Functional Analysis of Promoter Variants in Genes Involved in Sex Steroid Action, DNA Repair and Cell Cycle Control

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Promoter Polymorphisms
2.2. Accession Numbers
2.3. Linkage Disequilibrium Analysis and Haplotype Estimation
2.4. Subcloning and Reporter Plasmid Construction
2.5. In Silico Prediction of Putative Transcription Factor Binding Sites
2.6. Cell Culture
2.7. Transient Transfection and Luciferase Reporter Assays
2.8. Statistical Analysis
2.9. Electrophoretic Mobility Shift Assay
2.10. Functional Annotation Using Public Databases
3. Results
3.1. Identification of Regulatory Single Nucleotide Polymorphisms and Haplotype Estimation
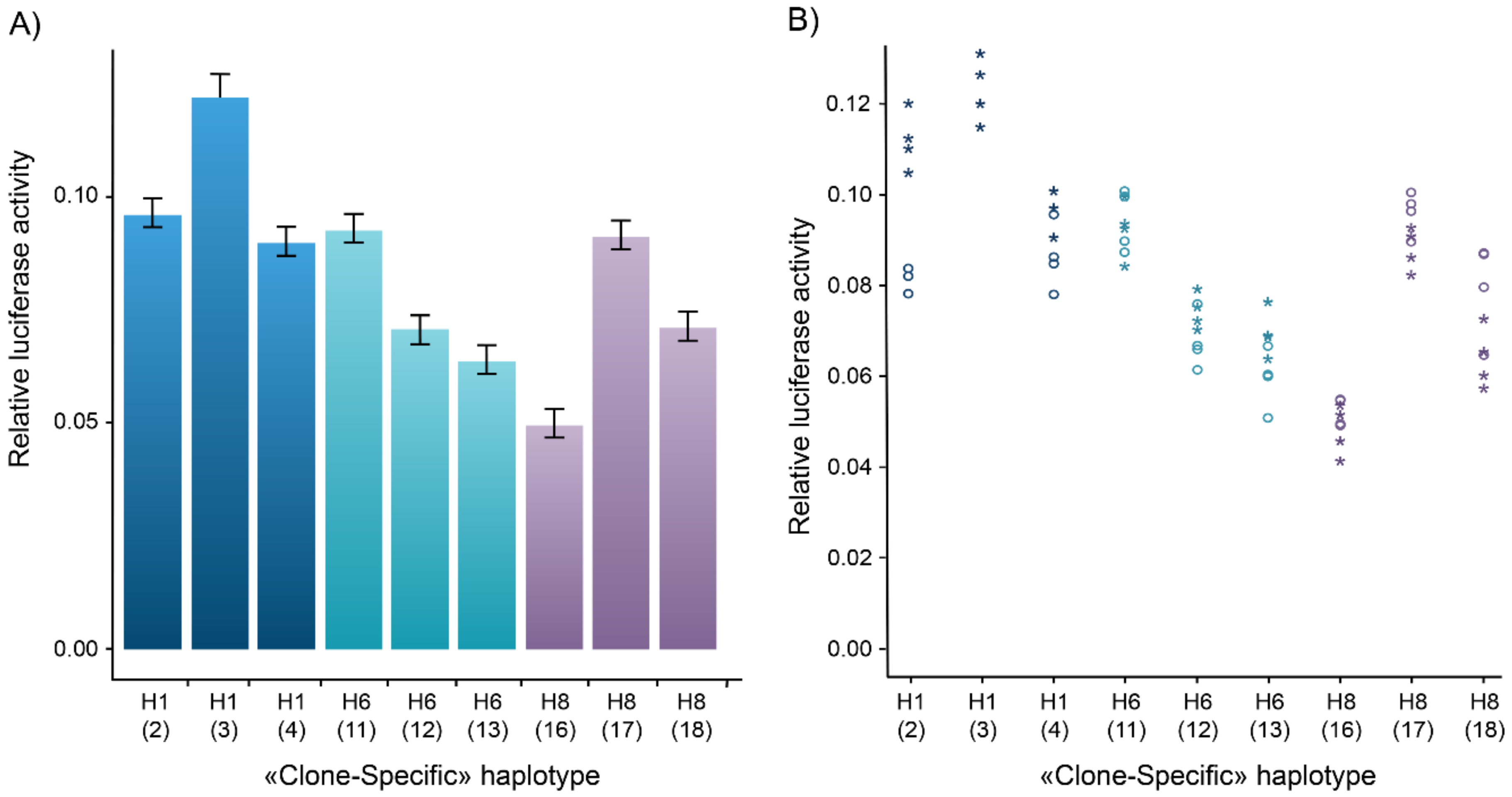
3.2. Gene Reporter Assays and Inter-Clone Variability
3.3. Transcriptional Activity of Major Regulatory Haplotypes
3.4. Sex Steroid Hormone Action Genes
3.5. DNA Repair and Cell Cycle Control Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Statements
References
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two Decades after BRCA: Setting Paradigms in Personalized Cancer Care and Prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Pharoah, P.D.; Michailidou, K.; Tyrer, J.; Brook, M.N.; Bolla, M.K.; Wang, Q.; Dennis, J.; Dunning, A.M.; Shah, M.; et al. Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst. 2015, 107, djv036. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Simard, J.; Rommens, J.; Couch, F.J.; Shattuck-Eidens, D.; Neuhausen, S.; Merajver, S.; Thorlacius, S.; Offit, K.; Stoppa-Lyonnet, D.; et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 1996, 12, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Børresen, A.L.; Andersen, T.; Garber, J.; Barbier-Piraux, N.; Thorlacius, S.; Eyfjörd, J.; Ottestad, L.; Smith-Sørensen, B.; Hovig, E.; Malkin, D.; et al. Screening for germline TP53 mutations in breast cancer patients. Cancer Res. 1992, 52, 3234–3236. [Google Scholar] [PubMed]
- Le Calvez-Kelm, F.; Lesueur, F.; Damiola, F.; Vallée, M.; Voegele, C.; Babikyan, D.; Durand, G.; Forey, N.; McKay-Chopin, S.; Robinot, N.; et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: Results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 2011, 13, R6. [Google Scholar] [CrossRef] [PubMed]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Foulkes, W.D.; Tischkowitz, M. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 1651–1652. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Tyrer, J.; Dunning, A.M.; Easton, D.F.; Ponder, B.A.; SEARCH Investigators. Association between common variation in 120 candidate genes and breast cancer risk. PLoS Genet. 2007, 3, e42. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; Fletcher, O.; Michailidou, K.; Turnbull, C.; Schmidt, M.K.; Dicks, E.; Dennis, J.; Wang, Q.; Humphreys, M.K.; Luccarini, C.; et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat. Genet. 2012, 44, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Pokholok, D.K.; Kwan, T.; Grundberg, E.; Morcos, L.; Verlaan, D.J.; Le, J.; Koka, V.; Lam, K.C.; Gagné, V.; et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat. Genet. 2009, 41, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.L.; Monteiro, A.N.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Orli, B. Functional annotation of susceptibility loci identified by COGS. Nat. Genet. 2013, 3. [Google Scholar] [CrossRef]
- Meyer, K.B.; O’Reilly, M.; Michailidou, K.; Carlebur, S.; Edwards, S.L.; French, J.D.; Prathalingham, R.; Dennis, J.; Bolla, M.K.; Wang, Q.; et al. Fine-Scale Mapping of the FGFR2 Breast Cancer Risk Locus: Putative Functional Variants Differentially Bind FOXA1 and E2F1. Am. J. Hum. Genet. 2013, 93, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- French, J.D.; Ghoussaini, M.; Edwards, S.L.; Meyer, K.B.; Michailidou, K.; Ahmed, S.; Khan, S.; Maranian, M.J.; O’Reilly, M.; Hillman, K.M.; et al. Functional Variants at the 11q13 Risk Locus for Breast Cancer Regulate Cyclin D1 Expression through Long-Range Enhancers. Am. J. Hum. Genet. 2013, 92, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, Y.; Soucy, P.; Adoue, V.; Michailidou, K.; Canisius, S.; Lemaçon, A.; Droit, A.; Andrulis, I.L.; Anton-Culver, H.; Arndt, V.; et al. Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget 2016, 7, 80140–80163. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, Y.; Soucy, P.; Kuchenbaeker, K.B.; Pastinen, T.; Droit, A.; Lemaçon, A.; Adlard, J.; Aittomäki, K.; Andrulis, I.L.; Arason, A.; et al. Association of breast cancer risk in BRCA1 and BRCA2 mutation carriers with genetic variants showing differential allelic expression: Identification of a modifier of breast cancer risk at locus 11q22.3. Breast Cancer Res. Treat. 2017, 161, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V.; Wray, G.A. Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol. 2002, 19, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 9, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Ulrich, C.M.; Potter, J.D. Polymorphisms in DNA Repair Genes and Associations with Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1513–1530. [Google Scholar]
- Wang, B. BRCA1 tumor suppressor network: Focusing on its tail. Cell Biosci. 2012, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Dausset, J.; Cann, H.; Cohen, D.; Lathrop, M.; Lalouel, J.M.; White, R. Centre d’etude du polymorphisme humain (CEPH): Collaborative genetic mapping of the human genome. Genomics 1990, 6, 575–577. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- De Bakker, P.I. Selection and evaluation of Tag-SNPs using Tagger and HapMap. Cold Spring Harb. Protoc. 2009, 2009, pdb-ip67. [Google Scholar] [CrossRef] [PubMed]
- Matthew, S.; Smith, N.K.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 21, E2191–E2199. [Google Scholar] [CrossRef] [PubMed]
- Lemaçon, A.; Joly Beauparlant, C.; Soucy, P.; Allen, J.; Easton, D.; Kraft, P.; Simard, J.; Droit, A. VEXOR: An integrative environment for prioritization of functional variants in fine-mapping analysis. Bioinformatics 2017, 33, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Hoogendoorn, B.; Coleman, S.L.; Guy, C.A.; Smith, K.; Bowen, T.; Buckland, P.R.; O’Donovan, M.C. Functional analysis of human promoter polymorphisms. Hum. Mol. Genet. 2003, 12, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Hoogendoorn, B.; Coleman, S.L.; Guy, C.A.; Smith, S.K.; O’Donovan, M.C.; Buckland, P.R. Functional analysis of polymorphisms in the promoter regions of genes on 22q11. Hum. Mutat. 2004, 24, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Menendez, D.; Resnick, M.A.; French, J.E.; Janardhan, K.S.; Jetten, A.M. RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer Res. 2012, 72, 5080–5090. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A. Genome wide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010, 363, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Bonte, D.; Lindvall, C.; Liu, H.; Dykema, K.; Furge, K.; Weinreich, M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 2008, 10, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Choschzick, M.; Lebeau, A.; Marx, A.H.; Tharun, L.; Terracciano, L.; Heilenkötter, U.; Jaenicke, F.; Bokemeyer, C.; Simon, R.; Sauter, G.; et al. Overexpression of cell division cycle 7 homolog is associated with gene amplification frequency in breast cancer. Hum. Pathol. 2010, 41, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, A.; Tenca, P.; Sola, F.; Carpani, D.; Brotherton, D.; Albanese, C.; Santocanale, C. Cdc7 Inhibition Reveals a p53-Dependent Replication Checkpoint That Is Defective in Cancer Cells. Cancer Res. 2004, 64, 7110–7116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Nakanishi, G.; Kurebayashi, S.; Yoshino, K.; Perantoni, A.; Kim, Y.S.; Jetten, A.M. Characterization of Glis2, a novel gene encoding a Gli-related, Kruppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J. Biol. Chem. 2002, 277, 10139–10149. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kang, H.S.; Jetten, A.M. The Krüppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/β-catenin signaling pathway. FEBS Lett. 2007, 581, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Ben-Ze’ev, A. Beta-Catenin signaling in biological control and cancer. J. Cell. Biochem. 2007, 102, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Barker, N.; Clevers, H. You Wnt some, you lose some: Oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 2003, 13, 28–33. [Google Scholar] [CrossRef]
- Akiyama, T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev. 2000, 11, 273–282. [Google Scholar] [CrossRef]
- Cowper-Sal lari, R.; Zhang, X.; Wright, J.B.; Bailey, S.D.; Cole, M.D.; Eeckhoute, J.; Moore, J.H.; Lupien, M. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012, 44, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Chenevix-Trench, G.; Milne, R.L.; Antoniou, A.C.; Couch, F.J.; Easton, D.F.; Goldgar, D.E. CIMBA; An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: The Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res. 2007, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Jetten, A.M. RAP80 and RNF8, key players in the recruitment of repair proteins to DNA damage sites. Cancer Lett. 2008, 271, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Domchek, S.M.; Wan, F.; Friebel, T.M.; Tran, T.V.; Singer, C.F.; Tea, M.K.; Blum, J.L.; Tung, N.; et al. Modification of BRCA1-Associated Breast and Ovarian Cancer Risk by BRCA1 Interacting Genes. Cancer Res. 2011, 71, 5792–5805. [Google Scholar] [CrossRef] [PubMed]
- Lenhart, J.S.; Schroeder, J.W.; Walsh, B.W.; Simmons, L.A. DNA Repair and Genome Maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 2012, 76, 3530–5641. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Brosh, R.M., Jr. Functional Analyses of Human DNA Repair Proteins Important for Aging and Genomic Stability Using Yeast Genetics. DNA Repair. (Amst.) 2012, 11, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Falvella, F.S.; Galvan, A.; Colombo, F.; Frullanti, E.; Pastorino, U.; Dragani, T.A. Promoter polymorphisms and transcript levels of nicotinic receptor CHRNA5. J. Natl. Cancer Inst. 2010, 102, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Idelman, G.; Taylor, J.G.; Tongbai, R.; Chen, R.A.; Haggerty, C.M.; Bilke, S.; Chanock, S.J.; Gardner, K. Functional profiling of uncommon VCAM1 promoter polymorphisms prevalent in African American populations. Hum. Mutat. 2007, 28, 824–829. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Haplotype Designation | Promotor Activity | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCF7 | HELA | |||||||||||||||||
| Renilla Luciferase Plasmid | Number of Experiments | Number of Clones | Activity Relative to Control | Relative Allele Activity to H1 | Renilla Luciferase Plasmid | Number of Experiments | Number of Clones | Activity Relative to Control | Relative Allele Activity to H1 | |||||||||
| Fold Induction | Fold Induction | p-value | Fold Induction | Fold Induction | p-Value | |||||||||||||
| PROMOTERS WITH DIFFERENTIAL HAPLOTYPE EXPRESSION IN TWO CELL LINES | ||||||||||||||||||
| SEX STEROID HORMONE ACTION GENES | ||||||||||||||||||
| ESR1 | pRL-null | 5 | pRL-null | 4 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 7.7 | 1.000 | - | 1 | 6.3 | 1.000 | - | ||||||||||
| H2 | 1 | 2.3 | 0.293 | <2.2 × 10−16 | ** | 1 | 3.6 | 0.564 | <2.2 × 10−16 | ** | ||||||||
| H3 | 1 | 2.5 | 0.326 | <2.2 × 10−16 | ** | 1 | 3.7 | 0.579 | <2.2 × 10−16 | ** | ||||||||
| H4 | 1 | 2.1 | 0.277 | <2.2 × 10−16 | ** | 1 | 3.1 | 0.496 | <2.2 × 10−16 | ** | ||||||||
| ESR2 | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 2.0 | 1.000 | - | 1 | 2.8 | 1.000 | - | ||||||||||
| H3 | 1 | 5.0 | 2.544 | 2.9 × 10−04 | ** | 1 | 7.3 | 2.582 | 8.3 × 10−11 | ** | ||||||||
| H6 | 1 | 6.0 | 3.053 | 9.0 × 10−06 | ** | 1 | 8.4 | 2.967 | 1.5 × 10−12 | ** | ||||||||
| FOXA1 | pRL-CMV | 1 | pRL-null | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 2 | 69.0 | 1.000 | - | 1 | 5.8 | 1.000 | - | ||||||||||
| H2 | 2 | 65.0 | 0.942 | 1.0 | 1 | 4.0 | 0.699 | 3.0 × 10−10 | ** | |||||||||
| H3 | 3 | 63.2 | 0.798 | 5.9 × 10−04 | ** | 1 | 3.6 | 0.619 | 3.1 × 10−13 | ** | ||||||||
| H4 | 2 | 64.5 | 0.935 | 1.0 | 1 | 5.4 | 0.938 | 3.8 × 10−01 | ||||||||||
| H5 | 2 | 52.6 | 0.762 | 3.7 × 10−06 | ** | 1 | 4.5 | 0.781 | 6.1 × 10−07 | ** | ||||||||
| H6 | 2 | 62.8 | 0.909 | 2.9 × 10−01 | 1 | 4.6 | 0.806 | 6.9 × 10−06 | ** | |||||||||
| DNA REPAIR & CELL CYCLE CONTROL GENES | ||||||||||||||||||
| ATR | pRL-CMV | 4 | pRL-CMV | 4 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 164.0 | 1.000 | - | 1 | 133.3 | 1.000 | - | ||||||||||
| H2 | 1 | 116.9 | 0.729 | 3.7 × 10−07 | ** | 1 | 103.4 | 0.775 | 1.2 × 10−05 | ** | ||||||||
| CDC7 | pRL-null | 1 | pRL-null | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 131.5 | 1.000 | - | 3 | 193.2 | 1.000 | - | ||||||||||
| H2 | 4 | 57.2 | 0.435 | <2.2 × 10−16 | ** | 4 | 146.0 | 0.756 | <2.2 × 10−16 | ** | ||||||||
| H3 | 3 | 132.8 | 1.011 | 1.0 | 3 | 168.3 | 0.871 | 1.6 × 10−12 | ** | |||||||||
| MRE11A | pRL-CMV | 1 | pRL-null | 1 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 5 | 116.7 | 1.000 | - | 3 | 7.8 | 1.000 | - | ||||||||||
| H4 | 5 | 115.5 | 0.990 | 1.0 | 3 | 5.9 | 0.753 | 2.5 × 10−12 | ** | |||||||||
| H6 | 3 | 115.0 | 0.985 | 1.0 | - | - | ||||||||||||
| H7 | 3 | 132.9 | 1.139 | 4.5 × 10−03 | * | 3 | 8.6 | 1.097 | 9.6 × 10−04 | ** | ||||||||
| H8 | 3 | 115.1 | 0.987 | 1.0 | 3 | 6.6 | 0.844 | 2.3 × 10−07 | ** | |||||||||
| PALB2 | pRL-CMV | 2 | pRL-CMV | 3 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 34.1 | 1.000 | - | 1 | 50.0 | 1.000 | - | ||||||||||
| H6 | 3 | 25.1 | 0.737 | 1.8 × 10−12 | ** | 1 | 40.1 | 0.803 | 9.9 × 10−06 | ** | ||||||||
| H8 | 3 | 23.5 | 0.689 | 2.7 × 10−15 | ** | 1 | 40.2 | 0.803 | 1.0 × 10−05 | ** | ||||||||
| RAD51c | pRL-null | 1 | pRL-CMV | 3 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 2 | 346.0 | 1.000 | - | 1 | 43.7 | 1.000 | - | ||||||||||
| H2 | 2 | 274.7 | 0.794 | 1.9 × 10−08 | ** | 1 | 56.0 | 1.280 | 1.4 × 10−06 | ** | ||||||||
| H3 | 1 | 240.5 | 0.695 | 6.4 × 10−10 | ** | 1 | 49.5 | 1.130 | 2.5 × 10−02 | |||||||||
| H4 | 2 | 256.4 | 0.741 | 3.1 × 10−10 | ** | 1 | 42.7 | 0.976 | 1.0 | |||||||||
| UIMC1 | pRL-null | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 38.3 | 1.000 | - | 1 | 25.9 | 1.000 | - | ||||||||||
| H2 | 1 | 43.1 | 1.125 | 4.0 × 10−01 | 1 | 26.8 | 1.035 | 9.2 × 10−01 | ||||||||||
| H3 | 1 | 26.5 | 0.693 | 2.3 × 10−03 | * | 1 | 16.9 | 0.651 | 1.9 × 10−10 | ** | ||||||||
| H4 | 1 | 31.7 | 0.828 | 1.3 × 10−01 | 1 | 23.0 | 0.889 | 8.3 × 10−03 | * | |||||||||
| PROMOTERS WITH DIFFERENTIAL HAPLOTYPE EXPRESSION IN ONE CELL LINE | ||||||||||||||||||
| SEX STEROID HORMONE ACTION GENES | ||||||||||||||||||
| PGR | pRL-CMV | 3 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 2.3 | 1.000 | - | 1 | 1.4 | 1.000 | - | ||||||||||
| H2 | 1 | 2.1 | 0.909 | 5.3 × 10−01 | 1 | 3.9 | 2.871 | <2.2 × 10−16 | ** | |||||||||
| H6 | 1 | 2.1 | 0.938 | 1.0 | 1 | 3.9 | 2.803 | <2.2 × 10−16 | ** | |||||||||
| H7 | 1 | 2.3 | 1.001 | 1.0 | 1 | 3.8 | 2.731 | <2.2 × 10−16 | ** | |||||||||
| H10 | 1 | 2.3 | 1.009 | 1.0 | 1 | 4.4 | 3.184 | <2.2 × 10−16 | ** | |||||||||
| DNA REPAIR & CELL CYCLE CONTROL GENES | ||||||||||||||||||
| BABAM1 | pRL-CMV | 4 | pRL-CMV | 4 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 100.0 | 1.000 | - | 1 | 19.0 | 1.000 | - | ||||||||||
| H2 | 1 | 127.0 | 1.271 | 5.6 × 10−02 | 1 | 17.1 | 0.896 | 4.2 × 10−02 | ||||||||||
| H3 | 1 | 134.8 | 1.348 | 1.1 × 10−02 | 1 | 16.2 | 0.852 | 2.8 × 10−03 | * | |||||||||
| BRCC3 | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 24.8 | 1.000 | - | 1 | 104.3 | 1.000 | - | ||||||||||
| H2 | 1 | 30.4 | 1.226 | 1.0 × 10−04 | ** | 1 | 128.7 | 1.234 | 5.0 × 10−02 | |||||||||
| H3 | 1 | 17.7 | 0.715 | 3.3 × 10−06 | ** | 1 | 90.5 | 0.867 | 4.8 × 10−01 | |||||||||
| H4 | 1 | 24.3 | 0.979 | 1.0 | 1 | 117.5 | 1.127 | 5.4 × 10−01 | ||||||||||
| BRE | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 175.9 | 1.000 | - | 1 | 10.9 | 1.000 | - | ||||||||||
| H2 | 2 | 91.8 | 0.522 | 7.8 × 10−08 | ** | 1 | 11.0 | 1.011 | 1.0 | |||||||||
| H3 | 3 | 117.9 | 0.670 | 2.7 × 10−05 | ** | 1 | 5.1 | 0.467 | 1.2 × 10−02 | |||||||||
| H4 | 2 | 64.3 | 0.365 | 4.4 × 10−12 | ** | 1 | 6.0 | 0.555 | 4.6 × 10−02 | |||||||||
| H5 | 2 | 160.8 | 0.915 | 1.0 | 1 | 3.4 | 0.311 | 8.9 × 10−04 | ** | |||||||||
| NELFB | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 1047.8 | 1.000 | - | 1 | 205.7 | 1.000 | - | ||||||||||
| H7 | 1 | 982.7 | 0.938 | 3.0 × 10−01 | 1 | 222.1 | 1.080 | 5.6 × 10−03 | * | |||||||||
| NBN | pRL-CMV | 3 | pRL-null | 1 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 68.5 | 1.000 | - | 3 | 8.5 | 1.000 | - | ||||||||||
| H2 | 3 | 75.3 | 1.099 | 2.6 × 10−02 | 3 | 13.4 | 1.577 | <2.2 × 10−16 | ** | |||||||||
| H3 | 3 | 67.1 | 0.979 | 1.0 | 3 | 14.7 | 1.729 | <2.2 × 10−16 | ** | |||||||||
| H4 | 3 | 41.3 | 0.603 | <2.2 × 10−16 | ** | 3 | 9.1 | 1.069 | 9.5 × 10−02 | |||||||||
| RAD51 | pRL-CMV | 1 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 235.4 | 1.000 | - | 1 | 163.5 | 1.000 | - | ||||||||||
| H5 | 2 | 259.8 | 1.103 | 3.0 × 10−03 | * | 1 | 162.1 | 0.992 | 1.0 | |||||||||
| H6 | 3 | 237.2 | 1.007 | 1.0 | 1 | 137.2 | 0.839 | 1.3 × 10−03 | * | |||||||||
| RNF8 | pRL-CMV | 2 | ||||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | pRL-CMV | 2 | 1.0 | - | |||||||||||
| H1 | 1 | 7.4 | 1.000 | - | 1 | 3.1 | 1.000 | - | ||||||||||
| H3 | 1 | 8.0 | 1.081 | 9.5 × 10−01 | 1 | 3.2 | 1.035 | 1.0 | ||||||||||
| H5 | 1 | 8.2 | 1.116 | 3.9 × 10−01 | 1 | 3.1 | 1.000 | 1.0 | ||||||||||
| H6 | 1 | 5.8 | 0.792 | 1.7 × 10−02 | 1 | 3.1 | 1.019 | 1.0 | ||||||||||
| H10 | 1 | 5.5 | 0.744 | 2.5 × 10−03 | * | 1 | 3.0 | 0.987 | 1.0 | |||||||||
| RPS6KA2 | pRL-null | 3 | pRL-null | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 6 | 9.3 | 1.000 | - | 6 | 9.0 | 1.000 | - | ||||||||||
| H2 | 5 | 8.5 | 0.920 | 1.0 | 5 | 8.2 | 0.908 | 3.9 × 10−05 | ** | |||||||||
| H3 | 6 | 10.0 | 1.088 | 9.7 × 10−01 | 6 | 10.8 | 1.194 | <2.2 × 10−16 | ** | |||||||||
| PROMOTERS WITH NO DIFFERENTIAL HAPLOTYPE EXPRESSION | ||||||||||||||||||
| DNA REPAIR & CELL CYCLE CONTROL GENES | ||||||||||||||||||
| ATM | pRL-CMV | 3 | pRL-CMV | 3 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 366.3 | 1.000 | - | 1 | 295.3 | 1.000 | - | ||||||||||
| H2 | 1 | 375.8 | 1.026 | 1.0 | 1 | 328.3 | 1.112 | 3.8 × 10−02 | ||||||||||
| H3 | 1 | 346.9 | 0.947 | 5.9 × 10−01 | 1 | 309.4 | 1.048 | 6.0 × 10−01 | ||||||||||
| BRIP1 | pRL-null | 1 | pRL-CMV | 1 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 3 | 68.8 | 1.000 | - | 3 | 7.3 | 1.000 | - | ||||||||||
| H2 | 3 | 69.9 | 1.016 | 1.0 | 3 | 6.9 | 1.065 | 1.1 × 10−02 | ||||||||||
| H4 | 3 | 63.7 | 0.926 | 3.6 × 10−01 | 3 | 6.9 | 0.944 | 2.5 × 10−02 | ||||||||||
| H2AFX | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 273.9 | 1.000 | - | 1 | 250.4 | 1.000 | - | ||||||||||
| H2 | 1 | 276.8 | 1.011 | 1.0 | 1 | 242.0 | 0.966 | 5.8 × 10−01 | ||||||||||
| H3 | 1 | 302.1 | 1.103 | 3.0 × 10−02 | 1 | 271.9 | 1.086 | 2.3 × 10−02 | ||||||||||
| UBE2N | pRL-CMV | 2 | pRL-CMV | 2 | ||||||||||||||
| pGL3-Basic | 1 | 1.0 | - | 1 | 1.0 | - | ||||||||||||
| H1 | 1 | 2.0 | 1.000 | - | 1 | 3.4 | 1.000 | - | ||||||||||
| H4 | 1 | 2.1 | 1.076 | 3.6 × 10−01 | 1 | 3.3 | 0.763 | 1.0 | ||||||||||
| H5 | 1 | 2.0 | 1.019 | 1.0 | 1 | 3.7 | 0.875 | 3.2 × 10−01 | ||||||||||
| H6 | 1 | 1.9 | 0.925 | 3.7 × 10−01 | 1 | 3.3 | 0.771 | 1.0 | ||||||||||
| * p < 0.01 | ||||||||||||||||||
| ** p < 0.001 | ||||||||||||||||||
| Gene Symbol | Variant # | Observed Variation | Rs ID * | EMSA Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Altered TFBS (MatInspector) | Gel Shift | Super Shift | ||||||||||
| Effect | Name of TFBS | Observed Binding | Antibody (Santa Cruz) | Observed Protein Binding | ||||||||
| SEX STEROID HORMONE ACTION GENES | ||||||||||||
| ESR1 | ||||||||||||
| v1 | insAA | rs75311867 | GAIN | Myelin Transcription factor | YES | Myt1 (N-13) | NO | |||||
| ESR2 | ||||||||||||
| v4 | T>G | rs1271572 | LOOSE | Poz (BTB) and AT hook containing zinc finger | YES | PaTZ1 (G-13) | NO | |||||
| v5 | insG | rs66615803 | LOOSE | Zebrafish PAX9 binding sites | NO | |||||||
| LOOSE | GLIS family zinc finger 3, Gli-similar 3 | NO | ||||||||||
| GAIN | MYC-associated zinc finger protein related transcription factor | NO | ||||||||||
| GAIN | Zinc finger transcription factor, Zic family member 2 (odd-paired homolog, Drosophila) | NO | ||||||||||
| FOXA1 | ||||||||||||
| v5 | delAGA | rs35237183 | GAIN | Forkhead domain factors | YES | HNF3-alpha | NO | |||||
| v6 | C>A | rs10145379 | GAIN | GC-Box factors SP1/GC | NO | |||||||
| LOOSE | DM domain-containing transcription factors | NO | ||||||||||
| LOOSE | Bromodomain and PHD domain transcription | NO | ||||||||||
| DNA REPAIR & CELL CYCLE CONTROL GENES | ||||||||||||
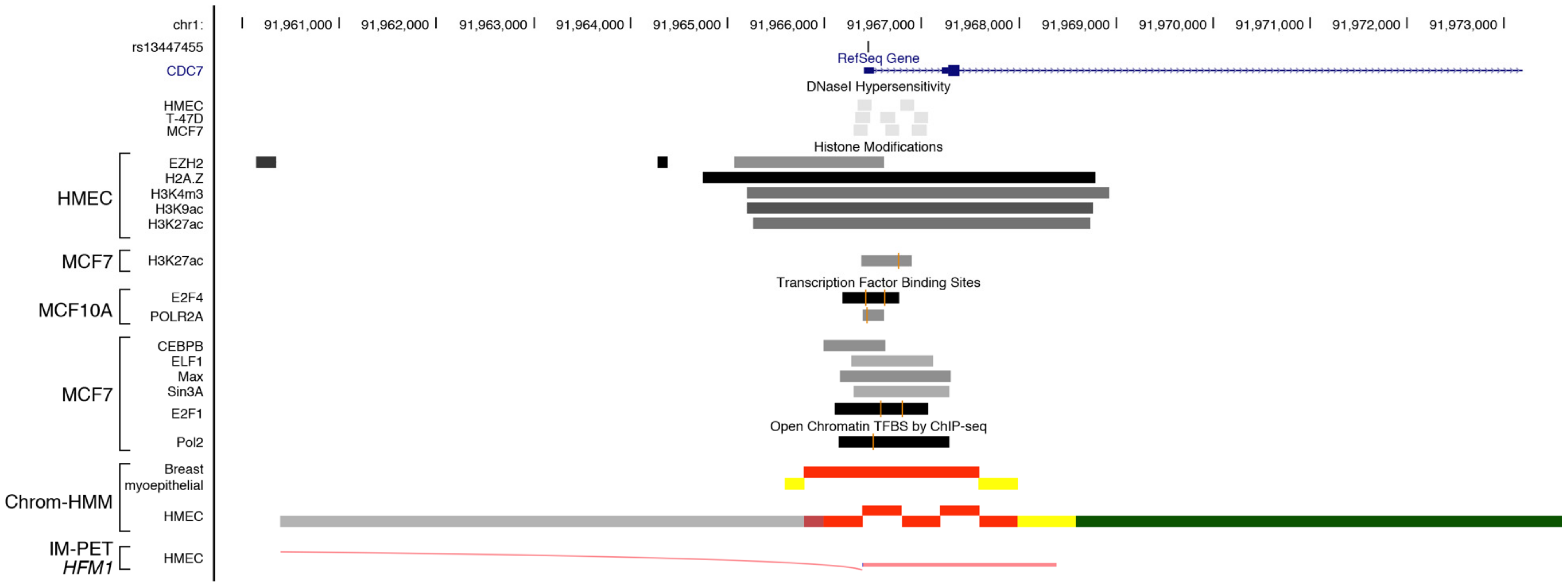
| CDC7 | ||||||||||||
| v7 | C>T | rs13447450 | NO CHANGE | |||||||||
| v10 | A>G | rs13447455 | GAIN | Paired box factor 5 | YES | PAX5 (C-20) | NO | |||||
| GAIN | Wilms tumor 1 | YES | WT1 (C-19) | NO | ||||||||
| GAIN | GLi-similar zinc finger | YES | GLIS2 (E-17) | YES | ||||||||
| LOOSE | Myeloid zinc finger | NO | ||||||||||
| UIMC1 | ||||||||||||
| v7 | T>G | rs7726380 | GAIN | p53 tumor suppressors | YES | P53 (Pab1801)X | NO | |||||
| LOOSE | POU Class 2 homeobox 1 | YES | OCT-1 (C-21) | NO | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdi, Y.; Leclerc, M.; Dumont, M.; Dubois, S.; Tranchant, M.; Reimnitz, G.; Soucy, P.; Cassart, P.; Ouimet, M.; Sinnett, D.; et al. Functional Analysis of Promoter Variants in Genes Involved in Sex Steroid Action, DNA Repair and Cell Cycle Control. Genes 2019, 10, 186. https://doi.org/10.3390/genes10030186
Hamdi Y, Leclerc M, Dumont M, Dubois S, Tranchant M, Reimnitz G, Soucy P, Cassart P, Ouimet M, Sinnett D, et al. Functional Analysis of Promoter Variants in Genes Involved in Sex Steroid Action, DNA Repair and Cell Cycle Control. Genes. 2019; 10(3):186. https://doi.org/10.3390/genes10030186
Chicago/Turabian StyleHamdi, Yosr, Martin Leclerc, Martine Dumont, Stéphane Dubois, Martine Tranchant, Guy Reimnitz, Penny Soucy, Pauline Cassart, Manon Ouimet, Daniel Sinnett, and et al. 2019. "Functional Analysis of Promoter Variants in Genes Involved in Sex Steroid Action, DNA Repair and Cell Cycle Control" Genes 10, no. 3: 186. https://doi.org/10.3390/genes10030186