Full-Length Multi-Barcoding: DNA Barcoding from Single Ingredient to Complex Mixtures
by
Peng Zhang
1,2,†, 
Chunsheng Liu
1,
Xiasheng Zheng
3,
Lan Wu
2,
Zhixiang Liu
2,
Baosheng Liao
2,
Yuhua Shi
2,
Xiwen Li
2,
Jiang Xu
2,* and
Shilin Chen
2,* 1
School of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing 102488, China
2
Key Laboratory of Beijing for Identification and Safety Evaluation of Chinese Medicine, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing 100700, China
3
Guangdong Provincial Key Laboratory of New Drug Development and Research of Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
*
Authors to whom correspondence should be addressed.
†
Present Address: Chinese Pharmacopoeia Commission, Beijing 100061, China.
Genes 2019, 10(5), 343; https://doi.org/10.3390/genes10050343
Submission received: 2 April 2019
/
Revised: 22 April 2019
/
Accepted: 29 April 2019
/
Published: 7 May 2019
(This article belongs to the Special Issue DNA Barcoding and Metabarcoding of Complex Matrices)
Abstract
:DNA barcoding has been used for decades, although it has mostly been applied to some single-species. Traditional Chinese medicine (TCM), which is mainly used in the form of combination-one type of the multi-species, identification is crucial for clinical usage. Next-generation Sequencing (NGS) has been used to address this authentication issue for the past few years, but conventional NGS technology is hampered in application due to its short sequencing reads and systematic errors. Here, a novel method, Full-length multi-barcoding (FLMB) via long-read sequencing, is employed for the identification of biological compositions in herbal compound formulas in adequate and well controlled studies. By directly sequencing the full-length amplicons of ITS2 and psbA-trnH through single-molecule real-time (SMRT) technology, the biological composition of a classical prescription Sheng-Mai-San (SMS) was analyzed. At the same time, clone-dependent Sanger sequencing was carried out as a parallel control. Further, another formula—Sanwei-Jili-San (SJS)—was analyzed with genes of ITS2 and CO1. All the ingredients in the samples of SMS and SJS were successfully authenticated at the species level, and 11 exogenous species were also checked, some of which were considered as common contaminations in these products. Methodology analysis demonstrated that this method was sensitive, accurate and reliable. FLMB, a superior but feasible approach for the identification of biological complex mixture, was established and elucidated, which shows perfect interpretation for DNA barcoding that could lead its application in multi-species mixtures.
1. Introduction
As DNA barcoding, a dominant method for species identification and discovery [1], emerges as a cost-effective standardized approach for rapid species identification [2,3], it has been widely used in almost all types of organisms. For traditional Chinese medicine (TCM), whose identification is crucial for its safety and effectiveness [4,5,6,7] in clinical practice, various methods have been applied, such as microscopic analysis, chromatography, spectroscopic methodology and molecular biology. Molecular biology methods, especially DNA barcoding, are relatively more precise and sensitive in general [8,9,10]. As a consequence, the DNA barcoding system for identifying herbal medicine (TcmBarcode system, http://www.tcmbarcode.cn/en/) [11] has been successfully established and widely used [12].
For Chinese herbal compounds, most of which are mixtures in the form of pills, powders or other dosage forms, the separation of different raw materials seems impossible, while parts of nucleic acid have been degraded, resulting in difficulties for biological identification [13]. Comparing with Sanger sequencing, in which separation and purification like cloning are commonly needed [14,15], next-generation sequencing (NGS) shows deeper sequencing depth and higher benefit-cost ratio [16] that has revealed legality issues and health safety concerns on TCM [17]. NGS, mainly second-generation sequencing, is considered a powerful approach for the analysis of biotic mixtures, such as the microorganism of the environment [18,19], the soil [20], the gut [21,22,23,24], and the food [25]. Several strategies, such as PCR-free metabarcoding [26], mass-PCR metabarcoding [27], multi-marker metabarcoding [28] and even metagenomics [29] have been employed to evaluate the biodiversity of different biotic communities. But some improvements are still needed, such as primers design [30], fractioning step [31] and algorithm optimization of datasets [32], due to its short sequencing reads; however, some errors, such as “false positives” [33], exist. The biological assessment of TCM preparation based on the NGS approach using both ITS2 and trnL biomarkers has been used to analyze Liuwei-Dihuang-Wan with an additional method as supplementary [34], but doubt regarding contamination existed and was not well explained. Since the lengths of some barcodes were out of the range of second-generation sequencing techniques, overlapping assembly was often used, from which algorithm errors might occur [26]. By contrast, long-read sequencing approaches, such as single-molecule real-time (SMRT) sequencing, which can output longer sequencing reads directly, have been shown to be competitive and also to exhibit more enrichment information and higher identification efficiency [35,36]. With the development of science and technology, they have emerged as potential approaches due to advantages of longer sequencing reads and deeper sequencing depth [37,38]. Long-read sequencing techniques have already been used in some aspects, such as microorganism identification [39,40] and taxonomic profiling [41], and have provided an economical way to monitor the legality and safety of traditional patent medicines [42,43]. Nevertheless, these studies have not yet elucidated this novel authentication approach for Chinese herbs in an adequate and well-controlled methodological analysis.
In the current study, basing on the TcmBarcode database and long-read sequencing technology, we put forward Full-length Multi-barcoding (FLMB), a method that capable of identifying the biological origins of multi-species mixture and could elucidate further details of DNA barcoding. This method was applied to the identification of Sheng-Mai-San (SMS), a classical prescription whose ingredients could be well controlled by hand-making, through SMRT sequencing of the amplicons of ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA) and psbA-trnH (a chloroplast gene), two barcodes which have been recommended as standard DNA markers that are widely used in herbal medicine [44,45]. Moreover, Sanwei-Jili-San (SJS), another herbal compound that contains animal ingredient, has been submitted to be analyzed with CO1 (cytochrome c oxidase subunit 1, a mitochondrial gene) and ITS2 as a verifying approach.
2. Materials and Methods
2.1. Sample Collection and Powders Preparation
Raw materials of Ginseng Radix et Rhizoma (dried roots and rhizomes of Panax ginseng C. A. Mey., RS1/2/3), Ophiopogonis Radix (dried roots of Ophiopogon japonicus (L. f) Ker-Gawl.; MD1/2/3/4), Schisandrae Chinensis Fructus (dried fruits of Schisandra chinensis (Turcz.) Baill., WW1/2/3), Malvae Fructus (dried fruits of Malva veriticillata L., DK) and Tribuli Fructus (dried fruits of Tribulus terrestris L., JL), were purchased from production places, medicinal materials markets or companies; Fresh crab of Eriocheir sinensis H. Miline–Edwalds (FH), was purchased from a local supermarket (Table 1). All raw materials were identified by their morphologies, according to the Chinese Pharmacopeia [46]. Biological origins of these decoction slices were further tested by DNA barcoding through Sanger sequencing with barcodes of ITS2 and psbA-trnH for the flora, while CO1 for the fauna.
Preparation of the powder Sheng-Mai-San (SMS): decoction slices of RS1, MD3 and WW2 were mixed by weight with a ratio of 3:3:2 and then ground into powder (SMS1/2/3). Mixed powder (SJS1/2/3) of Sanwei-Jili-San (SJS) was manufactured by decoction slices of DK, JL and FH with a weight ratio of 3:5:3. The reducing and samples collection (around 5g for each powder sample) of all these powders were performed by quartering according to the general principle ‘0211’ in Chinese Pharmacopeia [46].
2.2. DNA Preparation and Sanger Sequencing
DNA of the powders (200 mg per sample) and independent ingredients (50 mg per sample) in Table 1 were extracted using the Plant Genomic DNA Kit (Tiangen Biotech Co., Ltd, Beijing, China), respectively. PCR systems containing 1 × Taq MasterMix (Aidlab Biotechnologies Co., Ltd., Beijing, China), 1 μM of each primer [47,48,49,50] (primer information see in Supplementary Materials Table S1) and ~100 ng DNA templates, were performed using conditions as followed: 95 °C for 4 min; 94 °C for 30 s, 55 °C for 1 min, 72 °C for 1 min, 35 cycles; and 72 °C for 10 min for ITS2 and psbA-trnH [8,12,50], while 94 °C for 1 min; 94 °C for 1 min, 45 °C for 1.5 min, 72 °C for 1.5 min, 5 cycles; and 94 °C for 1 min, 50 °C for 1.5 min, 72 °C for 1 min, 35 cycles; 72 °C for 5 min for CO1 [47]. Sanger sequencing of those PCR products were performed to confirm their biological origin. Then DNA mixtures in Table 1 of both formulas were prepared from the DNA samples of their ingredients, all of which were identified by DNA barcoding.
At the same time, four PCR products of SMS’ ITS2 fragment were purified using the MinElute® Gel Extration Kit (Cat. No. 28606, Qiagen, Hilden, Germany). Then these purified fragments were inserted to the pMD 19-T vector (Takara, Beijing, China), transferred into competent cells of E. coli and selected through blue-white spot screening. Finally, a total of 81 white clones obtained were grown in liquid culture and then sequenced by Sanger method, as a comparison for SMRT sequencing.
2.3. Amplicon Libraries Preparation for SMRT Sequencing
As shown in Figure 1, DNA samples were prepared in three groups: (1) DNA extracted from raw materials; (2) mixtures of the DNA from raw materials, by a volume ratio of 3:3:2 (RS-MD-WW); (3) DNA extracted from the powders.
All DNA samples were used as templates for PCR amplification of ITS2 and psbA-trnH, respectively. Amplification for each amplicon were carried out using different pairs of tag-primers, to which several protective bases and labeling bases were attached to the 5′ end of the conventional primers (tag-primers’ sequences were shown in Supplementary Table S1, pairs of tag-primers corresponding to different amplicons were shown in Supplementary Materials Table S2). After electrophoresis, positive PCR products were purified using Agencourt® AMPure® XP beads (Beckman Coulter, Brea, CA, USA) by 0.8× volume and the concentrations of those purified amplicons was determined using a Qubit® 3.0 Fluorometer (Invitrogen by Life Technologies, Carlsbad, CA, USA). In the end, purified fragment amplicons were pooled together to form sequencing libraries by certain quantity of nucleic acids, 500 ng for per fragment amplicons amplified from group 2 and 3, and 200 ng for those from group 1 (distribution of those amplicon libraries in sequencing libraries are shown in Table 1 and Supplementary Table S2).
2.4. SMRT Sequencing and Data Analysis
The sequencing libraries underwent chemical process with the SMRTbellTM Template Prep Kit 1.0, and then were bound with V2 primers using the DNA/polymerase Binding Kit P6 V2 and P6- DNA polymerase, respectively. Next, each library was transferred to a 96-well PCR plate for real-time sequencing with C4 reagents on a PacBio II SMRT sequencing platform (Pacific Biosciences of California, Menlo Park, CA, USA).
Following the SMRT Analysis pipeline (v5.0.1), the resulting bas.h5 files were analyzed to generate Circular Consensus Sequence (CCS) passing reads. CCS parameters as follows: Minimum Full Passes = 7, Minimum Predicted Accuracy = 90, Minimum and Maximum Reads Length of Insert (In Bases) = 200 and 800). Resulting reads of each amplicon were extracted from CCS-pass reads according to corresponding tag-primer pairs. Along with the data analysis pipeline, data size, amount and length of the reads that remained available were carried out. Based on previous reports that error profile is about 13% for single pass in SMRT sequencing, the error profile of each locus in the resulting reads for 7-pass is estimated, in theory, to be around 0.72%
This will decline following an increase of the circular number. At the same time, the resulting reads in an amplicon that belonging to the same species were analyzed to define the profile and its influence on biological identification.
2.5. Clustering and Biological Identification of Resulting Reads
CodonCode Aligner (v5.1.5.3) was used to perform sequences alignment for resulting reads of each amplicon, with parameters set as followed: end to end alignments, Min. percent identity = 95.0, Min. overlap length = 200, Min. score = 150 (for CO1: Min. overlap length = 500, Min. score = 400). After verifying by labeling bases and trimming by tag-primers, the sequences of contigs and unassembled reads generated from assembly for each amplicon were submitted to perform BLAST (Basic Local Alignment Search Tool) against two public databases, the TcmBarcode system and the GenBank nucleotide Non-redundant database (https://www.ncbi.nlm.nih.gov/) [51], in which Max. score ≥ 400 and Identities ≥ 90% of the top hits for each sequence was defined as an effective one [52].
At the same time, four PCR products of SMS’ ITS2 fragment were purified using the MinElute® Gel Extration Kit (Cat. No. 28606, Qiagen). Then, these purified fragments were inserted to the pMD 19-T vector (Takara), transferred into competent cells of E. coli and selected through blue-white spot screening. Finally, a total of 81 white clones obtained were grown in liquid culture and then sequenced by Sanger method. Species identification for these clones was performed, as comparative analysis with SMRT sequencing.
The definitions of the resulting reads, in contigs or unassembled ones achieving from each same amplicon but being assigned to different species, are as below.
- Original: A resulting read is original if it was assigned to the biological species of a sample.
- Endogenous: A resulting read is endogenous if the species which it was assigned to could be found in the raw materials, though it was not original.
- Exogenous: A resulting read is exogenous if it was assigned to other species which was considered as contamination from the outside, but not the biological species of the raw material or the powder.
- Invalid: A resulting read is invalid if it could not be assigned to biological species due to a lower similarity with the sequences in both databases, which was also defined as noneffective, rather than the effective ones that were defined as original, endogenous or exogenous.
2.6. Method Testifying by SJS
In order to test the feasibility of our method, Sanwei-Jili-San (SJS) was involved in our study. SJS is a Mongolian proved recipe that contains both herbal and animal ingredients, including Malvae Fructus (dried fruits of Malva veriticillata L., DK), Tribuli Fructus (dried fruits of Tribulus terrestris L., JL), Chinese fresh-water crab (Eriocheir sinensis H. Miline-Edwalds, FH). Its ingredients and their DNA mixture (JH) were comparatively analyzed using the barcodes of ITS2 and CO1, while the latter was added for the identification of animal ingredient.
3. Results
3.1. Species Authentication by Sanger Sequencing
ITS2 and psbA-trnH regions of SMS and relevant independent raw materials were successfully amplified. Sanger sequencing of these PCR products demonstrated that all the raw materials were from correct original species. Interestingly, consensus sequences of both regions of SMS from Sanger sequencing were the same with those of Panax ginseng (RS). The raw materials of SJS was similarly tested with ITS2 and CO1.
The acquisition of ITS2 fragments of Ophiopogon japonicus (MD) was difficult but not impossible, due to its unsatisfactory amplifying efficiency. And 10 PCR cycles as well as two more parallel tests were individually added as a compensation to achieve sufficient concentration for SMRT sequencing. In total, five amplicon libraries of MD’s ITS2 were prepared and sequenced, four of which the PCR cycles were 35 + 10.
3.2. Analysis Results Via FLMB
3.2.1. Data Processing of SMRT Sequencing
A total of three sequencing libraries were sequenced by SMRT sequencing (Flow cell A, B and C for Library A, B and C respectively). For Library A as an example, which contained 12 purified amplicons of SMS and HSM, 57,147 raw reads were yield up with an average length of 31,217 bp. After CCS processing, 36,497 CCS-pass reads were produced, among which 25,877 resulting reads were extracted to their belonging amplicons according to corresponding sequences of tag-primer pairs. Reserving rate was about 45.3% (25,877/57,147). The datasets used during the current study are available via NCBI under the project number PRJNA419289 (SUB3240739). Details for the data processing information of Library A are shown in Table 2.
3.2.2. Species Identification of SMS by SMRT Sequencing
The resulting reads for each amplicon were clustered and aligned by CodonCode Aligner, and amplicon-corresponding labels were used to validate the correct belonging of these resulting reads. Then the species identifications were carried out through BLAST function in TcmBarcode System and NCBI. Species with nearest match and the number of resulting reads for each amplicon are shown in Figure 2.
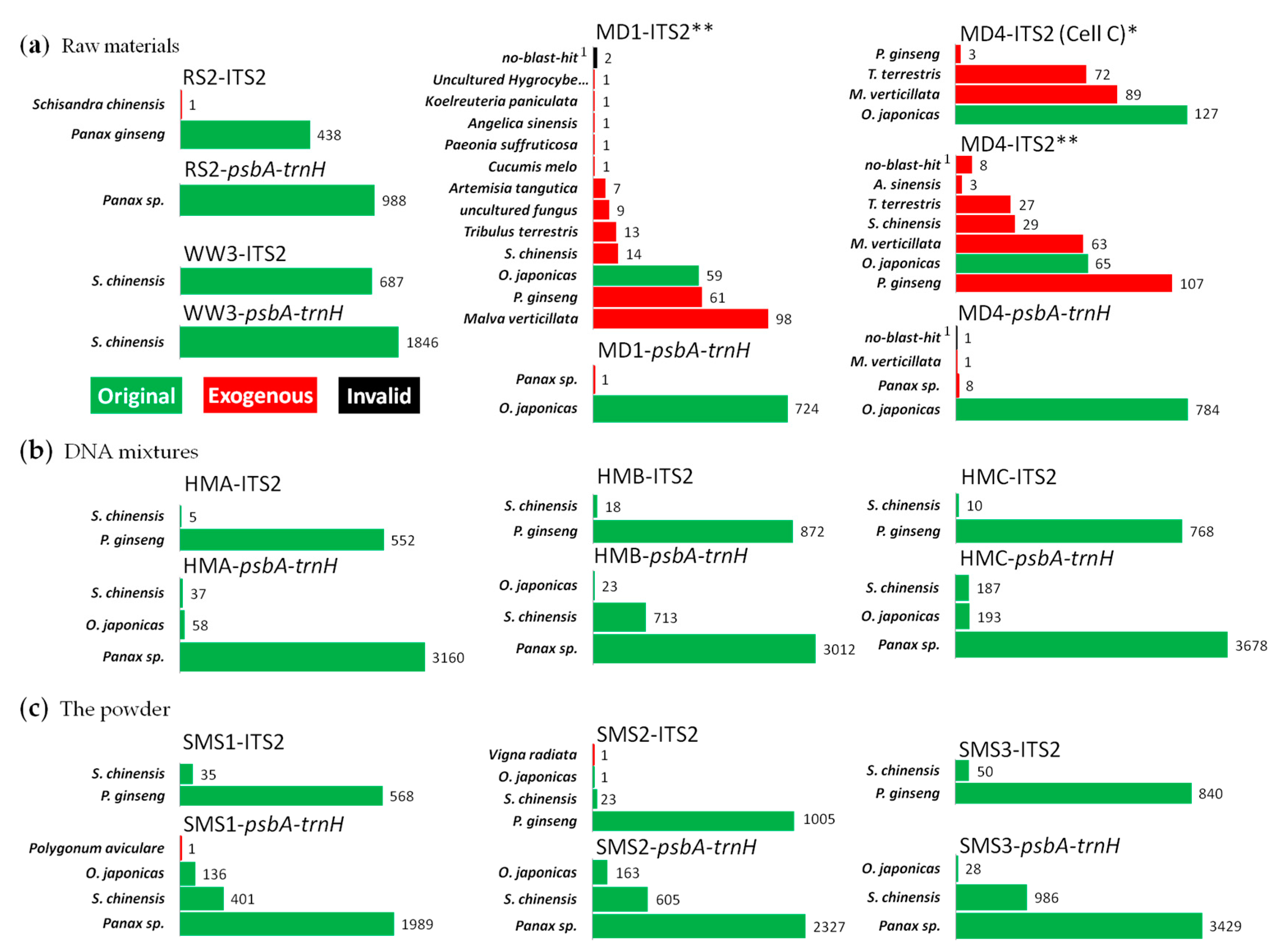
Species identification results of the reads from raw materials were as expected (Figure 2a), except for the ITS2 amplicons of MD, in which several exogenous reads were present (all results were shown in Supplementary Table S3). However, almost no exogenous reads were detected in their psbA-trnH amplicons and, in both ITS2 and psbA-trnH amplicons of the HSM samples, which were all prepared from each same DNA samples of MD as a raw material. No exogenous species were detected (Figure 2b). So, these contaminations in MD’s ITS2 amplicons were considered to be introduced during PCR amplification, thus an additional 10 PCR cycles were likely to bring in more exogenous reads or exogenous species.
For multi-species mixtures, ITS2 amplicons of SMS and HSM showed resulting reads identified as Panax ginseng and Schisandra chinensis, while psbA-trnH showed Panax sp., S. chinensis, and Ophiopogon japonicus, in summary (Figure 2b,c). For example, in ITS2 amplicons of SMS3, there were 840 and 50 resulting reads for P. ginseng and S. chinensis, while in psbA-trnH amplicons of SMS3, there were 3429, 986 and 28 resulting reads for Panax sp., S. chinensis and O. japonicus, respectively. The ITS2 sequence of O. japonicus nearly could not be found in results of all these mixtures, while the psbA-trnH sequence of P. ginseng could hardly be distinguished from those of some closely related species such as P. japonicus or P. quinquefolium (American ginseng).
On the other hand, amplicons of DNA mixtures (HSM) were extremely pure; therefore, no exogenous species were detected, while exogenous sequences such as Vigna radiata and Polygonum aviculare were found in those of SMS, whose ingredients were in higher cleanliness level after processing.
3.2.3. Result of Clone-Dependent Sanger Sequencing
A total of 81 clones from four PCR products of SMS’ ITS2 region were picked out for Sanger sequencing. Among them, 76 clones were identified as P. ginseng, and 5 as S. chinensis. Yet, no clone belonging to O. japonicus was detected in these selected clones, which was similar with the results of SMRT sequencing (Figure 3). Clone-dependent Sanger sequencing showed some randomness due to its low throughput that even 20 clones, such as Clone G2 (Figure 3), could not make a confident checkout of S. chinensis (its abundance in ITS2 amplicons of SMS was estimated to around 4.3% via the analysis of SMRT sequencing). SMRT sequencing allowed us to obtain ITS2 fragment of O. japonicus, in spite of its negligible abundance (below 0.1%).
3.2.4. Results for SJS
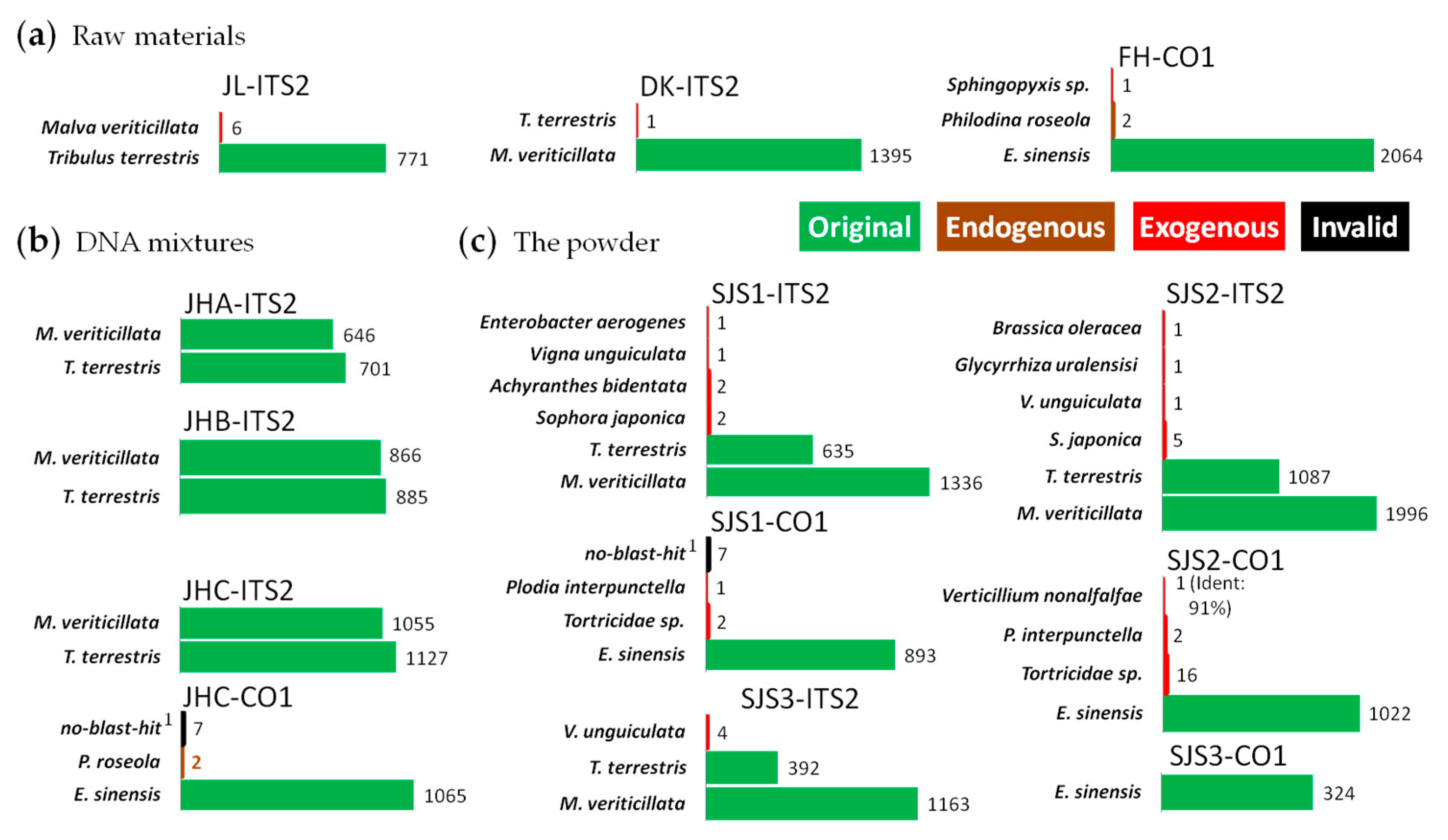
To further test the feasibility of this method, we performed our approach on Sanwei-Jili-San with barcodes of ITS2 and CO1. In summary, all the raw materials in the powder and their DNA mixtures could be successfully identified at species level (Figure 4). In the CO1 amplicons of the Eriocheir sinensis (FH) and JHC (DNA mixture), several reads were identified as Philodina roseola [53], which is often used in crab breeding and is considered as an endogenous species. In contrast, in a few reads from exogenous animal species, such as Plodia interpunctella [54], an insect commonly seen in storage spots, and Tortricidae sp. [55,56], a class of harmful insects in agriculture field, was found in the powders.
A total of nine kinds of exogenous biological sources, including five plant species, two animal species and two microorganism species, were found in the powders of Sanwei-Jili-San (SJS) by the combination of CO1 and ITS2 (Figure 4c). But no exogenous species were detected in these amplicons of its DNA mixtures (Figure 4b), which was similar with the results of HSM, leading to the conclusions that these contaminations were indeed existing in the powder. In conclusion, FLMB is an effective analysis method for biological mixtures.
4. Discussion
4.1. Methodological Analysis
Procedures’ quality control, optimized data-processing parameters, and other methodological analysis in these adequate and well controlled studies, all had provided more details on DNA barcoding that could contributed to a better understanding. And some more abundant but precise information could be carried out at the same time, via full-length multi-barcoding, a feasible, effective and accuracy method.
4.1.1. Precise Definitions from High Accuracy Resulting Reads
A relatively lower sequencing accuracy for single pass (about 13% error rate) [37] had once hindered the application of SMRT sequencing. But as the fragment lengths of frequently-used DNA barcodes were short (about 200 to 1500 bp), long-read sequencing strategy enables self-correction by CCS as the read length of SMRT sequencing could reach 20 kb in average [37], and thus, the sequencing accuracy could be improved [57]. In fact, the error properties of Pacific Biosciences sequencing technology was defined as free of the context-specific effects, which may affect other sequencing technologies [58], and it has already shown excellent utility in some aspects such as SNP discovery [59].
In this study, 462 resulting reads were screened out for the ITS2 amplicon from RS3. And a total of 812 bases (0.39%) with ‘mutations’ at 208 positions were found in 210,210 bases after primer trimming (Figure 5a), which should involve all ‘mutations’ from multi-copy gene, amplification mismatches and sequencing error. Therefore, the error profile was actually below 0.39%, and was significantly lower than the expectation (0.72% for 7 CCS passes), because the mean number of CCS passes had reached 40. Mutation percentage for each position shows in Figure 5d, which can be used to find homologous sequence reads from closely related species. Although there is a close taxonomy relationship between Panax ginseng and P. quinquefolium–only two different loci in their ITS2 regions [60], there were no resulting reads that might be assigned to P. quinquefolium (Figure 5e), reflecting a high accuracy rate of this method. And since there was a steady different site between the psbA-trnH sequences of “Zhe” Ophiopogon japonicus and “Chuan” O. japonicus—two different variance-types, genotype in resulting reads from different amplicons were verified (Supplementary Table S4). As a conclusion, high accuracy resulting reads generated by SMRT sequencing and CCS could lead to precise and reliable identification results.
4.1.2. Every Resulting Reads Counts
By using a combination of proper DNA barcodes, all the biological origins of raw materials in Sheng-Mai-San and Sanwei-Jili-San were validated with accurate identification, while some exogenous or endogenous species were detected at the same time. For all the 46 amplicons sequenced, only 7 of them had no-blast-hit (top hits with low Max. score or low Identities) resulting reads, that 85% (39/46) achieved resulting reads with 100% effective rate, which means all the resulting reads could be explained. And the effective rate of all resulting reads from the 46 amplicons was 99.93% (60,191/60,232), i.e., almost every resulting read was counted (Table 3). Although still unknown, some of these no-blast-hit resulting reads were believed to exist, but had not yet been submitted to both databases used in this study, because most of them distributed in amplicons of MD, while a few were homologous.
4.1.3. Recommended Sequencing Depth
Raw materials of the SMS were well cleaned before powdering. However, raw materials of Sanwei-Jili-San (SJS), like JL, were hard to clean in mass production because of their tiny size, i.e., lots of exogenous reads were found as a consequence. The quantitative relationship between the exogenous reads, the species detected with the total resulting reads were analyzed, i.e., more exogenous reads—or even more species before reaching a ceiling—were likely to be detected as the number of total resulting reads increasing (Figure 6). In general, 1000 resulting reads were recommended as a proper sequencing depth for each amplicon library of formula; thus, a single SMRT sequencing cell covered more than 20 amplicon libraries, and the cost per multi-species mixture rapidly decreased.
4.1.4. Combination Makers Makes Identification More Accuracy and Entirely
Markers combination could not only enhance the discrimination ability, but could also promote the possibility of successful amplification for templates from different biological original materials [61,62]. The psbA-trnH resulting reads of P. ginseng could hardly be distinguished from those of other Panax spp., while ITS2 reads showed the only biological origin, P. ginseng, which indicated the importance of appropriate regions for DNA barcoding. ITS2 sequences of O. japonicus could hardly be detected in amplicons of SMS or HSM due to its unsatisfactory PCR efficiency, while the psbA-trnH could be successfully amplified and detected, despite its different variance-types (Table S2). Moreover, a combination of CO1 and ITS2 revealed more species for the samples of Sanwei-Jili-San (SJS), not merely endogenous and exogenous, but also covering a vast number of species, ranging from plant, animal to microbial. For Chinese herbal formula that are preparation of plant, animal, fungi and minerals medicine, combination of DNA barcodes can facilitate more accurate and integrated authentication of biological origins.
4.1.5. Quality Control of the Procedures
Compared with a low effective rate of raw reads (~20.3%) in a former study [42], the effective rate of raw reads in this study was significantly enhanced to a much higher level (~45.3%), partly due to the protective bases designed in this study. And as suspect unauthentic contaminations or negative results of some species occurred in other studies [9,34], further research with adequate and well-controlled studies were needed. The method proposed in this study had provided technical support for the study on Danggui Buxue Formula [52], while the latter formed an overall quality evaluation system for herbal medicine, including subsequent quantitative analysis through high performance liquid chromatography.
To ensure the reliability of the identification results, several measurements for quality control of the library preparation and data analysis were taken. (1) All raw ingredients were purged before DNA extraction and their biological origins of herbs were confirmed by Sanger sequencing. (2) Analyses of the powders of SMS and SJS via FLMB were carried out by adequate and well-controlled studies, i.e., three batches for each formula were used, compared with a collateral test of the raw materials and their DNA mixture. (3) Tag-primers were designed, which contained labeling bases that were used to recognize which sample a particular read was derived from, and protective bases that were used to protect these labeling bases with which they joined due to damage. (4) Gel electrophoresis, beads purification and concentration determination were all used to achieve purified fragments with enough concentration. (5) After read extractions, all the resulting reads were examined by tag-primers to guarantee their correct ownership; the parameters of CCS, the damaged profile of protective bases and the error rate of resulting reads were all analyzed. (6) Two databases, TcmBarcode System, a professional identification system for herbs, and the GenBank sequence database which contains tremendous DNA sequences ranging from prokaryotes to eukaryotes, were simultaneously used to obtain creditable results.
4.2. Different Sequencing Technologies Applied to DNA Barcoding
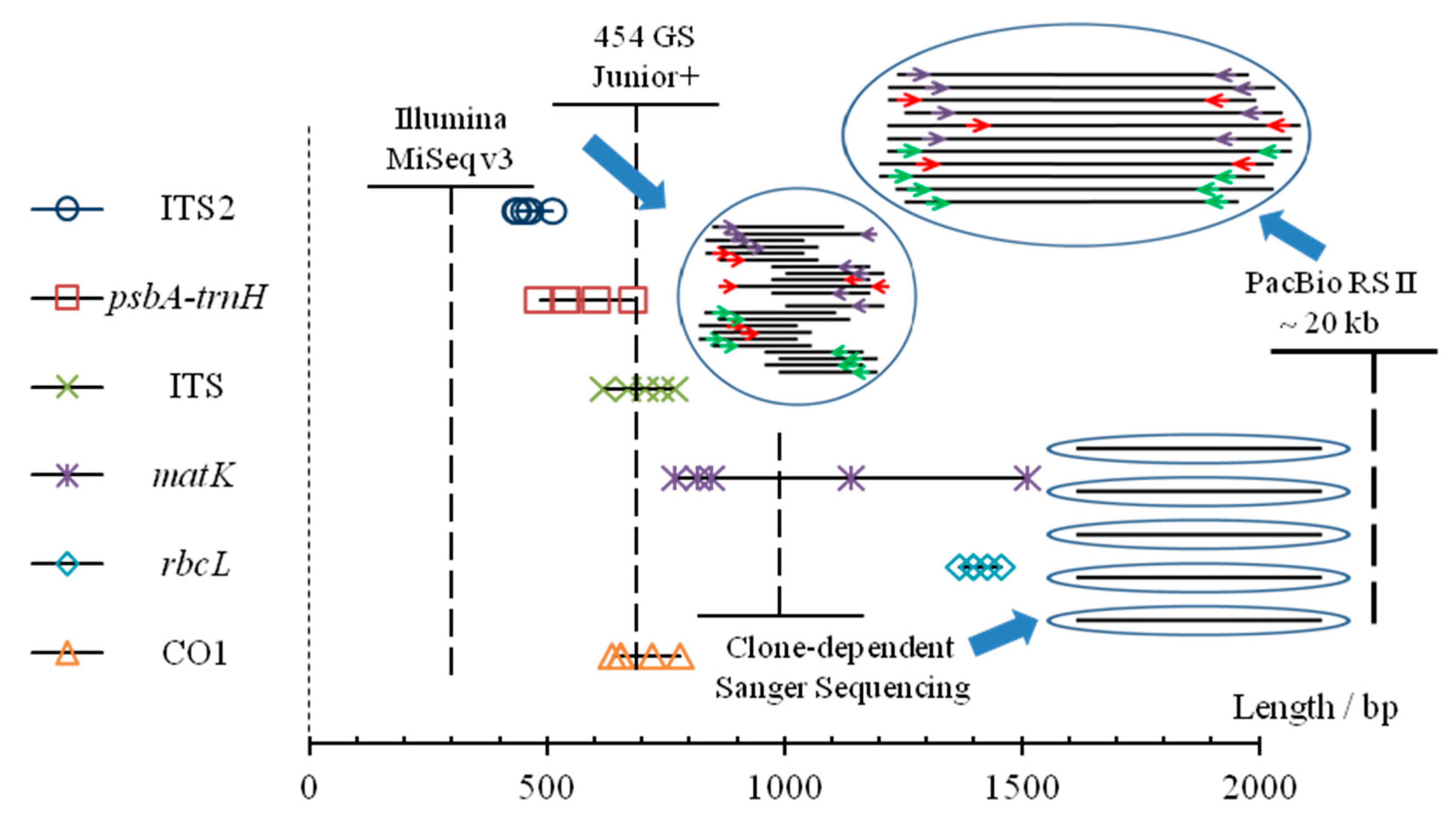
As shown in Figure 7, Sanger sequencing, though low in throughput but providing relatively long and accurate reads, has been widely used in authentication for single species sample [63]. As for complex biotic mixtures, high-throughput sequencing has eclipsed clone-dependent Sanger sequencing over the past decade [36]. For NGS with short read sequencing, identification results were suspicious sometimes, when problems emerged when overlapping was used [26]. This method, FLMB, is a perfect interpretation for DNA barcoding through adequate and well-controlled experiments (Table 4), that could achieve complete and accurate identification for biotic components [42,52], and it could shed some light on the negative or false positive results in some studies of DNA barcoding.
Long reads obtained from the SMRT sequencing could simplify the subsequent data analysis process by sequencing through the whole fragment of DNA barcodes repeatedly. Thus, the efficient rate of the resulting reads from SMRT sequencing could reach 100%, and a more reliable conclusion can be drawn in the end. Furthermore, the tag-primers designed in this study contain both labeling bases (6 bp) and protective bases (8 bp). The former ones, located in the middle of tag-primers, were group-specific, corresponding to different sample. The latter, at the 5′ end and all the same for different tag-primers in this study, could protect the labeling bases from damage by taking its outing place and maintain a high effective rate of the raw reads, that was similar but not the same with the sequences used to protect DNA from exonucleolytic degradation [64]. Besides, this strategy of multiplexing allows sequencing multiple samples simultaneously and guarantees a high-level retention rate from raw reads to resulting reads in the process of data manipulation, thus help to reduce the sequencing cost. On the contrary, the cost of Sanger sequencing for 20 clones is equivalent to one eighth the cost of a SMRT sequencing cell which may contain tens of amplicon libraries, not to mention the cumbersome and time-consuming process of cloning which was unlikely to find rare fragments [65].
4.3. Applications and Challenges
A comprehensive identification of biological origin for herbs is absolutely necessary because most herbal products are multi-species mixtures, such as the herbal compound formula and those being labeled as single ingredient but containing fillers or substitutions [9,66]. As shown in the results of SMS, the gene copy number from different species and PCR bias for different fragments was of varying significance, which might lead to the detection of those species with more gene copies or higher PCR efficiency. In contrast, parameters in the pipeline of DNA barcoding, especially barcode selections and PCR conditions, should be taken into account because they could make some difference to the final results, especially when samples tested were multi-species mixtures. Based on biological composition information as a qualitative analysis, a quantitative analysis could be carried out through other approaches such as chemical analysis. Thus, a more integrated quality evaluation system [4,52] could be established, from which a more accurate but credible identification result, including an objective definition of contamination, substitution and adulteration, will be obtained.
From these comparative analyses of the formula samples, the raw materials and their DNA mixtures by FLMB, a perfect interpretation on DNA barcoding for both single-species samples and multi-species samples was achieved, which will promote DNA barcoding from single-species ingredient to multi-species mixtures. As it can be used to detect most species of biotic components, a better assessment of Chinese medicine, not only for its safety clinical use but also for the conservation of protected species [67,68] could be carried out. What’s more, as some microbial species were also detected. This method could even be used to find possible microorganism that have potential toxicity, such as Aspergillus flavus and A. parasiticus fungi which may produce aflatoxins [69], by using proper barcodes. It could also provide a novel insight into the biodiversity analysis on other research areas [70]. While multigene was an effective way to promote the successfully identifying rate [71] for single-species, multi-barcoding could be used to find more species with more precisely identification for multi-species mixtures and in this study, a recommended sequencing depth was put forward.
In the current study, two powerful reference databases were employed to guarantee the reliability of identifying results which was considered as a challenge a few years ago [9], while another challenge exits, namely, the detection of the ingredients whose DNA was degraded, as parts of Chinese patent medicine covers procedures that may lead to DNA’s damage such as heating. Other than some species-specific methods [34,72], FLMB using short segments, though they might not have an equally distinguishing ability with the longer ones, as barcodes, should be taken into consideration for their potentially superior amplification.
5. Conclusions
A considerable number of classical preparations in TCM are still widely used [73], such as SMS and SJS [74,75]. Unlike raw materials and decoction slices whose biological identification is relatively simple because DNA barcoding and other approaches may easily be employed, biological identification of samples which contain multi-species ingredients is more challenging, and almost no methods have been adopted to address at this issue in Chinese Pharmacopeia. But the substitution and adulteration in herbs and other supplements [76], whether intentionally or unintentionally, leaves nonnegligible health risks for consumers, such as known organisms that have known toxicity, side effects, allergens and/or negatively interact with other herbs, supplements, or medications [9].
In this study, we established a novel method, Full-Length Multi-Barcoding (FLMB), for biological analyses of multi-species mixtures. The proposed methodology was carried out with adequate and well-controlled studies, by analyzing a classic formula Sheng-Mai-San with barcodes of ITS2 and psbA-trnH, comparing with its raw materials and their DNA mixtures. Extremely pure amplicons for both the single-species and the multi-species were successfully achieved, yielding more more scientific and believable results by DNA barcoding. The results of another formula, Sanwei-Jili-San using ITS2 and CO1, showed that this method was feasible and reproducible. In conclusion, the method could provide a powerful and credible approach for the biological analysis of complex biotical mixtures, covering a vast number species ranging from plants, to animals to microbes.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/10/5/343/s1, Table S1: Information of primers and tag-primers, Table S2: Distribution of amplicons in different sequencing Cells and their corresponding tag-primers pairs, Table S3: Species identifying results on Ophiopogon japonicus by FLMB, Table S4: Validation of different locus between two types of Ophiopogon japonicus in psbA-trnH region.
Author Contributions
Conceptualization, P.Z., J.X. and S.C.; methodology, P.Z.; software, B.L. and J.X.; validation, L.W. and Z.L.; formal analysis, B.L.; investigation, C.L. and S.C.; resources, X.L.; data curation, P.Z.; writing—original draft preparation, P.Z. and X.Z.; writing—review and editing, J.X. and Y.S.; visualization, P.Z. and X.Z.; supervision, L.W.; project administration, J.X.; funding acquisition, J.X., Y.S. and S.C.
Funding
This research was funded by the Major Scientific and Technological Special Project for Significant New Drugs Creation, grant number 2014ZX09304307, and grants from the National Natural Science Foundation of China, grant number 81403053 & 81603248.
Acknowledgments
We thank the teams at Institute of Chinese Matria Medica, China Academy of Chinese Medical Sciences and Softgene biological technology company (Beijing, China) for their essential work on this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hao, D.-C.; Chen, S.L.; Xiao, P.-G. Authentication of medicinal plants based on molecular biology and genomics. Pharm. Biotechnol. 2009, 16, 490–494. [Google Scholar]
- Miller, S.E. DNA barcoding and the renaissance of taxonomy. Proc. Natl. Acad. Sci. USA 2007, 104, 4775–4776. [Google Scholar] [CrossRef]
- Wallace, L.J.; Boilard, S.M.; Eagle, S.H.; Spall, J.L.; Shokralla, S.; Hajibabaei, M. DNA barcodes for everyday life: Routine authentication of natural health products. Food Res. Int. 2012, 49, 446–452. [Google Scholar] [CrossRef]
- Wu, L.; Sun, W.; Wang, B.; Zhao, H.; Li, Y.; Cai, S.; Xiang, L.; Zhu, Y.; Yao, H.; Song, J.; et al. An integrated system for identifying the hidden assassins in traditional medicines containing aristolochic acids. Sci. Rep. 2015, 5, 11318. [Google Scholar] [CrossRef]
- Vanherweghem, J.L.; Tielemans, C.; Abramowicz, D.; Depierreux, M.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Dratwa, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; et al. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- Chen, C.-H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.-R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pang, X.; Liao, B.; Yao, H.; Song, J.; Chen, S. An authenticity survey of herbal medicines from markets in China using DNA barcoding. Sci. Rep. 2016, 6, 18723. [Google Scholar] [CrossRef]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 2010, 5, e8613. [Google Scholar]
- Newmaster, S.G.; Grguric, M.; Shanmughanandhan, D.; Ramalingam, S.; Ragupathy, S. DNA barcoding detects contamination and substitution in North American herbal products. BMC Med. 2013, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yao, H.; Li, Y.; Li, X.; Lin, Y.; Liu, C.; Jianping, H.; Xie, C.; Chen, S. Authentication of the family polygonaceae in Chinese pharmacopoeiaby DNA barcoding technique. J. Ethnopharmacol. 2009, 124, 434–439. [Google Scholar] [CrossRef]
- DNA Barcoding System for Identifying Herbal Medicine. Available online: http://www.tcmbarcode.cn/en/ (accessed on 22 November 2017).
- Chen, S.; Pang, X.; Song, J.; Shi, L.; Yao, H.; Han, J.; Leon, C. A renaissance in herbal medicine identification: From morphology to DNA. Biotechnol. Adv. 2014, 32, 1237–1244. [Google Scholar] [CrossRef]
- Cheng, K.-T.; Tsay, H.-S.; Chen, C.-F.; Chou, T.-W. Determination of the components in a Chinese prescription, Yu-Ping-Feng San, by RAPD analysis. Planta Med. 1998, 64, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Shi, L.-C.; Xu, Z.-C.; Xin, T.-Y.; Song, J.-Y.; Chen Shi, L. Identification of antler powder components based on DNA barcoding technology. Yao Xue Xue Bao Acta Pharm. Sin. 2015, 50, 1356–1361. [Google Scholar]
- Cui, Z.-H.; Jiang, C.; Li, M.-H.; Chen, M.; Zhou, L.-S.; Yuan, Y. Molecular identification of raw materials from lian qiao bai du wan. Yao Xue Xue Bao Acta Pharm. Sin. 2013, 48, 590–596. [Google Scholar]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Coghlan, M.L.; Haile, J.; Houston, J.; Murray, D.C.; White, N.E.; Moolhuijzen, P.; Bellgard, M.I.; Bunce, M. Deep sequencing of plant and animal DNA contained within traditional Chinese medicines reveals legality issues and health safety concerns. PLoS Genet. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Cummings, L.A.; Kurosawa, K.; Hoogestraat, D.R.; SenGupta, D.J.; Candra, F.; Doyle, M.; Thielges, S.; Land, T.A.; Rosenthal, C.A.; Hoffman, N.G.; et al. Clinical next generation sequencing outperforms standard microbiological culture for characterizing polymicrobial samples. Clin. Chem. 2016, 62, 1465–1473. [Google Scholar] [CrossRef]
- Korshunov, S.O.; Gorovtsov, A.V.; Faleeva, T.G.; Gorbov, S.N.; Kornienko, I.V. Comparison of biochemical and molecular genetic approaches for identification of environmental strains. Microbiology 2014, 83, 376–380. [Google Scholar] [CrossRef]
- Sarikhani, E.; Sagova-Mareckova, M.; Omelka, M.; Kopecky, J. The effect of peat and iron supplements on the severity of potato common scab and bacterial community in tuberosphere soil. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.; Bassis, C.M.; Hein, R.; Assari, S.; Flowers, S.A.; Kelly, M.B.; Young, V.B.; Ellingrod, V.E.; McInnis, M.G. The gut microbiome composition associates with bipolar disorder and illness severity. J. Psychiatr. Res. 2017, 87, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2017, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2017, 359, 91–97. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Pompanon, F.; Deagle, B.E.; Symondson, W.O.C.; Brown, D.S.; Jarman, S.N.; Taberlet, P. Who is eating what: Diet assessment using next generation sequencing. Mol. Ecol. 2012, 21, 1931–1950. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Liu, S.; Yang, Q.; Su, X.; Zhou, L.; Tang, M.; Fu, R.; Li, J.; Huang, Q. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience 2013, 2, 4. [Google Scholar] [CrossRef]
- Yu, D.W.; Ji, Y.; Emerson, B.C.; Wang, X.; Ye, C.; Yang, C.; Ding, Z. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012, 3, 613–623. [Google Scholar] [CrossRef]
- Marcelino, V.R.; Verbruggen, H. Multi-marker metabarcoding of coral skeletons reveals a rich microbiome and diverse evolutionary origins of endolithic algae. Sci. Rep. 2016, 6, 31508. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Tyson, G.W. Microbiology: Metagenomics. Nature 2008, 455, 481–483. [Google Scholar] [CrossRef]
- Alanio, A.; Gits-Muselli, M.; Mercier-Delarue, S.; Dromer, F.; Bretagne, S. Diversity of pneumocystis jirovecii during infection revealed by ultra-deep pyrosequencing. Front. Microbiol. 2016, 7, 733. [Google Scholar] [CrossRef]
- Kermarrec, L.; Franc, A.; Rimet, F.; Chaumeil, P.; Humbert, J.F.; Bouchez, A. Next-generation sequencing to inventory taxonomic diversity in eukaryotic communities: A test for freshwater diatoms. Mol. Ecol. Resour. 2013, 13, 607–619. [Google Scholar] [CrossRef]
- Nguyen, T.-D.; Schmidt, B.; Zheng, Z.; Kwoh, C.-K. Efficient and accurate OTU clustering with GPU-based sequence alignment and dynamic dendrogram cutting. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 1060–1073. [Google Scholar] [CrossRef]
- Blanckenhorn, W.U.; Rohner, P.T.; Bernasconi, M.V.; Haugstetter, J.; Buser, A. Is qualitative and quantitative metabarcoding of dung fauna biodiversity feasible? Environ. Toxicol. Chem. 2016, 35, 1970–1977. [Google Scholar] [CrossRef]
- Cheng, X.; Su, X.; Chen, X.; Zhao, H.; Bo, C.; Xu, J.; Bai, H.; Ning, K. Biological ingredient analysis of traditional Chinese medicine preparation based on high-throughput sequencing: The story for Liuwei Dihuang Wan. Sci. Rep. 2014, 4, 5147. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Bowers, R.M.; Levy, A.; Gies, E.A.; Cheng, J.-F.; Copeland, A.; Klenk, H.-P.; et al. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Mosher, J.J.; Bowman, B.; Bernberg, E.L.; Shevchenko, O.; Kan, J.; Korlach, J.; Kaplan, L.A. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing. J. Microbiol. Methods 2014, 104, 59–60. [Google Scholar] [CrossRef]
- George, N.; Flamiatos, E.; Kawasaki, K.; Kim, N.; Carriere, C.; Phan, B.; Joseph, R.; Strauss, S.; Kohli, R.; Choi, D.; et al. Oral microbiota species in acute apical endodontic abscesses. J. Oral Microbiol. 2016, 8, 30989. [Google Scholar] [CrossRef]
- Franzén, O.; Hu, J.; Bao, X.; Itzkowitz, S.H.; Peter, I.; Bashir, A. Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome 2015, 3, 43. [Google Scholar] [CrossRef]
- Jia, J.; Xu, Z.; Xin, T.; Shi, L.; Song, J. Quality control of the traditional patent medicine Yimu Wan based on SMRT sequencing and DNA barcoding. Front. Plant Sci. 2017, 8, 926. [Google Scholar] [CrossRef]
- Xin, T.; Xu, Z.; Jia, J.; Leon, C.; Hu, S.; Lin, Y.; Ragupathy, S.; Song, J.; Newmaster, S.G. Biomonitoring for traditional herbal medicinal products using DNA metabarcoding and single molecule, real-time sequencing. Acta Pharm. Sin. B 2017, 8, 488–497. [Google Scholar] [CrossRef]
- Xin, T.; Li, X.; Yao, H.; Lin, Y.; Ma, X.; Cheng, R.; Song, J.; Ni, L.; Fan, C.; Chen, S. Survey of commercial Rhodiola products revealed species diversity and potential safety issues. Sci. Rep. 2015, 5, 8337. [Google Scholar] [CrossRef]
- Chen, X.; Xiang, L.; Shi, L.; Li, G.; Yao, H.; Han, J.; Lin, Y.; Song, J.; Chen, S. Identification of crude drugs in the Japanese pharmacopoeia using a DNA barcoding system. Sci. Rep. 2017, 7, 42325. [Google Scholar] [CrossRef]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Tate, J.; Simpson, B. Paraphyly of tarasa (malvaceae) and diverse origins of the polyploid species. Syst. Bot. 2003, 28, 723–738. [Google Scholar]
- Sang, T.; Crawford, D.; Stuessy, T. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120. [Google Scholar] [CrossRef]
- Cheng, T.; Xu, C.; Lei, L.; Li, C.; Zhang, Y.; Zhou, S. Barcoding the kingdom Plantae: New PCR primers forITSregions of plants with improved universality and specificity. Mol. Ecol. Resour. 2016, 16, 138–149. [Google Scholar] [CrossRef]
- Standard Nucleotide BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome (accessed on 1 April 2019).
- Zheng, X.; Zhang, P.; Liao, B.; Li, J.; Liu, X.; Shi, Y.; Cheng, J.; Lai, Z.; Xu, J.; Chen, S. A Comprehensive quality evaluation system for complex herbal medicine using PacBio sequencing, PCR-Denaturing gradient gel electrophoresis, and several chemical approaches. Front. Plant. Sci. 2017, 8, 1578. [Google Scholar] [CrossRef]
- Lebedeva, L.I.; Gerasimova, T.N. Peculiarities of Philodina roseola (Ehrbg.) (Rotatoria, Bdelloida)—Growth and reproduction under various temperature conditions. Int. Rev. Gesamten Hydrobiol. Hydrogr. 1985, 70, 509–525. [Google Scholar] [CrossRef]
- Scheff, D.S.; Sehgal, B.; Subramanyam, B. Evaluating penetration ability of Plodia interpunctella (Hübner) (Lepidoptera: Pyralidae) larvae into multilayer polypropylene packages. Insects 2018, 9, 42. [Google Scholar] [CrossRef]
- McMahan, E.E.; Guédot, C. Development of Sparganothis sulfureana (Lepidoptera: Tortricidae) on cranberry cultivars. Insects 2018, 9, 4. [Google Scholar] [CrossRef]
- Amarasekare, K.G.; Shearer, P.W. Stability of Cacopsylla pyricola (Hemiptera: Psyllidae) populations in Pacific Northwest pear Orchards managed with long-term mating disruption for Cydia pomonella (Lepidoptera: Tortricidae). Insects 2017, 8, 105. [Google Scholar] [CrossRef]
- Orkunoglu-Suer, F.; Harralson, A.F.; Frankfurter, D.; Gindoff, P.; O’Brien, T.J. Targeted single molecule sequencing methodology for ovarian hyperstimulation syndrome. BMC Genom. 2015, 16, 264. [Google Scholar] [CrossRef]
- Carneiro, M.O.; Russ, C.; Ross, M.G.; Gabriel, S.B.; Nusbaum, C.; DePristo, M.A. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genom. 2012, 13, 375. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Song, J.; Xu, H.; Xu, J.; Zhu, Y.; Li, X.; Gao, H.; Dong, L.; Qian, J.; et al. High-accuracyde novoassembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef]
- Chen, X.; Liao, B.; Song, J.; Pang, X.; Han, J.; Chen, S. A fast SNP identification and analysis of intraspecific variation in the medicinal Panax species based on DNA barcoding. Gene 2013, 530, 39–43. [Google Scholar] [CrossRef]
- Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; Yang, J.B.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar]
- Pang, X.; Liu, C.; Shi, L.; Liu, R.; Liang, D.; Li, H.; Cherny, S.S.; Chen, S. Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: A meta-analysis. PLoS ONE 2012, 7, e48833. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Biswas, I.; Maguin, E.; Ehrlich, S.D.; Gruss, A. A 7-base-pair sequence protects DNA from exonucleolytic degradation in Lactococcus lactis. Proc. Natl. Acad. Sci. USA 1995, 92, 2244–2248. [Google Scholar] [CrossRef]
- Liu, W.; Kim, H.J.; Lucchetta, E.M.; Du, W.; Ismagilov, R.F. Isolation, incubation, and parallel functional testing and identification by FISH of rare microbial single-copy cells from multi-species mixtures using the combination of chemistrode and stochastic confinement. Lab Chip 2009, 9, 2153–2162. [Google Scholar] [CrossRef]
- Stoeckle, M.Y.; Gamble, C.C.; Kirpekar, R.; Young, G.; Ahmed, S.; Little, D.P. Commercial teas highlight plant DNA barcode identification successes and obstacles. Sci. Rep. 2011, 1, 42. [Google Scholar] [CrossRef]
- Chang, C.-H.; Jang-Liaw, N.-H.; Lin, Y.-S.; Fang, Y.-C.; Shao, K.-T. Authenticating the use of dried seahorses in the traditional Chinese medicine market in Taiwan using molecular forensics. J. Food Drug Anal. 2013, 21, 310–316. [Google Scholar] [CrossRef]
- Yan, D.; Luo, J.Y.; Han, Y.M.; Peng, C.; Dong, X.P.; Chen, S.L.; Sun, L.G.; Xiao, X.H. Forensic DNA barcoding and bio-response studies of animal horn products used in traditional medicine. PLoS ONE 2013, 8, e55854. [Google Scholar] [CrossRef]
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of toxicology, exposure, potential health consequences, and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef]
- Pawlowski, J.; Audic, S.; Adl, S.; Bass, D.; Belbahri, L.; Berney, C.; Bowser, S.S.; Cepicka, I.; Decelle, J.; Dunthorn, M.; et al. CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 2012, 10, e1001419. [Google Scholar] [CrossRef]
- Newmaster, S.G.; Fazekas, A.J.; Ragupathy, S. DNA barcoding in land plants: Evaluation of rbcL in a multigene tiered approach. Can. J. Bot. 2006, 84, 335–341. [Google Scholar] [CrossRef]
- Kumeta, Y.; Maruyama, T.; Asama, H.; Yamamoto, Y.; Hakamatsuka, T.; Goda, Y. Species identification of Asini Corii Collas (donkey glue) by PCR amplification of cytochrome b gene. J. Nat. Med. 2014, 68, 181–185. [Google Scholar] [CrossRef]
- Chen, S. I19 Major achievements of traditional medicine in treating epidemic and chronic diseases. Biochem. Pharmacol. 2017, 139, 110. [Google Scholar] [CrossRef]
- Ni, Q.; Wang, J.; Li, E.-Q.; Zhao, A.-B.; Yu, B.; Wang, M.; Huang, C.-R. Study on the protective effect of the mixture of Shengmai powder and danshen decoction on the myocardium of diabetic cardiomyopathy in the rat model. Chin. J. Integr. Med. 2011, 17, 116–125. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, H.; Yuan, H.; Zhang, C.; Bao, G.; Zhao, Z.; Song, B.; Bao, L. Anti-inflammatory, diuretic and removing urinary calculus actions of Sanweijili powder. Mod. J. Integr. Tradit. Chin. West. Med. 2009, 23, 2766–2767. [Google Scholar]
- Ko, R. Safety of ethnic & imported herbal and dietary supplements. Clin. Toxicol. 2006, 44, 611–616. [Google Scholar]
Figure 1.
Pipeline of Full-length Multi-barcoding, analysis of SMS samples as an example. (a) sample preparation, (b) data analysis. (This figure was drawn by Peng Zhang).
Figure 1.
Pipeline of Full-length Multi-barcoding, analysis of SMS samples as an example. (a) sample preparation, (b) data analysis. (This figure was drawn by Peng Zhang).

Figure 2.
Analysis results on Sheng-Mai-San by full-length multi-barcoding. (a) Representative results of the raw materials; (b,c), results of the HSM and SMS, respectively. **, PCR cycles were 35 + 10; *, PCR cycles were 35. Panax sp., reads that cannot BLAST to certain species in genus of Panax; no-blast-hit 1, resulting reads that with Max. score < 400 or Identities < 90% for top hits in BLAST.
Figure 2.
Analysis results on Sheng-Mai-San by full-length multi-barcoding. (a) Representative results of the raw materials; (b,c), results of the HSM and SMS, respectively. **, PCR cycles were 35 + 10; *, PCR cycles were 35. Panax sp., reads that cannot BLAST to certain species in genus of Panax; no-blast-hit 1, resulting reads that with Max. score < 400 or Identities < 90% for top hits in BLAST.

Figure 3.
Percentage of different raw materials’ reads in ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA) amplicons of SMS. HMA/B/C and SMS1/2/3 were analyzed by FLMB, while Clone G1/G2/G3/G4 were analyzed by clone-dependent Sanger sequencing.
Figure 3.
Percentage of different raw materials’ reads in ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA) amplicons of SMS. HMA/B/C and SMS1/2/3 were analyzed by FLMB, while Clone G1/G2/G3/G4 were analyzed by clone-dependent Sanger sequencing.

Figure 4.
Analysis results on Sanwei-Jili-San by full-length multi-barcoding.

Figure 5.
Analysis of 462 resulting reads from the ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA) amplicon of RS3. (a) Part of stacked resulting reads; red boxes mark degenerate bases in tag-primers whose constitution shows in the smaller block below. (b) Damaged profile of protective bases, as the bases missing at both ends in (a). (c) Length distribution after primers trimming. (d) Mutation percentage at each loci (only shows those with percentage higher than 0.5%). (e) Species assignment results.
Figure 5.
Analysis of 462 resulting reads from the ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA) amplicon of RS3. (a) Part of stacked resulting reads; red boxes mark degenerate bases in tag-primers whose constitution shows in the smaller block below. (b) Damaged profile of protective bases, as the bases missing at both ends in (a). (c) Length distribution after primers trimming. (d) Mutation percentage at each loci (only shows those with percentage higher than 0.5%). (e) Species assignment results.

Figure 6.
Number of exogenous reads and exogenous species rise along with the increasing of sequencing depth. (a) Relations between exogenous reads and total resulting reads. (b) Relations between species detected and total resulting reads. ITS2: the second internal transcribed spacer of nuclear ribosomal DNA), CO1: cytochrome c oxidase subunit 1.
Figure 6.
Number of exogenous reads and exogenous species rise along with the increasing of sequencing depth. (a) Relations between exogenous reads and total resulting reads. (b) Relations between species detected and total resulting reads. ITS2: the second internal transcribed spacer of nuclear ribosomal DNA), CO1: cytochrome c oxidase subunit 1.

Figure 7.
Comparative analysis of different sequencing platforms applied to DNA barcoding. Lengths of amplicon fragments of five barcodes regions (ITS2, psbA-trnH, ITS, matK, rbcL) for five vegetable materials in two studied formulas, and lengths of CO1 for five animal materials (including crabs) have been shown as marks. For each sequencing platform, the dotted vertical line represents the read length while the solid transverse line represents the sequencing throughput. Though the number of raw reads obtained from the platform of PacBio is less than those of the other two NGS platforms, its advantages of long-read sequencing could maintain more original and effective information.
Figure 7.
Comparative analysis of different sequencing platforms applied to DNA barcoding. Lengths of amplicon fragments of five barcodes regions (ITS2, psbA-trnH, ITS, matK, rbcL) for five vegetable materials in two studied formulas, and lengths of CO1 for five animal materials (including crabs) have been shown as marks. For each sequencing platform, the dotted vertical line represents the read length while the solid transverse line represents the sequencing throughput. Though the number of raw reads obtained from the platform of PacBio is less than those of the other two NGS platforms, its advantages of long-read sequencing could maintain more original and effective information.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Preparation of the DNA samples and their information.
| Formula | Samples (and Its Biological Origin) | DNA ID | Notes |
|---|---|---|---|
| Sheng-Mai-San | Ginseng Radix et Rhizoma (Panax ginseng) | RS1 | Company A |
| RS2 | Company A | ||
| RS3 | Company A | ||
| Ophiopogonis Radix (Ophiopogon japonicus) | MD1 | ‘Zhe’ O. japonicus; Cixi, Zhejiang province; producing area | |
| MD2 | ‘Zhe’ O. japonicus; Xiangshan, Zhejiang province; wildness | ||
| MD3 | ‘Chuan’ O. japonicus; Santai, Sichuan province; Market B | ||
| MD4 | ‘Chuan’ O. japonicus; Sichuan province; Company C | ||
| Schisandrae Chinensis Fructus (Schisandra chinensis) | WW1 | Company C | |
| WW2 | Company C | ||
| WW3 | Company C | ||
| Sheng-Mai-San (SMS) | SMS1 | Mixed powder | |
| SMS2 | Mixed powder | ||
| SMS3 | Mixed powder | ||
| DNA mixture (HSM) | HMA | DNA Volume of MD1-RS1-WW1 = 3:3:2 | |
| HMB | DNA Volume of MD2-RS2-WW2 = 3:3:2 | ||
| HMC | DNA Volume of MD3-RS3-WW3 = 3:3:2 | ||
| Sanwei-Jili-San | Malvae Fructus (Malva veriticillata) | DK | Company D |
| Tribuli Fructus (Tribulus terrestris) | JL | Company D | |
| Chinese fresh-water crab (Eriocheir sinensis) | FH | A local supermarket | |
| Sanwei-Jili-San (SJS) | SJS1 | Mixed powder | |
| SJS2 | Mixed powder | ||
| SJS3 | Mixed powder | ||
| DNA mixture (JH) | JHA | DNA Volume of DK-JL-FH = 3:5:3 | |
| JHB | DNA Volume of DK-JL-FH = 3:5:3 | ||
| JHC | DNA Volume of DK-JL-FH = 3:5:3 |
Table 2.
Processing information for library A’s data generated by SMRT sequencing.
| Raw Data | Circular Consensus Sequence Filtration | Extraction | |
|---|---|---|---|
| Reads | 57,147 | 36,497 | 25,877 |
| Total bases | 1.784 × 109 | 19,308,858 | 14,117,133 |
| Data size | 24.4 GB | 21.3 MB | 13.7 MB |
| Mean length | 31,217 bp | 529 bp | 546 bp |
| Length range | 0~70 kb | 200~799 bp | 348~799 bp |
Table 3.
Amplicons’ information in different SMRT cells and effective rate for each amplicon.
| SMRT Cell | DNA ID | Pair of Tag-Primers # | Resulting Reads | No-Blast-Hit1 Resulting Reads | Effective Rate |
|---|---|---|---|---|---|
| Cell A | HM1 | T17 | 3255 | 0 | 100.00% |
| HM2 | T18 | 3748 | 0 | 100.00% | |
| HM3 | T19 | 4058 | 0 | 100.00% | |
| SMS1 | T13 | 2527 | 0 | 100.00% | |
| SMS2 | T14 | 3095 | 0 | 100.00% | |
| SMS3 | T15 | 4443 | 0 | 100.00% | |
| SMS1 | i13 | 603 | 0 | 100.00% | |
| SMS2 | i14 | 1030 | 0 | 100.00% | |
| SMS3 | i15 | 890 | 0 | 100.00% | |
| HMA | i17 | 557 | 0 | 100.00% | |
| HMB | i18 | 890 | 0 | 100.00% | |
| HMC | i19 | 778 | 0 | 100.00% | |
| Cell B | RS1 | i5 | 279 | 0 | 100.00% |
| RS2 | i6 | 439 | 0 | 100.00% | |
| RS3 | i7 | 462 | 0 | 100.00% | |
| WW1 | i9 | 431 | 0 | 100.00% | |
| WW2 | i10 | 706 | 0 | 100.00% | |
| WW3 | i11 | 686 | 0 | 100.00% | |
| MD1 | T1 | 725 | 0 | 100.00% | |
| MD2 | T2 | 578 | 0 | 100.00% | |
| MD3 | T3 | 749 | 0 | 100.00% | |
| MD4 | T4 | 794 | 1 | 99.87% | |
| MD1 ** | i1 | 268 | 2 | 99.25% | |
| MD2 ** | i2 | 512 | 7 | 98.63% | |
| MD3 ** | i3 | 568 | 14 | 97.54% | |
| MD4 ** | i4 | 302 | 8 | 97.35% | |
| WW1 | T9 | 1276 | 0 | 100.00% | |
| WW2 | T10 | 445 | 0 | 100.00% | |
| WW3 | T11 | 1846 | 0 | 100.00% | |
| RS1 | T5 | 911 | 0 | 100.00% | |
| RS2 | T6 | 988 | 0 | 100.00% | |
| RS3 | T7 | 1617 | 0 | 100.00% | |
| Cell C | MD3 * | i3 | 291 | 0 | 100.00% |
| JL | i39 | 777 | 0 | 100.00% | |
| DK | i40 | 1396 | 0 | 100.00% | |
| FH | C9 | 2067 | 0 | 100.00% | |
| SJS1 | i23 | 1977 | 0 | 100.00% | |
| SJS2 | i22 | 3091 | 0 | 100.00% | |
| SJS3 | i21 | 1559 | 0 | 100.00% | |
| SJS1 | C1 | 898 | 2 | 99.78% | |
| SJS2 | C2 | 1041 | 0 | 100.00% | |
| SJS3 | C3 | 324 | 0 | 100.00% | |
| JHC | C6 | 1074 | 7 | 99.35% | |
| JHC | i32 | 2182 | 0 | 100.00% | |
| JHB | i31 | 1751 | 0 | 100.00% | |
| JHA | i24 | 1348 | 0 | 100.00% | |
| Total | 60,232 | 41 | 99.93% | ||
Note: no-blast-hit 1, top hits with low Max. score (≤400) or low Identities (≤90%). **, PCR cycles were 35 + 10; *, PCR cycles were 35. #,‘T’ as psbA-trnH, ‘i’ as ITS2 (the second internal transcribed spacer of nuclear ribosomal DNA), ‘C’ as CO1 (cytochrome c oxidase subunit 1).
Table 4.
Cases of different sequencing approaches applied to DNA barcoding of herbs.
| Briefing | Reference | |
|---|---|---|
| Sanger sequencing | The trnH-psbA could distinguish 18 species of Polygonaceae and their adulterants including 10 species that recorded in Chinese pharmacopoeia. | [10] |
| The discrimination ability of ITS2, a most suitable region for DNA barcoding, were tested by more than 6600 plant samples and its successful identification rate was 92.7% at the species level. | [8] | |
| Most (26/44) of the North American herbal products tested contained DNA barcodes from plant species not listed on the labels. Sequences of different species had yield up from the same sample product. | [9] | |
| DNA barcoding was used to authenticate the components of antler powder in the market, while a few samples containing multi-species were analyzed by cloning method. | [14] | |
| Short-read NGS | Amplicons of trnL and 16S from 15 TCM samples were sequenced in platform of Roche GS Junior, and over 49,000 amplicon sequence reads were generated. Many trnL sequence reads could not been precisely identified (most were just assigned to genera level), due to the limitation of read-length and reference database. | [17] |
| HTS data for 30 Liuwei-Dihuang-Wan samples were generated based on 454 GS FLX Titanium sequencing. On averages, 3 and 2.4 prescribed species could be detected from a sample based on ITS2 and trnL, respectively. Vigna genus, a possible contaminated species, was detected in all three reference samples. | [34] | |
| Long-read NGS | A total of 3703 and 4810 CCS reads from two reference and three commercial Yimu-Wan samples were mapped to the ITS2 and psbA-trnH regions, respectively. SMRT sequencing provides an affordable way to monitor the legality and safety of traditional patent medicines. | [42] |
| A comprehensive quality evaluation system for herbal medicine was established, by combining two genetic-based approaches—third-generation sequencing and denaturing gradient gel electrophoresis, with analytical chemistry approaches. | [52] | |
| SMS and SJS have been analyzed through FLMB by an adequate and well-controlled methodological analysis, which shows perfect interpretation for DNA barcoding. | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, P.; Liu, C.; Zheng, X.; Wu, L.; Liu, Z.; Liao, B.; Shi, Y.; Li, X.; Xu, J.; Chen, S. Full-Length Multi-Barcoding: DNA Barcoding from Single Ingredient to Complex Mixtures. Genes 2019, 10, 343. https://doi.org/10.3390/genes10050343
AMA Style
Zhang P, Liu C, Zheng X, Wu L, Liu Z, Liao B, Shi Y, Li X, Xu J, Chen S. Full-Length Multi-Barcoding: DNA Barcoding from Single Ingredient to Complex Mixtures. Genes. 2019; 10(5):343. https://doi.org/10.3390/genes10050343
Chicago/Turabian StyleZhang, Peng, Chunsheng Liu, Xiasheng Zheng, Lan Wu, Zhixiang Liu, Baosheng Liao, Yuhua Shi, Xiwen Li, Jiang Xu, and Shilin Chen. 2019. "Full-Length Multi-Barcoding: DNA Barcoding from Single Ingredient to Complex Mixtures" Genes 10, no. 5: 343. https://doi.org/10.3390/genes10050343
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.