Protein Folding Absent Selection

Abstract
:1. Introduction
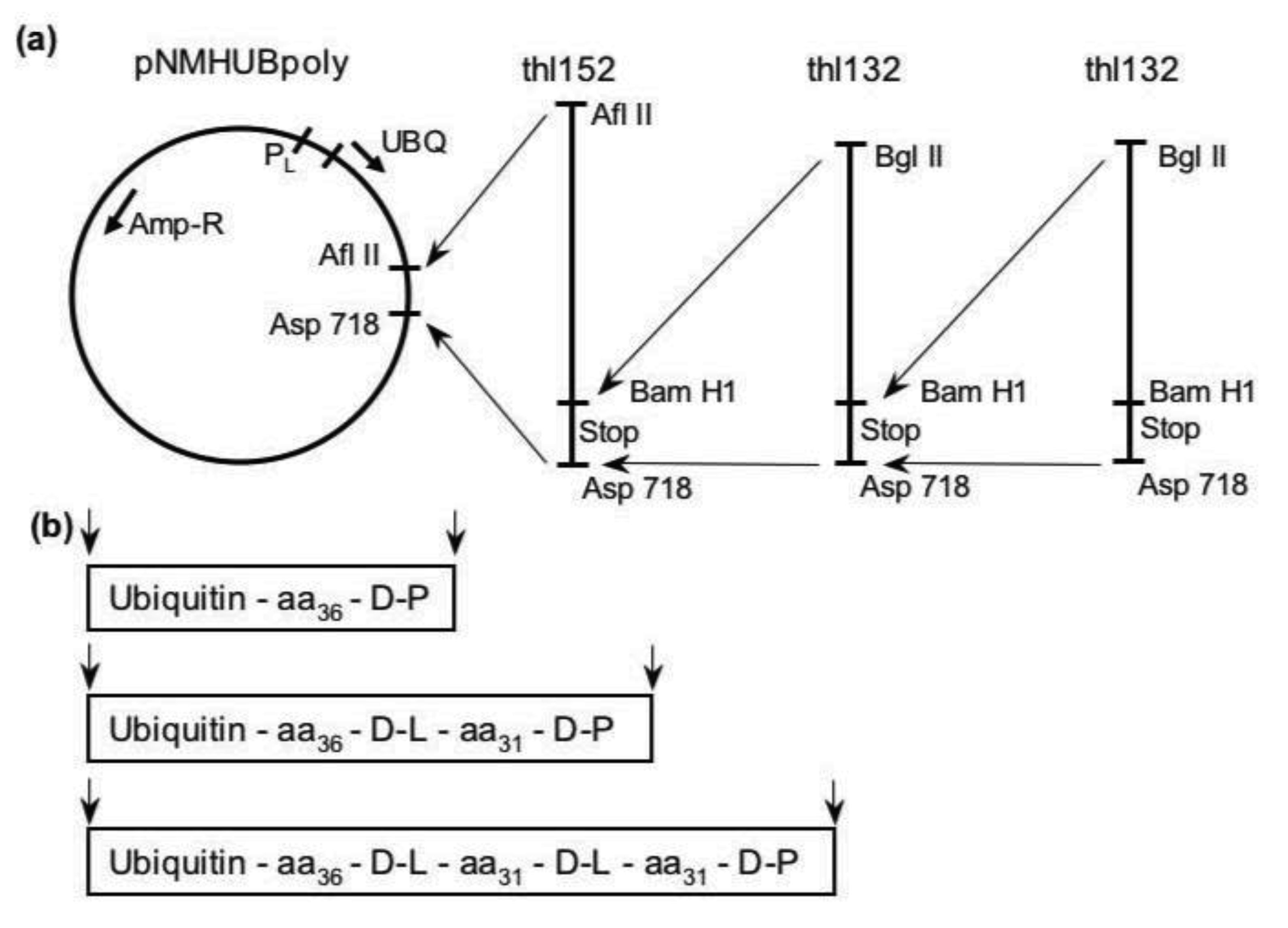
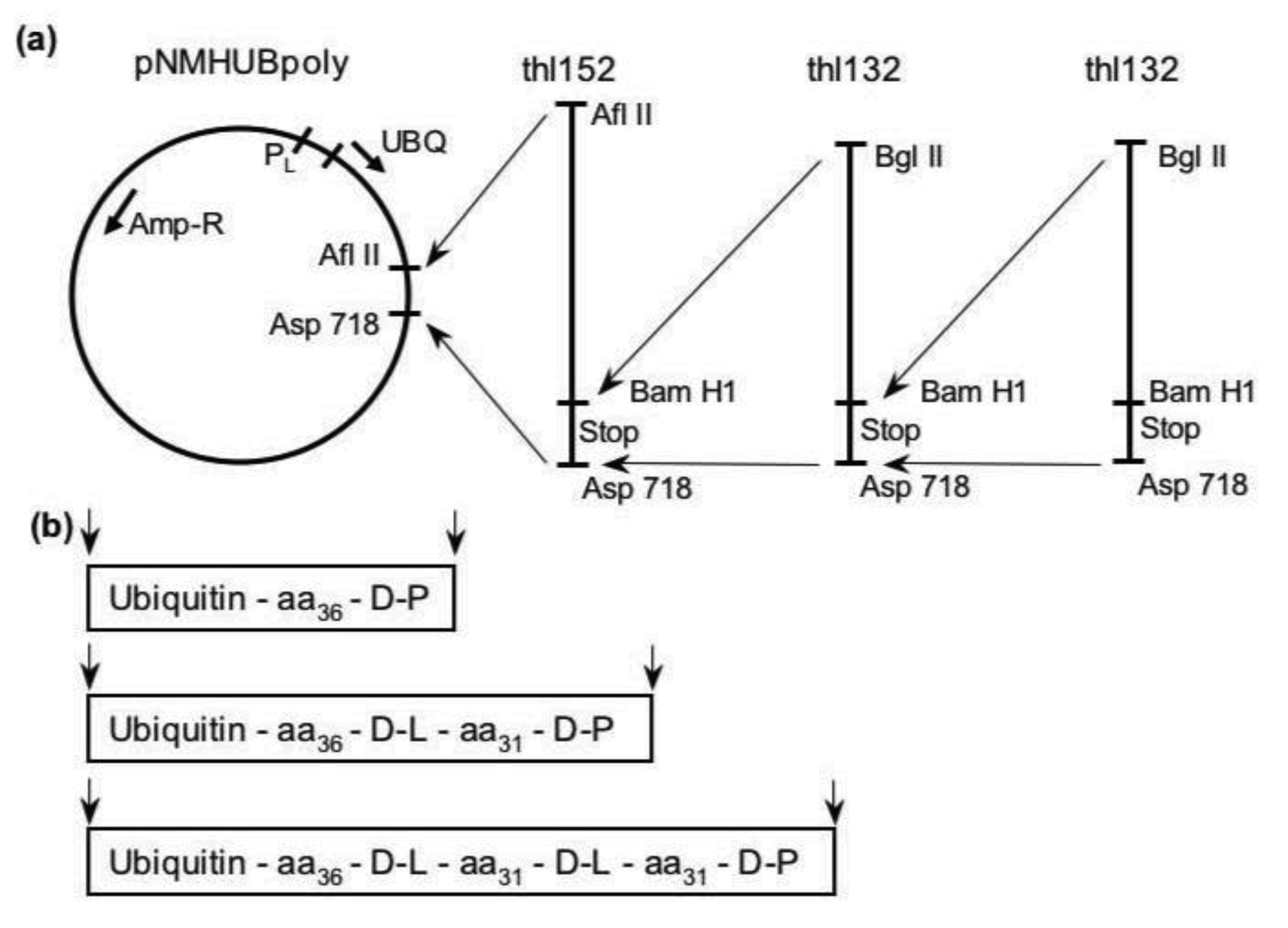
2. Results and Discussion
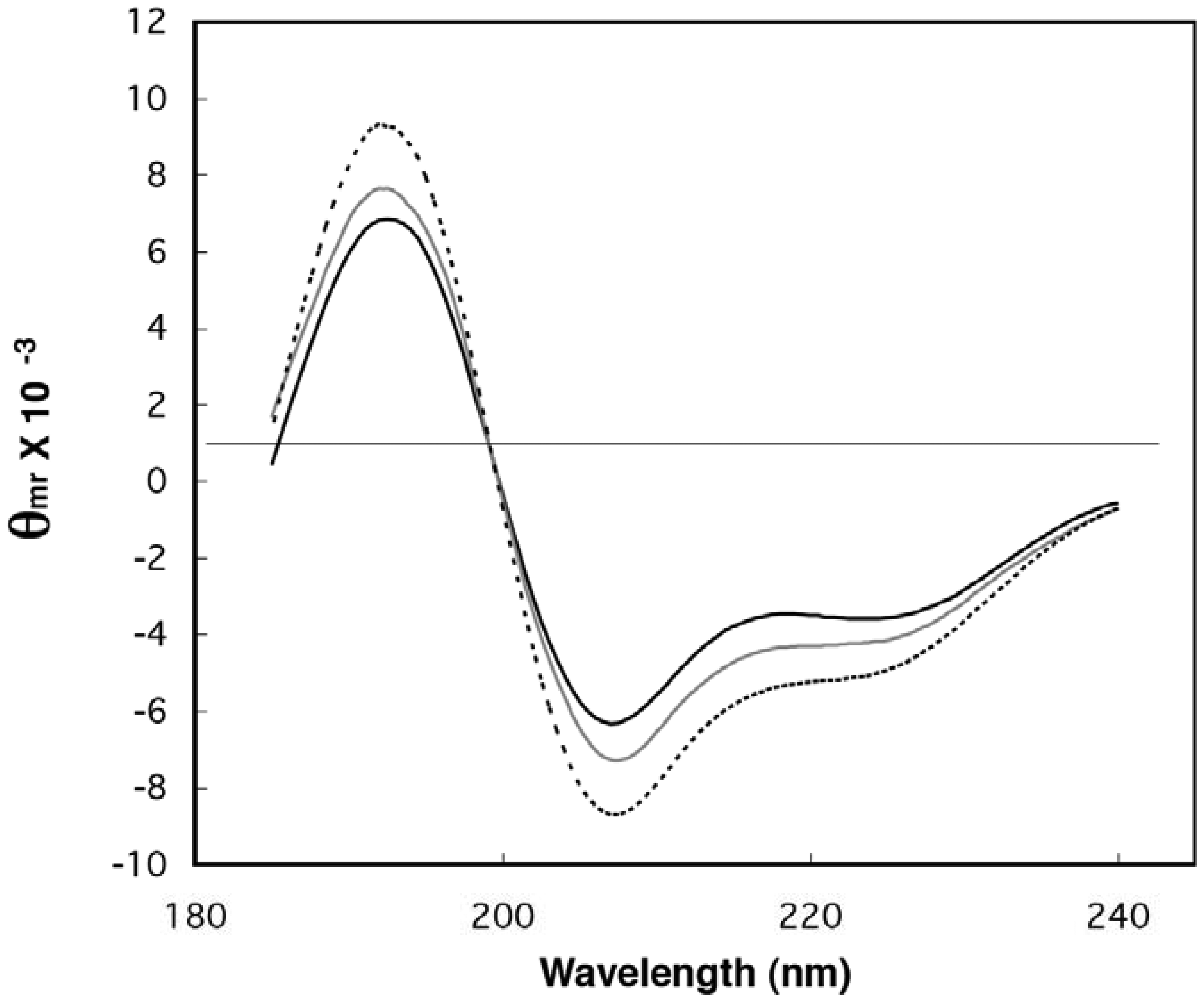
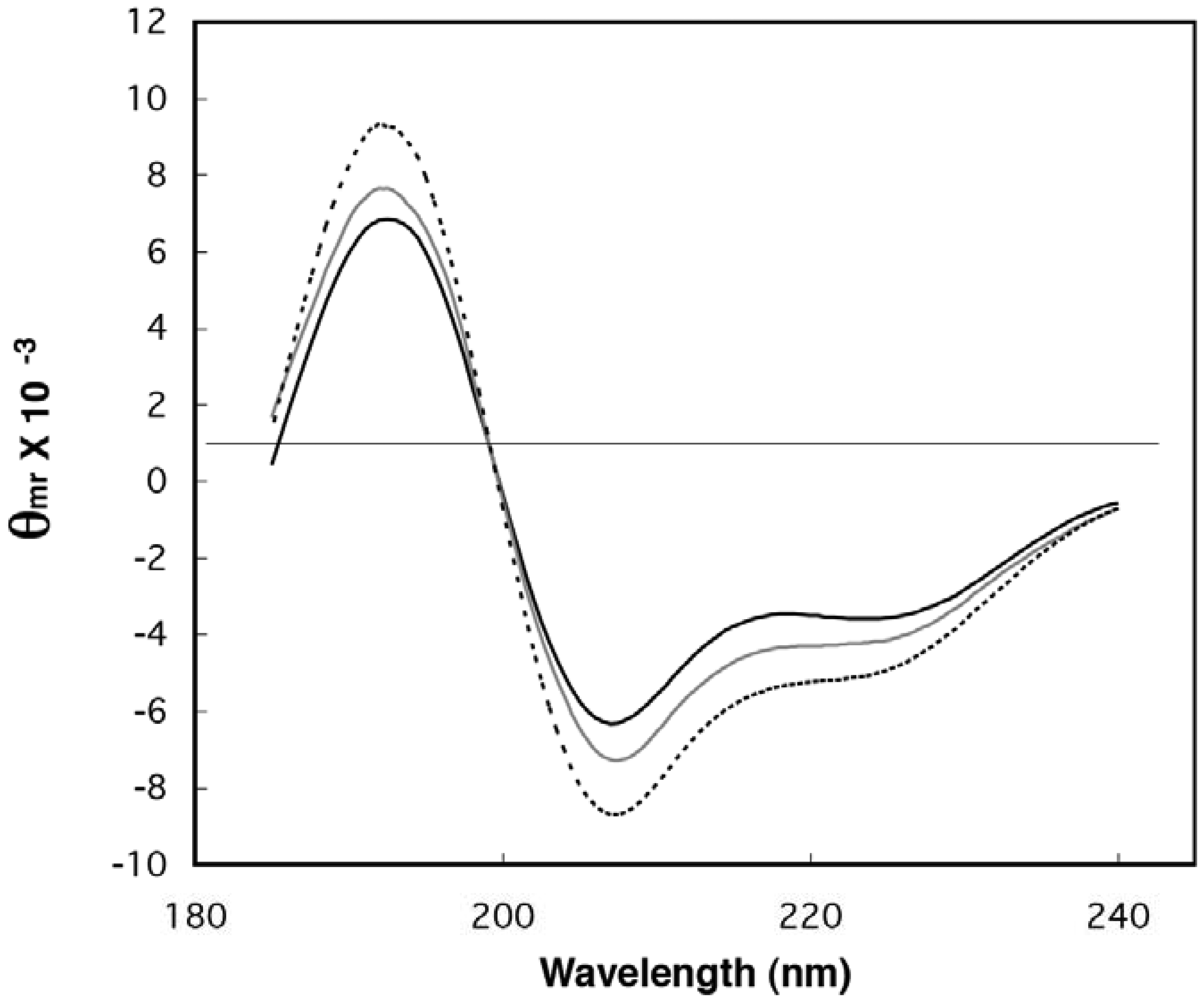
2.1. Demonstration of Secondary Structure in Random-Sequence Fusion Protein Pools
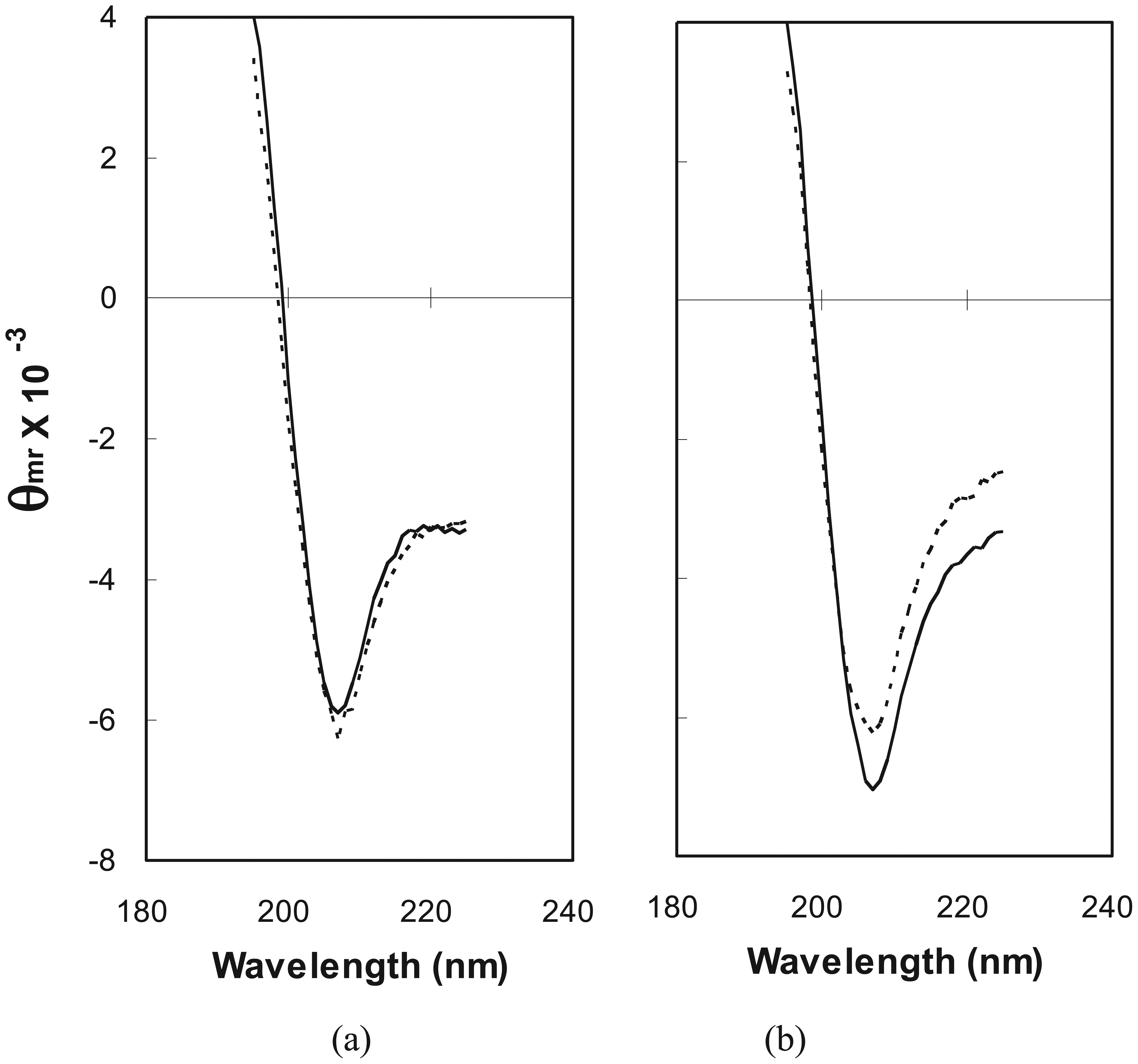
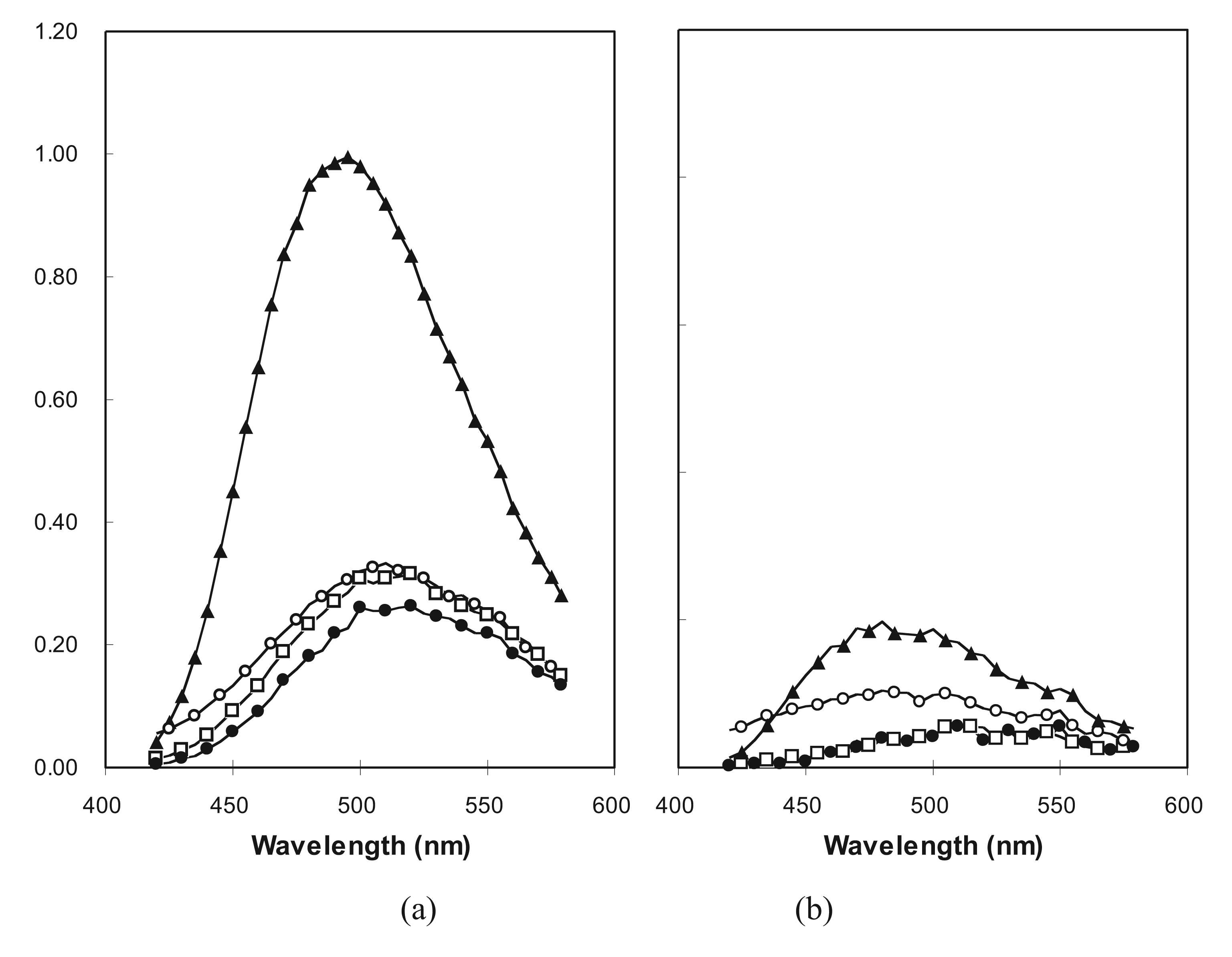
2.2. Estimation of Folded Structure in Individual Random-Sequence Fusion Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool | Isolates | Helix content | Isolate averages b |
|---|---|---|---|
| LIB38 | 18.6* | ||
| 38 mm | 14.7* | 16.6 ± 1.9 | |
| 38a | 16.4 | ||
| 38c | 15.7 | ||
| 38e | 16.4 | ||
| 38j | 16.2 | ||
| 38k | 13.8* | ||
| 38m | 19.3* | ||
| 38o | 17.6* | ||
| 38p | 19.6* | ||
| LIB71 | 22.4* | ||
| 71c | 19.5* | 17.0 ± 1.8 | |
| 71d | 18.2* | ||
| 71e | 14.4* | ||
| 71h | 17.0* | ||
| 71j | 15.9 | ||
| 71L | 17.1* | ||
| Ubiquitin | 16.1 ± 0.04 | ||
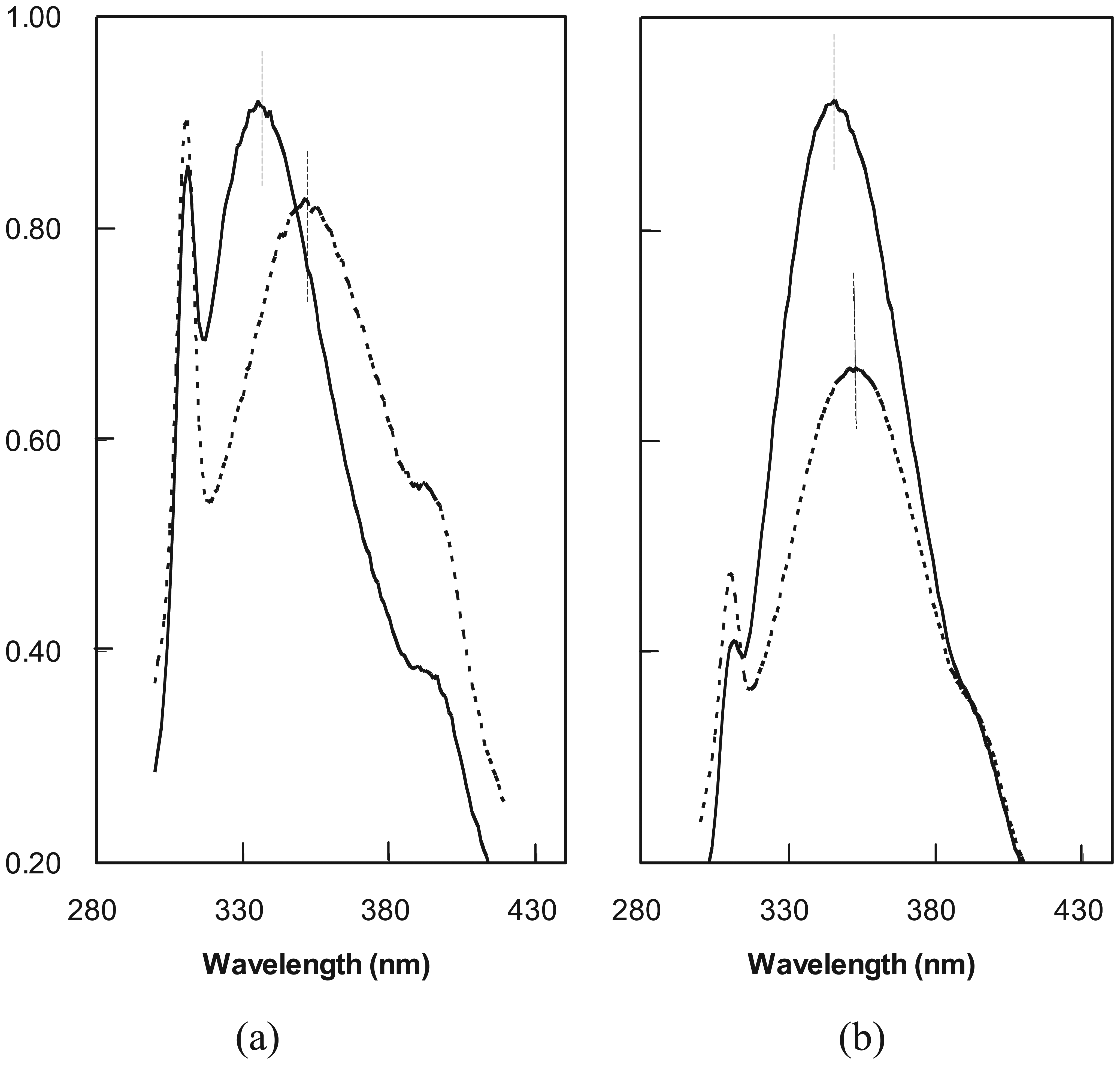
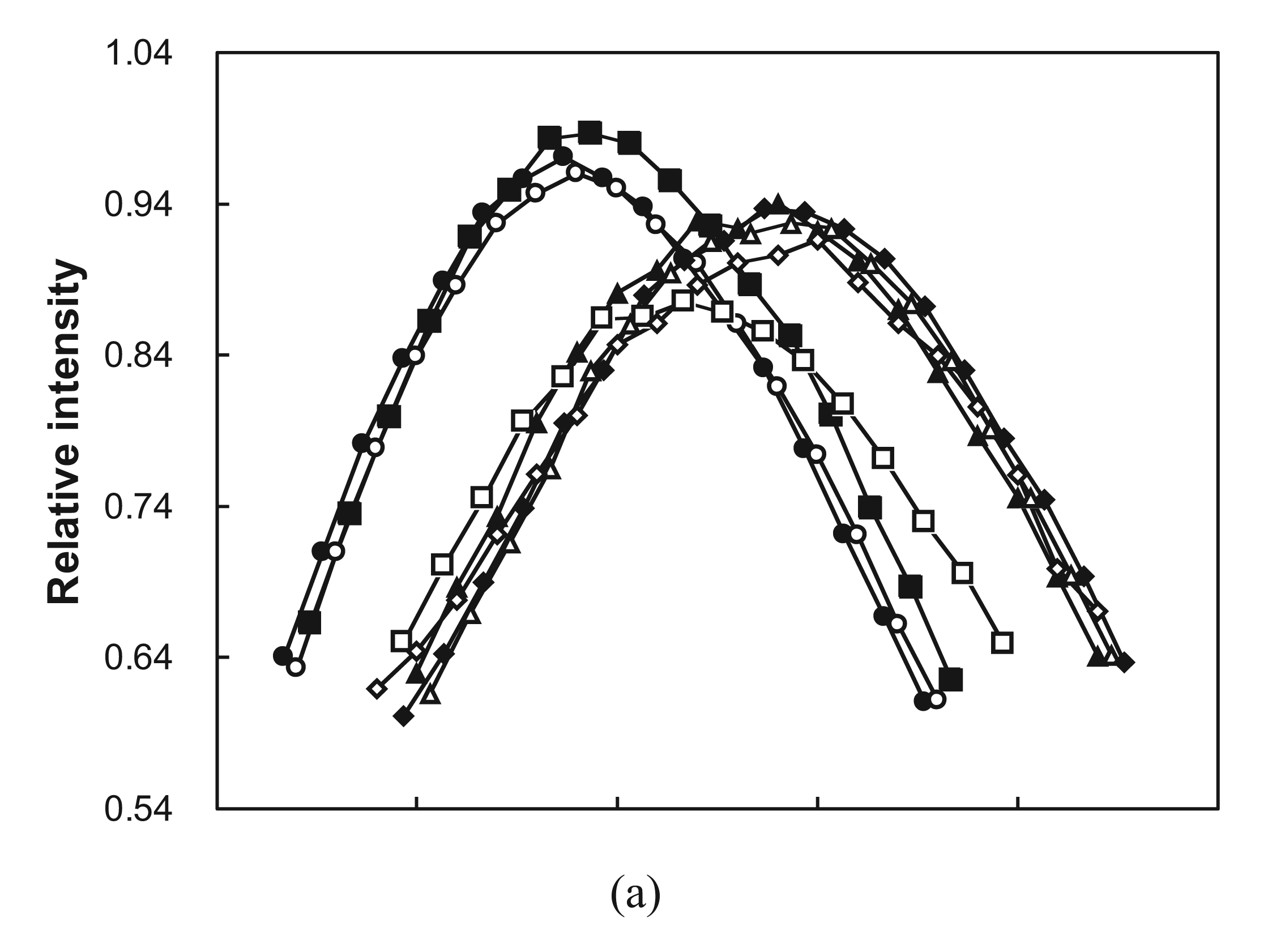
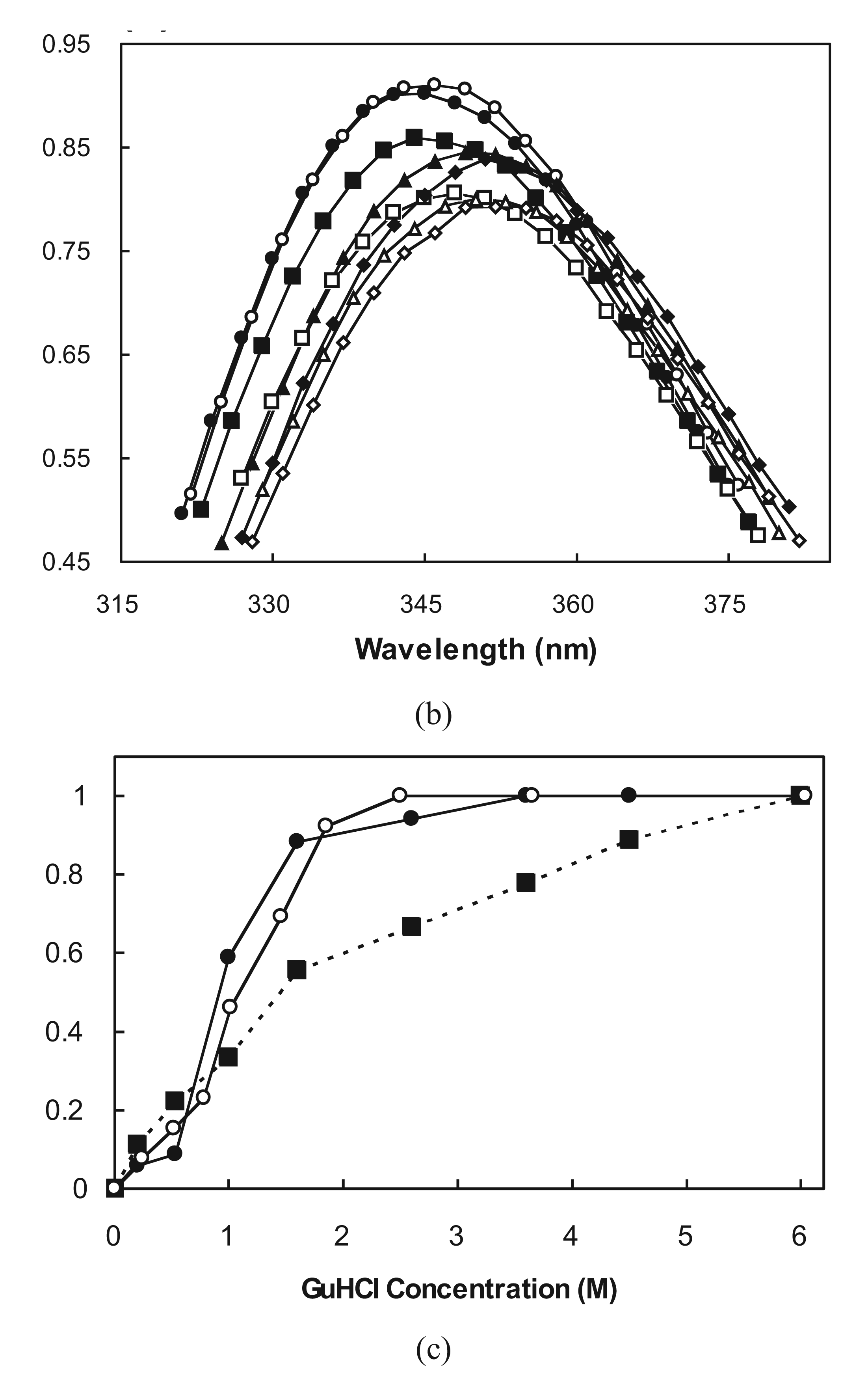
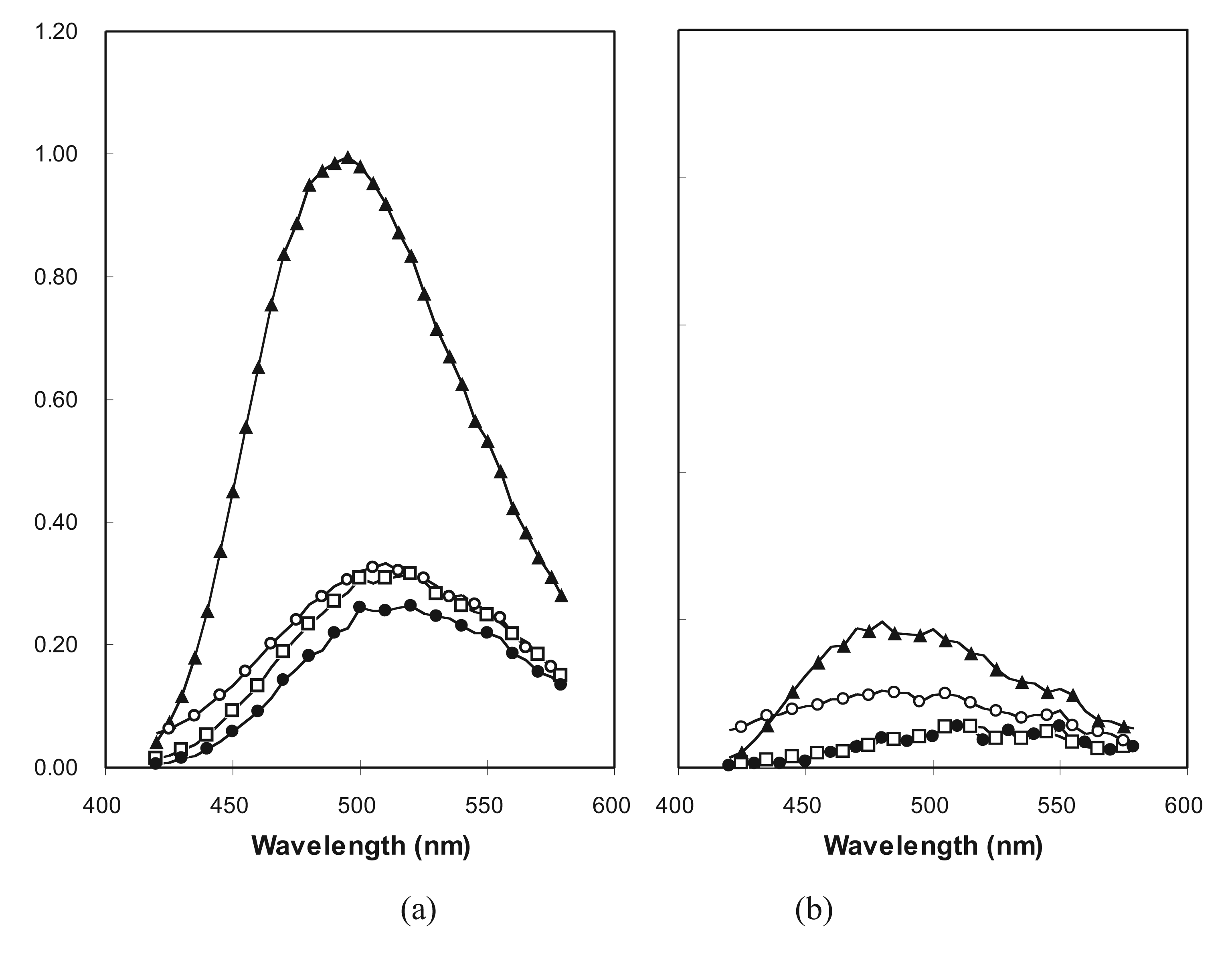
2.3. Cooperative Unfolding Shown by Intrinsic Fluorescence Emission
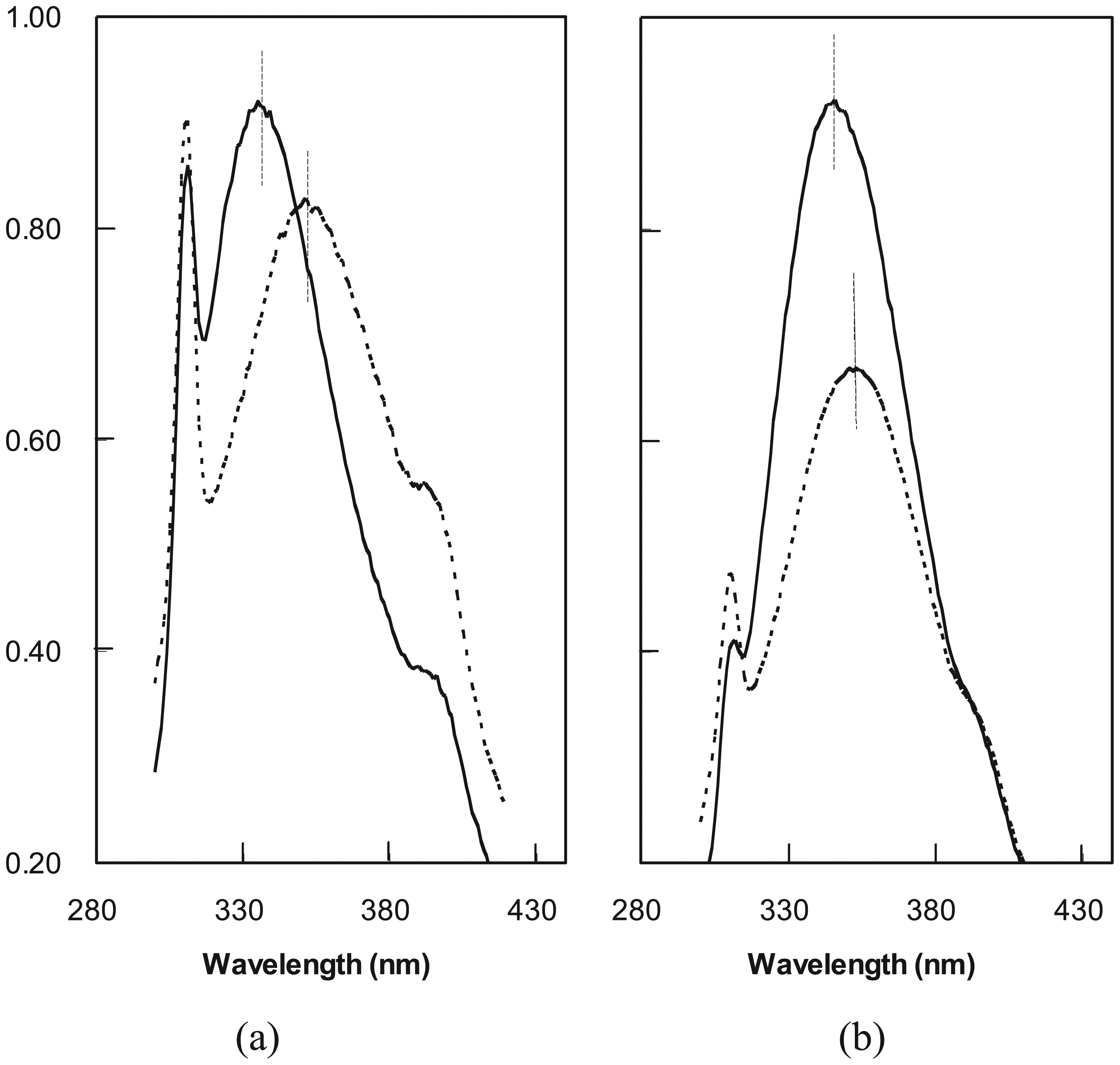
2.4. Ubiquitin Independent Unfolding and Refolding
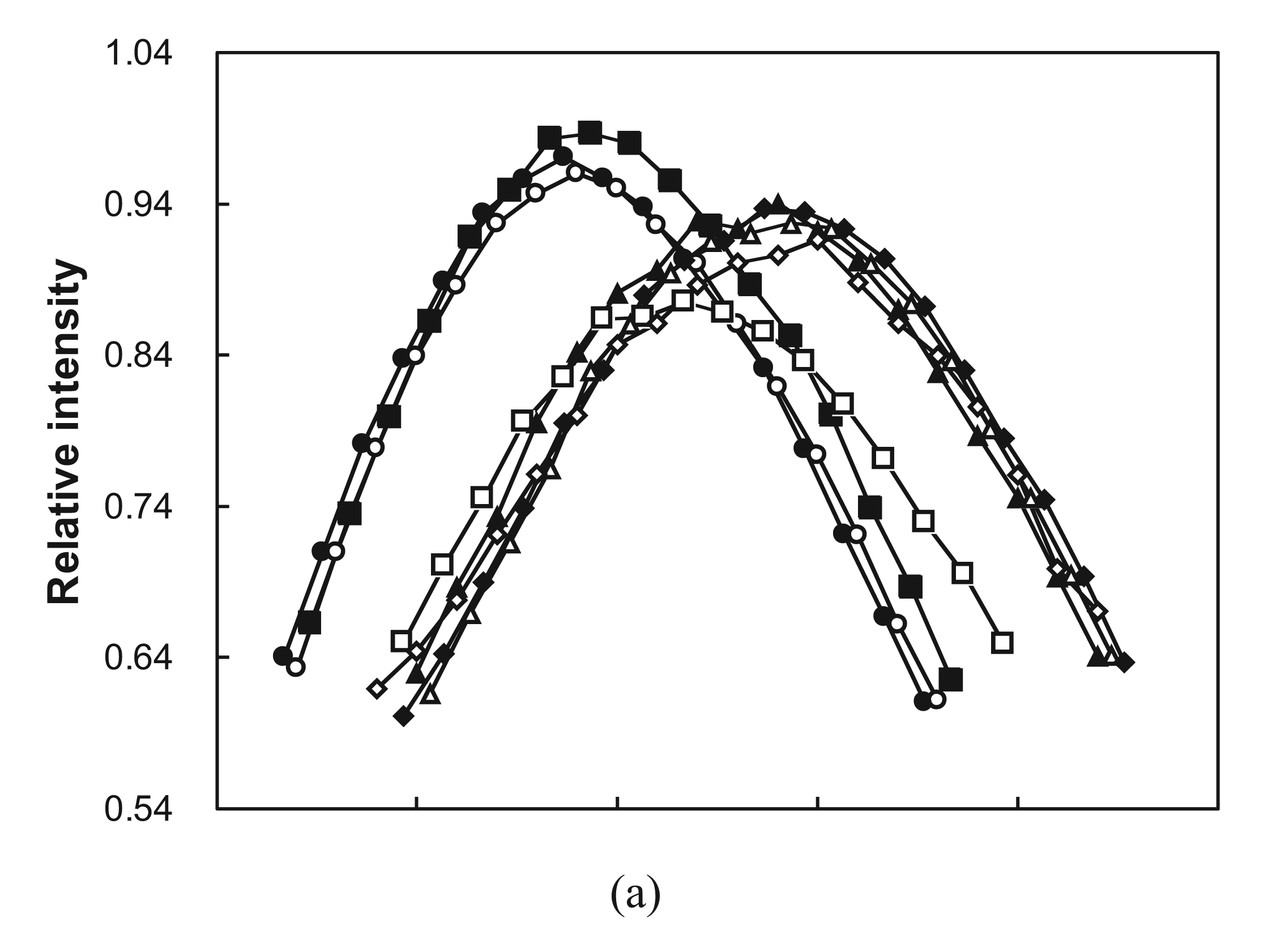
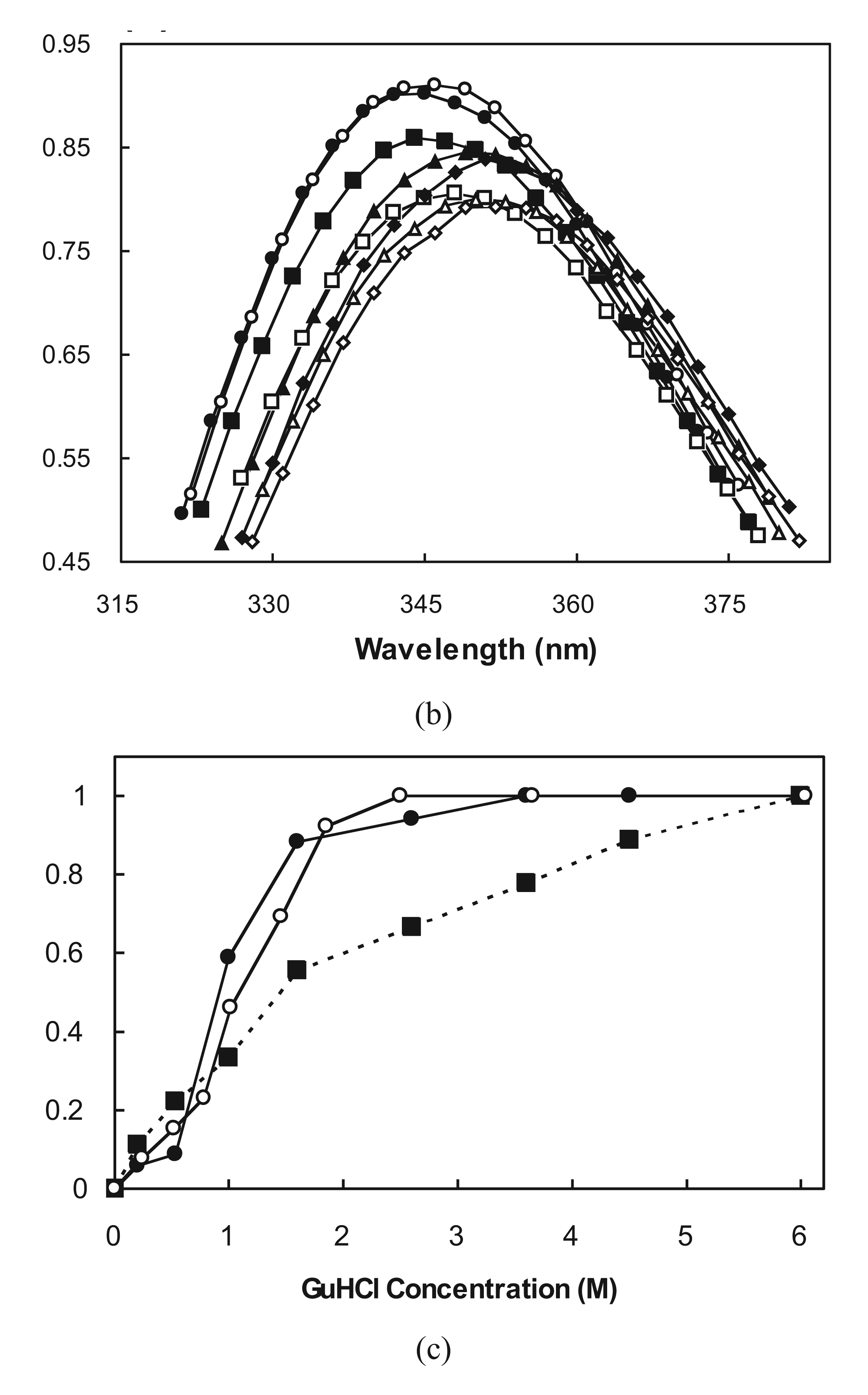
2.5. Molten Globule-Like and Native-Like Behavior Suggested by ANS Binding Data
2.6. The Statistical Validity of a Random Sample of Molecular Sequences in vivo and in vitro
2.7. Rudimentary Folds
3. Experimental Section
3.1. Library Construction
3.2. Protein Purification
3.3. Fusion Protein Hydrolysis
3.4. Spectroscopy
4. Conclusions
Acknowledgments
References
- Van Holde, K.E. Reflections on a century of protein chemistry. Biophys. Chem. 2003, 100, 71–79. [Google Scholar]
- Frauenfelder, H.; Wolynes, P.G. Biomolecules: Where the physics of complexity and simplicity meet. Phys. Today 1994, 47, 58–64. [Google Scholar]
- Meier, S.; Özbek, S. A Biological cosmos of parallel universes: Does protein structural plasticity facilitate evolution? BioEssays 2007, 29, 1095–1104. [Google Scholar]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar]
- King, J.L.; Jukes, T.H. Non-Darwinian evolution. Science 1969, 164, 788–98. [Google Scholar]
- Salisbury, F.B. Natural selection and the complexity of the gene. Nature 1969, 224, 342–343. [Google Scholar]
- Smith, J.M. Natural Selection and the Concept of Protein Space. Nature 1970, 225, 563–564. [Google Scholar]
- Keefe, A.D.; Szostak, J.W. Functional proteins from a random-sequence library. Nature 2001, 410, 715–718. [Google Scholar]
- Meyerguz, L.; Kleinberg, J.; Elber, R. The network of sequence flow between protein structures. Proc. Natl. Acad. Sci. USA 2007, 104, 11627–11632. [Google Scholar]
- Prymula, K.; Piwowar, M.; Kochanczyk, M.; Flis, L.; Malawski, M.; Szepieniec, T.; Evangelista, G.; Minervini, G.; Polticelli, F.; Wiśniowski, Z. In silico structural study of random amino acid sequence proteins not present in nature. Chem. Biodiv. 2009, 6, 2311–2336. [Google Scholar]
- Minervini, G.; Evangelista, G.; Villanova, L.; Slanzi, D.; de Lucrezia, D.; Poli, I.; Luisi, P.L.; Polticelli, F. Massive non-natural proteins structure prediction using grid technologies. BMC Bioinf. 2009, 10, S22. [Google Scholar]
- Cunningham, B.C.; Wells, J.A. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 1989, 244, 1081–1085. [Google Scholar]
- Hermes, J.D.; Blacklow, S.C.; Knowles, J.R. Searching sequence space by definably random mutagenesis: Improving the catalytic potency of an enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 696–700. [Google Scholar]
- Lim, W.A.; Sauer, R.T. The role of internal packing interactions in determining the structure and stability of a protein. J. Mol. Biol. 1991, 219, 359–376. [Google Scholar]
- Hellinga, H.W.; Wynn, R.; Richards, F.M. The hydrophobic core of Escherichia coli thioredoxin shows a high tolerance to nonconservative single amino acid substitutions. Biochemistry 1992, 31, 11203–11209. [Google Scholar]
- Sondek, J.; Shortle, D. Accommodation of single amino acid insertions by the native state of staphylococcal nuclease. Proteins 1990, 7, 299–305. [Google Scholar]
- Urfer, R.; Kirschner, K. The importance of surface loops for stabilizing an eightfold beta alpha barrel protein. Protein Sci. 1992, 1, 31–45. [Google Scholar]
- Bloom, J.D.; Raval, A.; Wilke, C.O. Thermodynamics of neutral protein evolution. Genetics 2007, 175, 255–266. [Google Scholar]
- Yeates, T.O. Protein structure: Evolutionary bridges to new folds. Curr. Biol. 2007, 17, R48–R50. [Google Scholar]
- Schultes, E.; Bartel, D.P. One sequence, two ribozymes: Implications for the emergence of new ribozyme folds. Science 2000, 289, 448–452. [Google Scholar]
- Xia, Y.; Levitt, M. Simulating protein evolution in sequence space and structure space. Curr. Opin. Struct. Biol. 2004, 14, 202–207. [Google Scholar]
- Katchelski, E.; Sela, M. Synthesis and chemical properties of poly-alpha-amino acids. Adv. Prot. Chem. 1958, 13, 243–492. [Google Scholar]
- Rao, S.P.; Carlstrom, D.E.; Miller, W.G. Collapsed structure polymers: A scattergun approach to amino acid copolymers. Biochemistry 1974, 13, 943–952. [Google Scholar]
- Anufrieva, E.V.; Bychkova, V.E.; Krakovyak, M.G.; Pautov, V.D.; Ptitsyn, O.B. A synthetic polypeptide with a compact structure and its self-organization. FEBS Lett. 1975, 55, 46–49. [Google Scholar]
- Davidson, A.R.; Sauer, R.T. Folded Proteins Occur Frequently in Libraries of Random Amino Acid Sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 2146–2150. [Google Scholar]
- Davidson, A.R.; Lumb, K.J.; Sauer, R.T. Cooperatively folded proteins in random sequence libraries. Nat. Struct. Biol. 1995, 2, 856–864. [Google Scholar]
- Doi, N.; Kakukawa, K.; Oishi, Y.; Yanagawa, H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Prot. Eng. Des. Sel. 2005, 18, 279–284. [Google Scholar]
- Prijambada, I.D.; Yomo, T.; Tanaka, F.; Kawama, T.; Yamamoto, K.; Hasegawa, A.; Shima, Y.; Negoro, S.; Urabe, I. Solubility of artificial proteins with random sequences. FEBS Lett. 1996, 382, 21–25. [Google Scholar]
- Chiarabelli, C.; Vrijbloed, J.W.; de Lucrezia, D.; Thomas, R.M.; Stano, P.; Polticelli, F.; Ottone, T.; Papa, E.; Luisi, P.L. Investigation of de novo totally random biosequences. Part II. On the folding frequency in a totally random library of de novo proteins obtained by phage display. Chem. Biodiv. 2006, 3, 840–859. [Google Scholar]
- LaBean, T.H.; Kauffman, S.A. Design of synthetic gene libraries encoding random sequence proteins with desired ensemble characteristics. Prot. Sci. 1993, 2, 1249–1254. [Google Scholar]
- Monia, B.P.; Ecker, D.J.; Jonnalagadda, S.; Marsh, J.; Gotlib, J.L.; Butt, T.R. Gene synthesis, expression and processing of human ubiquitin carboxy extension proteins in bacteria and yeast. J. Biol. Chem. 1989, 264, 4093–4103. [Google Scholar]
- Butt, T.R.; Jonnalagadda, S.; Monia, B.P.; Sternberg, E.J.; Marsh, J.A.; Stadel, J.M.; Ecker, D.J.; Crooke, S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2540–2544. [Google Scholar]
- Yoo, Y.; Rote, K.; Rechsteiner, M. Synthesis of peptides as cloned ubiquitin extensions. J. Biol. Chem. 1989, 264, 17078–17083. [Google Scholar]
- LaBean, T.H.; Kauffman, S.A.; Butt, T.R. Libraries of random-sequence polypeptides produced with high yield as carboxy-terminal fusions with ubiquitin. Mol. Div. 1995, 1, 29–38. [Google Scholar]
- Wilkinson, K.D.; Lee, K.M.; Deshpande, S.; Duerksen-Hughes, P.; Boss, J.M.; Pohl, J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 1989, 246, 670–673. [Google Scholar]
- Perczel, A.; Hollosi, M.; Tusnady, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Prot. Eng. 1991, 4, 669–679. [Google Scholar]
- Kabsch, W.; Sander, C. Dictonary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Jenson, J.; Goldstein, G.; Breslow, E. Physical-chemical properties of ubiquitin. Biochim. Biophys. Acta 1980, 624, 378–385. [Google Scholar]
- Cary, P.D.; King, D.S.; Crane-Robinson, C.; Bradbury, E.M.; Rabbani, A.; Goodwin, G.H. Structural studies on two high-mobility-group proteins from calf thymus, HMG-14 and HMG-20 (ubiquitin), and their interaction with DNA. Eur. J. Biochem. 1980, 112, 577–580. [Google Scholar]
- Johnson, W.C.J. Secondary structure of proteins through circular dichroism spectroscopy. Annu. Rev. Biophys. Biophys. Chem 1988, 17, 145–166. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Pearce, S.F.; Hawrot, E. Intrinsic fluorescence of binding-site fragments of the nicotinic acetylcholine receptor: Perturbations produced upon binding alpha-bungarotoxin. Biochemistry 1990, 29, 10649–10659. [Google Scholar]
- Saito, Y.; Wada, A. Comparative study of GuHCl denaturation of globular proteins. II. A phenomenological classification of denaturation profiles of 17 proteins. Biopolymers 1983, 22, 2123–2132. [Google Scholar]
- Hecht, M.H.; Richardson, J.S.; Richardson, D.C.; Ogden, R.C. De novo design, expression, and characterization of Felix: A four-helix bundle protein of native-like sequence. Science 1990, 249, 884–891. [Google Scholar]
- Regan, L.; DeGrado, W.F. Characterization of a helical protein designed from first principles. Science 1988, 241, 976–978. [Google Scholar]
- Uversky, V.N.; Semisotnov, G.V.; Pain, R.H.; Ptitsyn, O.B. ‘All-or-none’ mechanism of the molten globule unfolding. FEBS Lett. 1992, 314, 89–92. [Google Scholar]
- Semisotnov, G.V.; Rodionova, N.A.; Razgulyaev, O.I.; Uversky, V.N.; Gripas, A.F.; Gilmanshin, R.I. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 1991, 31, 119–128. [Google Scholar]
- Lenkinski, R.E.; Chen, D.M.; Glickson, J.D.; Goldstein, G. Nuclear magnetic resonance studies of the denaturation of ubiquitin. Biochim. Biophys. Acta 1977, 494, 126–130. [Google Scholar]
- Butt, T.R.; Jonnalagadda, S.; Monia, B.P.; Sternberg, E.J.; Marsh, J.A.; Stadel, J.M. Ubiquitin fusion augments the yield of cloned gene products in. Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2540–2544. [Google Scholar]
- Ptitsyn, O.B. Molten globule and protein folding. Adv. Prot. Chem. 1995, 47, 83–229. [Google Scholar]
- Schultes, E.; Hraber, P.T.; LaBean, T.H. Global similarities in nucleotide base composition among disparate functional classes of single-stranded RNA imply adaptive evolutionary convergence. RNA 1997, 3, 792–806. [Google Scholar]
- Schultes, E.; Hraber, P.T.; LaBean, T.H. A parameterization of RNA sequence space. Complexity 1999, 4, 61–71. [Google Scholar]
- Schultes, E.; Hraber, P.T.; LaBean, T.H. Estimating the contributions of selection and self-organization in RNA secondary structures. J. Mol. Evol. 1999, 49, 76–83. [Google Scholar]
- Smit, S.; Yarus, M.; Knight, R. Natural selection is not required to explain universal compositional patterns in rRNA secondary structure categories. RNA 2006, 12, 1–14. [Google Scholar]
- Smit, S.; Yarus, M.; Knight, R. RNA structure prediction from evolutionary patterns of nucleotide composition. Nucl. Acids Res. 2009, 37, 1378–1386. [Google Scholar]
- Kennedy, R.; Lladser, M.E.; Wu, Z.; Zhang, C.; Yarus, M.; de Sterck, H.; Knight, R. Natural and artificial RNAs occupy the same restricted region of sequence space. RNA 2010, 16, 280–289. [Google Scholar]
- Schultes, E.A.; Spasic, A.; Mohanty, U.; Bartel, D.P. Compact and ordered collapse in randomly generated RNA sequences. Nat. Struct. Mol. Bio. 2005, 12, 1130–1136. [Google Scholar]
- Bartel, D.P.; Szostak, J.W. Isolation of new ribozymes from a large pool of random sequences. Science 1993, 261, 1411–1418. [Google Scholar]
- Zon, G.; Gallo, K.A.; Samson, C.J.; Shao, K.L.; Summers, M.F.; Byrd, R.A. Analytical studies of ‘mixed sequence’ oligodeoxyribonucleotides synthesized by competitive coupling of either methyl- or beta-cyanoethyl-N,N-diisopropylamino phosphoramidite reagents, including 2′-deoxyinosine. Nucl. Acids Res. 1985, 13, 8181–8196. [Google Scholar]
- Mandecki, W.A. A method for construction of long randomized open reading frames and polypeptides. Prot. Eng. 1990, 3, 221–226. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Mott, J.E.; Grant, R.A.; Ho, Y.S.; Platt, T. Maximizing gene expression from plasmid vectors containing the lambda PL promoter: Strategies for overproducing transcription termination factor rho. Proc. Natl. Acad. Sci. USA 1985, 82, 88–92. [Google Scholar]
- Wilkinson, K.D.; Cox, M.J.; Mayer, A.N.; Frey, T. Synthesis and characterization of ubiquitin ethyl ester, a new substrate for ubiquitin carboxyl–terminal hydrolase. Biochemistry 1986, 25, 6644–6649. [Google Scholar]
- Nadiger, G.S.; Bhat, N.V.; Padhye, M.R. Investigation of amino acid composition in the crystalline region of silk fibroin. J. App. Pol. Sci. 1985, 30, 221–225. [Google Scholar]
- Tusnády, G.E.; Simon, I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Bio. 1998, 283, 489–506. [Google Scholar]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
LaBean, T.H.; Butt, T.R.; Kauffman, S.A.; Schultes, E.A. Protein Folding Absent Selection. Genes 2011, 2, 608-626. https://doi.org/10.3390/genes2030608
LaBean TH, Butt TR, Kauffman SA, Schultes EA. Protein Folding Absent Selection. Genes. 2011; 2(3):608-626. https://doi.org/10.3390/genes2030608
Chicago/Turabian StyleLaBean, Thomas H., Tauseef R. Butt, Stuart A. Kauffman, and Erik A. Schultes. 2011. "Protein Folding Absent Selection" Genes 2, no. 3: 608-626. https://doi.org/10.3390/genes2030608