or not characterized (S and X), are the most represented in both plant related subsets. The categories related to regulation of gene expression (K and J) and energy production and conversion (C) show a slightly higher proportion in the Plant-Associated subset. Regarding the Plant Symbionts subset, a slightly higher over-representation of the carbohydrate transport and metabolism category (G) was observed, suggesting the key role of carbohydrate metabolism for establishing nitrogen fixing symbiosis (i.e., for the formation of the so-called Nod factors as well as for bacteroid trophism [23]). Indeed, some plant symbionts, as for instance Sinorhizobium meliloti, contain large genomic regions or replicons mainly devoted to carbohydrate transport and metabolism [24-26]. However, the percentages of COG categories represented in the different subsets are not statistically different (Spearman Rank Correlation and Chi-square test with Monte Carlo simulation, data not shown).
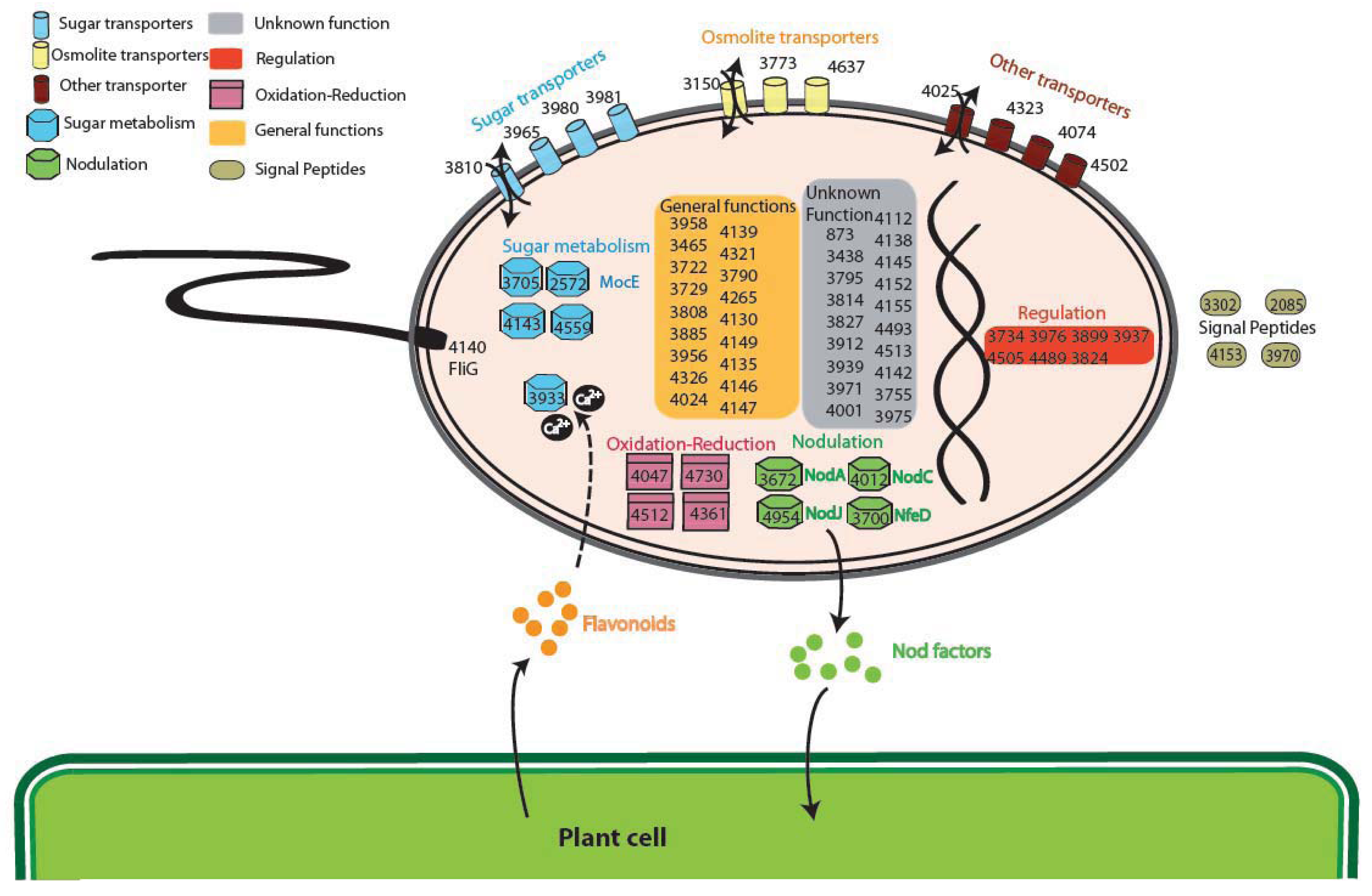
The analysis of the GO (Gene Ontology) categories in the Plant-Associated subset (Supplementary Material S3) shows that the most represented biological process is related to electron carrier activity (4 groups), while for the Plant Symbionts the functions encoded appear to be more heterogeneous (Figure 4). In particular, proteins involved in many process were found, ranging from symbiosis specific functions, like nodulation (4 orthologous groups: 3672, 4012 and 4954 encoding NodA, NodC, and the NodJ protein respectively, plus group 3700 also encoding NfeD another protein necessary for nodulation [27]) to trascriptional regulation and to more general biological functions (especially oxidation-reduction), with a slightly higher presence of transport-related functions (11 groups, with 3 of them probably involved in osmolarity control). As reported in Figure 4, sugar transport (4 groups out of 11 transporter) and metabolism (5 groups), are highly represented in the symbiont subset. Within this category, of particular interest is group 2572 encoding for MocE, a Rieske non-heme iron oxygenase essential in the catabolism of rhizopines (3-O-methylscyllo-inosamine, 3-O-MSI) a nodule-specific compounds that confer an intraspecies competitive nodulation advantage to strains able to utilize them [28]. Another intriguing group is 3933 which encodes for a protein belonging to the senescence marker protein 30 (SMP-30)/gluconolaconase superfamily which contains many mammalian sequences [29]; this protein was found to accommodate multiple functions [30], among which calcium regulation (as a regucalcin) [31]; that is particularly intriguing as the involvement of Ca2+ in the symbiotic signaling pathway activated by flavonoids was found in Rhizobium leguminosarum bv. viciae [32]. Another interesting function which could be related to the plant symbiosis is cell motility, encoded by orthologous group 4140 (protein FliG). No nitrogen-fixation related proteins were found as exclusive, due to the presence in alphas of free-living nitrogen-fixing species (Xanthobacter autotrophicus [22]) and to the presence of the fix signaling module also in Caulobacter [33].
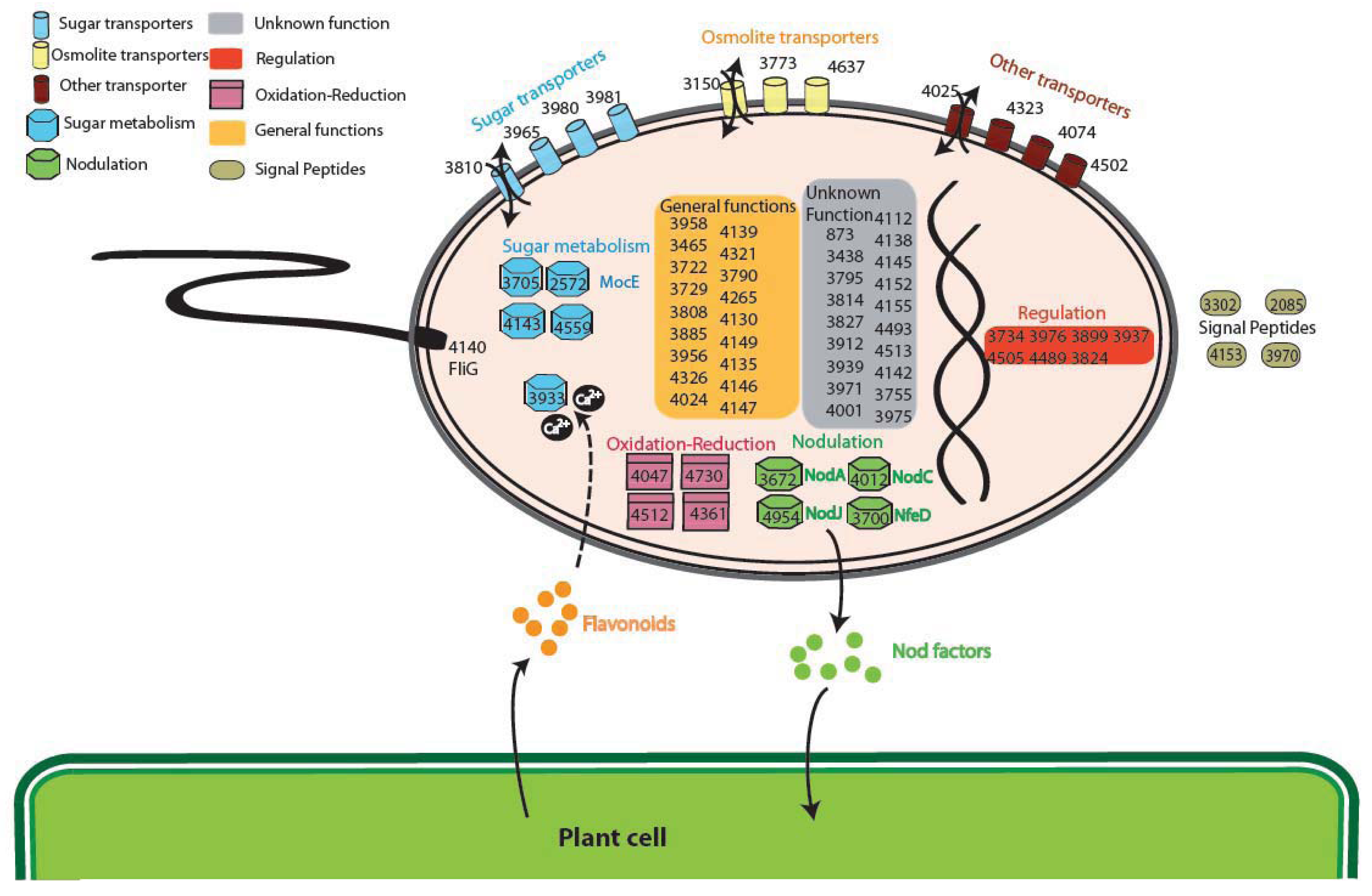
Interestingly, none of the functions previously putatively associated with endophytic life-style was detected, as type IV pili [34] or other metabolic or hormonal-related activities [35]. This is possibly due to the wide range of associations of our Plant-Associated subset, which includes also symbiotic and rhizospheric interaction; absence of type IV pili could be also linked to their involvement in many other processes in other species not engaged in plant interactions.
2.4. Taxonomic Range of the Life-Style Associated Genes Outside Alphaproteobacteria
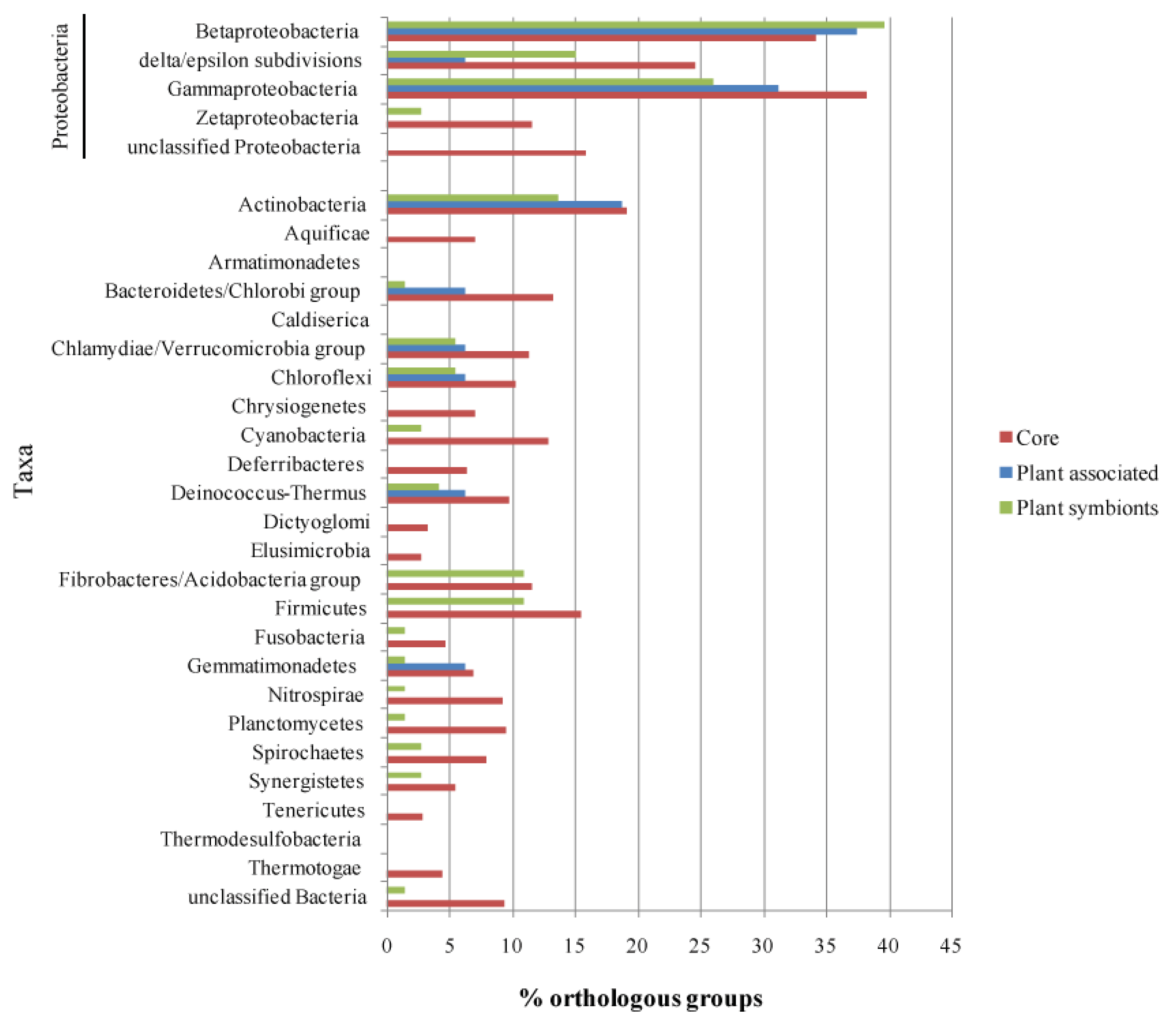
Once the list of life-style related orthologous groups was defined, we looked for their presence in the other branches of the bacterial taxonomic tree, in order to understand if such functions are specific for alphas or are widespread in other taxas, thus giving an insight into the evolutionary pathways of those functions. Each life-style associated orthologous group, was then used as a query on the GenBank database to find homologous sequences in all bacterial taxa; results of the analysis are reported in Figure 5 and Supplementary Material S4. Figure 5 offers an overview of the proportion of the orthologous groups occurring in each subset which have hits in the different bacterial classes. As expected, most of the hits were scored within Proteobacteria, in particular in the classes Beta and Gamma, possibly reflecting both a higher phylogenetic proximity and a general bias of the database which is abundant in sequences from members of such classes (Alphaproteobacteria were excluded from the analysis).
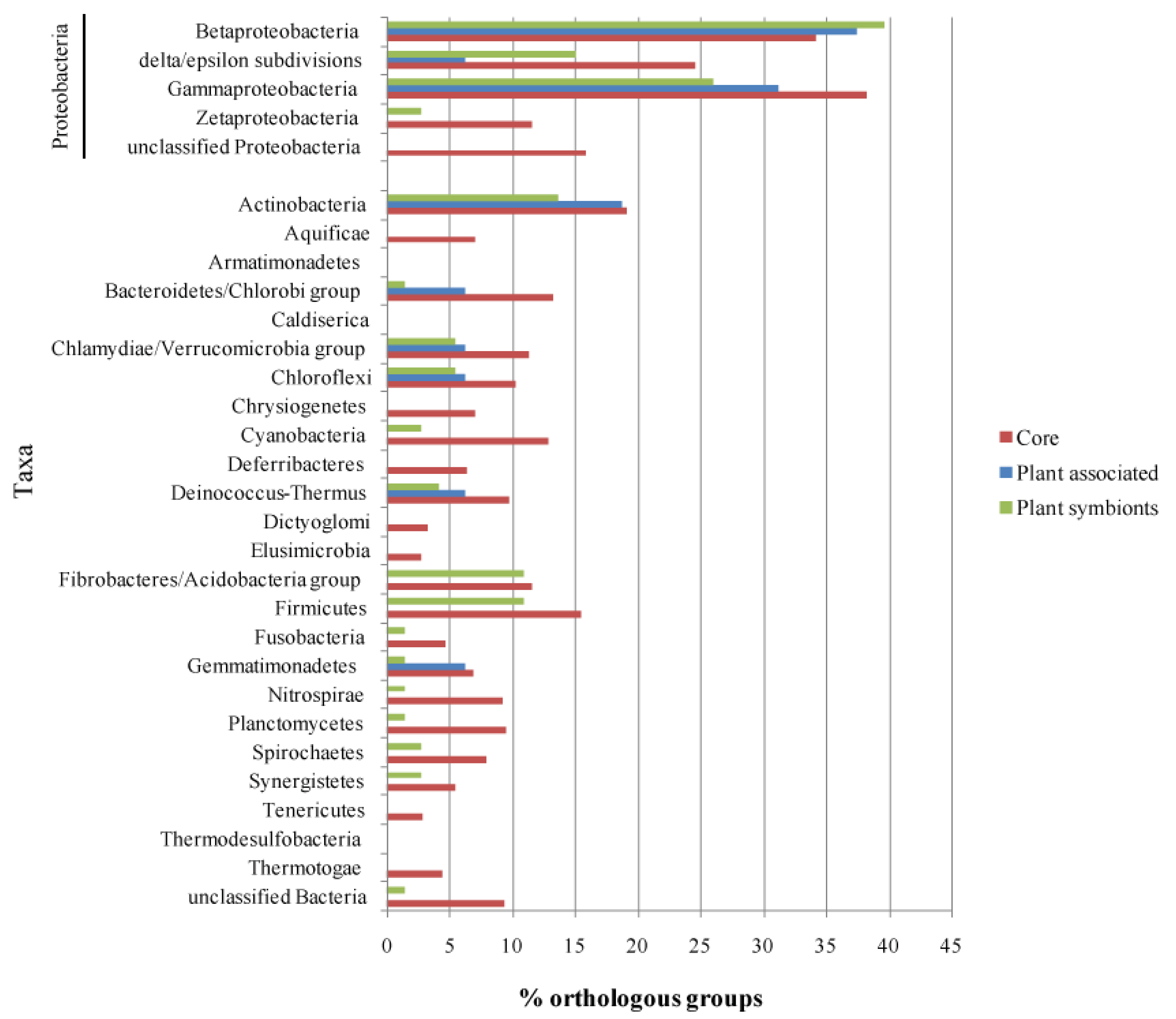
A high proportion of Alpha Core genes have at least one hit in almost all the taxa probed by the analysis, with an average of 10.5% of the Alpha Core subset having an hit in the selected taxa, while on average, only 4.2% and 5.0% of the Plant Associated and Plant Symbionts orthologous groups have at least a hit in each taxa; this observation suggests that plant association genes tend to be conserved only inside Alphaproteobacteria and to a lesser extent inside Beta- and Gamma-Proteobacteria (36% average), while just few genes have an homolog in phylogenetically distant species, where they might not be related to a plant association behavior.
To further elucidate this point, all the taxa found by this approach were investigated for their plant-association life-style, according to the GOLD database annotation; 33.3% of the Plant-Associated orthologous groups have at least one hit in species associated with plants, followed by the Plant Symbionts (30.1%) and the Alpha Core (24.6%). The two plant related subsets have plant association hits only inside the Protebacteria class (Beta- and Gamma-Proteobacteria), while the Alpha Core hits are distributed in a broader range, including Actinobacteria, Cyanobacteria and Firmicutes (Supplementary Material S5); the results of this analysis then imply that the plant association related genes are rather specific of the Proteobacteria class, while the housekeeping genes exhibit an higher degree of sequence conservation across all the bacterial phylogenetic tree.
To shed some light on the possibility that Plant-associated specific genes are involved in plant association at a broader taxonomic level, the protein coding genes found as exclusively present in Plant-Associated alphas were checked for their presence in the genomes of four known and fully-sequenced plant-associated Proteobacteria, in particular in the class of Beta-Proteobacteria the strains Cuprividus taiwanensis [17] and Azoarcus sp. BH72 [34] and in the class of Gamma-Proteobacteria the species Enterobacter sp. 638 [35] and Klebsiella pneumoniae 342 [36]. Results of the comparison are shown in Table 1. Interestingly, two out of 15 orthologous groups are present in all the four strains selected, namely orthologous group 2149 (Transcriptional regulator) and 2774 (endoribonuclease l-psp), suggesting that regulation (either by transcriptional regulation and RNA stability) may play pivotal roles in establishing the association with plant. Moreover 5 other genes were found to be present in at least 2 of the 4 species investigated. A previous work dealing with the description of the genome sequence of the β-rhizobium Cuprividus taiwanensis [17], found no gene both common and specific to all rhizobia, suggesting that symbiotic association with plants evolved with multiple strategies, even though genes preferentially associated (on a statistical basis) with plant symbiosis were detected. However, our findings suggest that these two genes, putatively needed in Alphaproteobacteria to establish a successful interaction with plants, are also present in the Beta and Gamma-Proteobacteria model organisms for plant association. Endoribonuclease L-PSP is involved in single-stranded mRNA cleavage in Leishmania infantum, a parasite that in its life cycle alternates two stages, and is hypothesized to be involved in specific post-transcriptional regulation of gene expression [37]; characterization of mutants for those orthologs is however necessary to fully elucidate their role in plant association in such a broad taxonomic background.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orthologous group | Function | Azoarcus BH72 | Cupriavidus taiwanensis | Enterobacter 638 | Klebsiella pneumoniae |
|---|---|---|---|---|---|
| 2149 | Transcriptional regulator | YP_932298 | YP_002005188 YP_002007781 | YP_001177142 | YP_002237096 YP_002237759 |
| YP_001177763 | |||||
| YP_001177947 | |||||
| 2248 | Transcriptional regulator | YP_001795747 | YP_001177733 | YP_002239091 YP_002240771 | |
| YP_001177763 | |||||
| YP_001177947 | |||||
| 2654 | Adenylate cyclase | YP_932132 | YP_002008552 | ||
| 2734 | ABC transporter | YP_001175837 | YP_002237478 | ||
| YP_001177423 | YP_002239806 | ||||
| 2737 | Unknown | YP_002005759 | |||
| 2774 | Endoribonuclease l-psp | YP_931980 | YP_002008711 | YP_001178228 | YP_002237662 |
| YP_002008874 | YP_002238064 | ||||
| 2791 | Phosphoesterase | ||||
| 2853 | Electron transfer flavoprotein, beta subunit | YP_001796225 | |||
| 2898 | Unknown | YP_002236173 | |||
| 2908 | Electron transfer flavoprotein, alpha subunit | YP_001796224 | |||
| 2912 | Methyltransferase | YP_935409 | YP_001176000 | YP_002237443 | |
| YP_001177854 | YP_002239590 | ||||
| 2927 | Aminoacid aldolase or racemase | YP_002007445 | |||
| 2981 | Mg2+ and Co2+ transporters | YP_002006900 | YP_001176870 | YP_002238775 | |
| YP_002238859 | |||||
| 3082 | Ferredoxin-like protein | YP_001796222 | |||
| 3137 | Unknown | YP_002236905 |
3. Experimental Section
3.1. Phylogenetic Tree
To construct our reference phylogenetic tree, all 16S rRNA gene sequences were aligned using MUSCLE [38], alignment was manually checked. The alignment was used with the software Mega 5.05 [39] to generate a phylogenetic tree. A Model test (Supplementary Material S6) was performed before running the Maximum Likelihood algorithm, with 1,000 bootstrap replicates and the Tamura-Nei model of evolution.
3.2. Genomes Clusterization
The 92 genomes were clustered together using the approach proposed by Kim and collaborators [40], using the PanGenomer software (available upon request); a total number of 8,464 pairwise InParanoid analyses with no thresholds were generated and the results were merged in a single file as an input for MCL [41], using an inflation factor of 5.0, a pruning threshold of 30,000 and a selection number of 5,000. To test the clusterization the presence of 5 well known orthologous genes, involved in cell cycle regulation and DNA replication (ctrA, dnaA, rpoE, gyrB, dnaQ), was used as a positive control (referred to as group 4, 315, 158, 524 and 26 respectively).
3.3. Fuzzy Orthologs-Species Clusterization
The obtained orthologous groups were mapped to the four species subsets (Figure 1) looking at the species from which each protein belonging to that group came from, using a so-called “Fuzzy” approach: an orthologous group was regarded as specific for one of the four subsets when its species list was comprised between 80% and 110% of the subset list. The biological value of each subset was tested generating 10 random organism lists with the same length of the subset and looking at how many orthologous groups were then retrieved.
3.4. Orthologous Groups Annotation
The orthologous groups belonging to the four subsets were annotated, using ten proteins from each group (selected randomly) to speed-up the analysis. Each protein was mapped to the COG database [42] using rpsblast 2.2.25+ and an e-value threshold of 1e-10; the domain content and the GO [43] annotation were obtained using Iprscan 4.8 [44] with the InterPro database release 33.0.
3.5. Taxonomic Analysis
The protein sequence similarity across the bacterial kingdom of each orthologous group was inspected using TaxonomyBlaster (available upon request); the same proteins used for the annotation were analyzed using a series of taxonomically-restricted portions of the NCBI nr database (downloaded on 1 July 2011): all the taxonomic classes (excluding the “environmental samples”) inside the Bacteria kingdom were iteratively used; the Proteobacteria class was further divided into the distinct classes, excluding the Alphaproteobacteria and the Proteobacterial “environmental samples”. BLAST was run using the BLOSUM45 matrix, the soft masking option, a fixed database size of 500,000,000 and the Smith-Waterman local optimal alignments option. Those hits showing an e-value below 1e-10, a query coverage above 66% and an homology index above 0.33, were retained. The obtained species were marked as Plant-Associated looking at the available information in the GOLD database [20].
4. Conclusions
As vector-borne intracellular Alphaproteobacteria have evolved towards smaller genome [4], the trend in plant-associated Alphaproteobacteria seems to be headed in the opposite direction towards an increase of genome size, probably due to the different habitats colonized including soil and plant tissues [19]. Here, for the first time to our best knowledge, we report an investigation of the genomic features, as different genes, which could be related, on a genomic basis, to the symbiotic and nonsymbiotic plant-association in Alphaproteobacteria. This analysis was carried out with a novel orthologs-species clusterization approach that was able to take into account the natural horizontal gene transfer dynamics, allowing us to also identify those genes that are (partially) shared with other species or that are not present in all the life-style related species.
Interestingly, a relatively large set of genes shared by an exclusive symbiotic alphaproteobacterial species was found, suggesting that a common genomic base is indeed present; tough multiple “recipes” for plant association are present [15]. This set includes functions previously known to be linked to the symbiotic interaction, but also others, which were previously unsuspected. In particular, genes necessary for plant-bacteria communication were retrieved as well as an enrichment in protein coding genes involved in sugar transport and metabolism. Most of these orthologs could likely be associated with metabolic exchanges and communication between plant cells and bacteroids; interestingly an ortholog of SMP30/gluconolaconase family (regucalcin) was also found suggesting a link between nodulation and calcium spiking in the rhizobial cell, in agreement with recent experimental findings[32].
Contrary to what could be expected by this highly heterogeneous phenotype, concerning all plant-associated species obtained, results showed a numerically low, but computationally consistent, set of genes which could account for their ability to associate with plants as both symbiont and nonsymbiont (i.e., rhizospheric, pathogen, endophyte). Interestingly, several functions were related to the regulation of gene expression, which makes sense considering the pivotal role of the perception of environmental signals for association with plants. This set of putatively plant-associated genes showed two apparently contradictory properties: a relatively high degree of conservation of these few genes inside Proteobacteria (when compared to the other branches of the bacterial tree) but also a certain degree of conservation across phylogenetically distant plant-associated species. This evidence could mean that even though there are no common genetic traits that distinguish this ecologically heterogeneous group of species, single genetic “pieces” may be shared, in a vast phylogenetic range, with other plant-associated species. We can then speculate that association with plants is therefore addressed using several pathways and mechanisms (which mirror the different types of association), even within a relatively narrow taxonomic range.
In conclusion, while symbiotic lifestyle needs a defined gene set, nonsymbiotic plant-bacteria association can occur through multiple strategies with functions specific for the single interaction.
Acknowledgments
This work has been partially supported by the Italian Ministry of Research (PRIN 2008 research grant contract No. TCKNJL, “Il pangenoma di Sinorhizobium meliloti: L'uso della genomica per il miglioramento agronomico dell'erba medica”) and intramural funding of the University of Florence to M.B. and A.M. F.P. is supported by a research fellowship of Ente Cassa di Risparmio di Firenze charity trust. M.G. is supported by a PhD fellowship by the University of Florence.
Supplementary Material
S1. Alphaproteobacteria dataset.
S2. Clusterization of orthologous groups in the four subset analyzed (Alphas, NonPlant-Associated, Plant-Associated, Symbionts).
S3. COG names list, Gene Ontology and COGs categories for Plant-associated and Symbionts.
S4. Taxonomic sharing of life-style associated genes.
S5. Taxonomic sharing of life-style associated genes in other plant-associated species.
S6. Model test.
References
- Ettema, T.J.; Andersson, S.G. The α-proteobacteria: The Darwin finches of the bacterial world. Biol. Lett. 2009, 5, 429–432. [Google Scholar]
- van Rhijn, P.; Vanderleyden, J. The Rhizobium-plant symbiosis. Microbiol. Rev. 1995, 59, 124–142. [Google Scholar]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar]
- Sallstrom, B.; Andersson, S.G. Genome reduction in the α-Proteobacteria. Curr. Opin. Microbiol. 2005, 8, 579–585. [Google Scholar]
- Harrison, P.W.; Lower, R.P.; Kim, N.K.; Young, J.P. Introducing the bacterial ‘chromid’: Not a chromosome, not a plasmid. Trends Microbiol. 2010, 18, 141–148. [Google Scholar]
- Moreno, E. Genome evolution within the alpha-Proteobacteria: Why do some bacteria not possess plasmids and others exhibit more than one different chromosome? FEMS Microbiol. Rev. 1998, 22, 255–275. [Google Scholar]
- Danhorn, T.; Fuqua, C. Biofilm formation by plant-associated bacteria. Annu. Rev. Microbiol. 2007, 61, 401–422. [Google Scholar]
- Whipps, J.M.; Hand, P.; Pink, D.; Bending, G.D. Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 2008, 105, 1744–1755. [Google Scholar]
- Singh, B.K.; Millard, P.; Whiteley, A.S.; Murrell, J.C. Unravelling rhizosphere-microbial interactions: Opportunities and limitations. Trends Microbiol. 2004, 12, 386–393. [Google Scholar]
- Ryan, R.P.; Germaine, K.; Franks, A.; Ryan, D.J.; Dowling, D.N. Bacterial endophytes: Recent developments and applications. FEMS Microbiol. Lett. 2008, 278, 1–9. [Google Scholar]
- Rajkumar, M.; Ae, N.; Freitas, H. Endophytic bacteria and their potential to enhance heavy metal phytoextraction. Chemosphere 2009, 77, 153–160. [Google Scholar]
- Sadowsky, M.; Graham, P. Root and Stem Nodule Bacteria of Legumes. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 2, pp. 818–841. [Google Scholar]
- Bashan, Y.; Holguin, G.; de-Bashan, L.E. Azospirillum-plant relationships: Physiological, molecular, agricultural, and environmental advances (1997-2003). Can. J. Microbiol. 2004, 50, 521–577. [Google Scholar]
- Chi, F.; Shen, S.H.; Cheng, H.P.; Jing, Y.X.; Yanni, Y.G.; Dazzo, F.B. Ascending migration of endophytic rhizobia, from roots to leaves, inside rice plants and assessment of benefits to rice growth physiology. Appl. Environ. Microbiol. 2005, 71, 7271–7278. [Google Scholar]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar]
- Giraud, E.; Moulin, L.; Vallenet, D.; Barbe, V.; Cytryn, E.; Avarre, J.C.; Jaubert, M.; Simon, D.; Cartieaux, F.; Prin, Y.; et al. Legumes symbioses: Absence of nod genes in photosynthetic bradyrhizobia. Science 2007, 316, 1307–1312. [Google Scholar]
- Amadou, C.; Pascal, G.; Mangenot, S.; Glew, M.; Bontemps, C.; Capela, D.; Carrere, S.; Cruveiller, S.; Dossat, C.; Lajus, A.; et al. Genome sequence of the β-rhizobium Cupriavidus taiwanensis and comparative genomics of rhizobia. Genome Res. 2008, 18, 1472–1483. [Google Scholar]
- Taghavi, S.; Garafola, C.; Monchy, S.; Newman, L.; Hoffman, A.; Weyens, N.; Barac, T.; Vangronsveld, J.; van der Lelie, D. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 2009, 75, 748–757. [Google Scholar]
- Batut, J.; Andersson, S.G.; O'Callaghan, D. The evolution of chronic infection strategies in the alpha-Proteobacteria. Nat. Rev. Microbiol. 2004, 2, 933–945. [Google Scholar]
- Bernal, A.; Ear, U.; Kyrpides, N. Genomes OnLine Database (GOLD): A monitor of genome projects world-wide. Nucleic Acids Res. 2001, 29, 126–127. [Google Scholar]
- GOLD database. Available online: http://www.genomesonline.org/cgi-bin/index.cgi/ (accessed on 8 November 2011).
- Krieg, N.R.; Holt, J.G.; Bergey, D.H. Bergey's Manual of Systematic Bacteriology; Williams & Wilkins: Baltimore, MD, USA, 1984; Volume 2, pp. 1–574. [Google Scholar]
- Gibson, K.E.; Kobayashi, H.; Walker, G.C. Molecular determinants of a symbiotic chronic infection. Annu. Rev. Genet. 2008, 42, 413–441. [Google Scholar]
- Galardini, M.; Mengoni, A.; Brilli, M.; Pini, F.; Fioravanti, A.; Lucas, S.; Lapidus, A.; Cheng, J.F.; Goodwin, L.; Pitluck, S.; et al. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genomics 2011, 12, 235. [Google Scholar]
- Finan, T.M.; Weidner, S.; Wong, K.; Buhrmester, J.; Chain, P.; Vorholter, F.J.; Hernandez-Lucas, I.; Becker, A.; Cowie, A.; Gouzy, J.; et al. The complete sequence of the 1,683-kb pSymB megaplasmid from the N2-fixing endosymbiont Sinorhizobium meliloti. Proc. Natl. Acad. Sci. USA 2001, 98, 9889–9894. [Google Scholar]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar]
- Garcia-Rodriguez, F.M.; Toro, N. Sinorhizobium meliloti nfe (nodulation formation efficiency) genes exhibit temporal and spatial expression patterns similar to those of genes involved in symbiotic nitrogen fixation. Mol. Plant Microbe Interact. 2000, 13, 583–591. [Google Scholar]
- Bahar, M.; de Majnik, J.; Wexler, M.; Fry, J.; Poole, P.S.; Murphy, P.J. A model for the catabolism of rhizopine in Rhizobium leguminosarum involves a ferredoxin oxygenase complex and the inositol degradative pathway. Mol. Plant Microbe Interact. 1998, 11, 1057–1068. [Google Scholar]
- Chen, C.N.; Chin, K.H.; Wang, A.H.; Chou, S.H. The first crystal structure of gluconolactonase important in the glucose secondary metabolic pathways. J. Mol. Biol. 2008, 384, 604–614. [Google Scholar]
- Fujita, T. Senescence marker protein-30 (SMP30): Structure and biological function. Biochem. Biophys. Res. Commun. 1999, 254, 1–4. [Google Scholar]
- Yamaguchi, M. Role of regucalcin in calcium signaling. Life Sci. 2000, 66, 1769–1780. [Google Scholar]
- Moscatiello, R.; Squartini, A.; Mariani, P.; Navazio, L. Flavonoid-induced calcium signalling in Rhizobium leguminosarum bv. viciae. New Phytol. 2010, 188, 814–823. [Google Scholar]
- Crosson, S.; McGrath, P.T.; Stephens, C.; McAdams, H.H.; Shapiro, L. Conserved modular design of an oxygen sensory/signaling network with species-specific output. Proc. Natl. Acad. Sci. USA 2005, 102, 8018–8023. [Google Scholar]
- Krause, A.; Ramakumar, A.; Bartels, D.; Battistoni, F.; Bekel, T.; Boch, J.; Bohm, M.; Friedrich, F.; Hurek, T.; Krause, L.; et al. Complete genome of the mutualistic, N2-fixing grass endophyte Azoarcus sp. strain BH72. Nat. Biotechnol. 2006, 24, 1385–1391. [Google Scholar]
- Taghavi, S.; van der Lelie, D.; Hoffman, A.; Zhang, Y.-B.; Walla, M.D.; Vangronsveld, J.; Newman, L.; Monchy, S. Genome sequence of the plant growth promoting endophytic bacterium Enterobacter sp. 638. PLoS Genet. 2010, 6, e1000943. [Google Scholar]
- Fouts, D.E.; Tyler, H.L.; DeBoy, R.T.; Daugherty, S.; Ren, Q.; Badger, J.H.; Durkin, A.S.; Huot, H.; Shrivastava, S.; Kothari, S.; et al. Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet. 2008, 4, e1000141. [Google Scholar]
- Alcolea, P.J.; Alonso, A.; Gomez, M.J.; Moreno, I.; Dominguez, M.; Parro, V.; Larraga, V. Transcriptomics throughout the life cycle of Leishmania infantum: High down-regulation rate in the amastigote stage. Int. J. Parasitol. 2010, 40, 1497–1516. [Google Scholar]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar]
- Kim, S.; Jung, K.; Ryu, K. Automatic Orthologous-Protein-Clustering from Multiple Complete-Genomes by the Best Reciprocal BLAST Hits. In Data Mining for Biomedical Applications; Springer: Berlin/Heidelberg, Germany, 2006; Volume 3916, pp. 60–70. [Google Scholar]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar]
- Tatusov, R.; Fedorova, N.; Jackson, J.; Jacobs, A.; Kiryutin, B.; Koonin, E.; Krylov, D.; Mazumder, R.; Mekhedov, S.; Nikolskaya, A.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinformatics 2003, 4, 41. [Google Scholar]
- The Gene Ontology Consortium; Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pini, F.; Galardini, M.; Bazzicalupo, M.; Mengoni, A. Plant-Bacteria Association and Symbiosis: Are There Common Genomic Traits in Alphaproteobacteria? Genes 2011, 2, 1017-1032. https://doi.org/10.3390/genes2041017
Pini F, Galardini M, Bazzicalupo M, Mengoni A. Plant-Bacteria Association and Symbiosis: Are There Common Genomic Traits in Alphaproteobacteria? Genes. 2011; 2(4):1017-1032. https://doi.org/10.3390/genes2041017
Chicago/Turabian StylePini, Francesco, Marco Galardini, Marco Bazzicalupo, and Alessio Mengoni. 2011. "Plant-Bacteria Association and Symbiosis: Are There Common Genomic Traits in Alphaproteobacteria?" Genes 2, no. 4: 1017-1032. https://doi.org/10.3390/genes2041017
APA StylePini, F., Galardini, M., Bazzicalupo, M., & Mengoni, A. (2011). Plant-Bacteria Association and Symbiosis: Are There Common Genomic Traits in Alphaproteobacteria? Genes, 2(4), 1017-1032. https://doi.org/10.3390/genes2041017