An Examination of KCNE1 Mutations and Common Variants in Chronic Tinnitus

Abstract
:1. Introduction
2. Results and Discussion

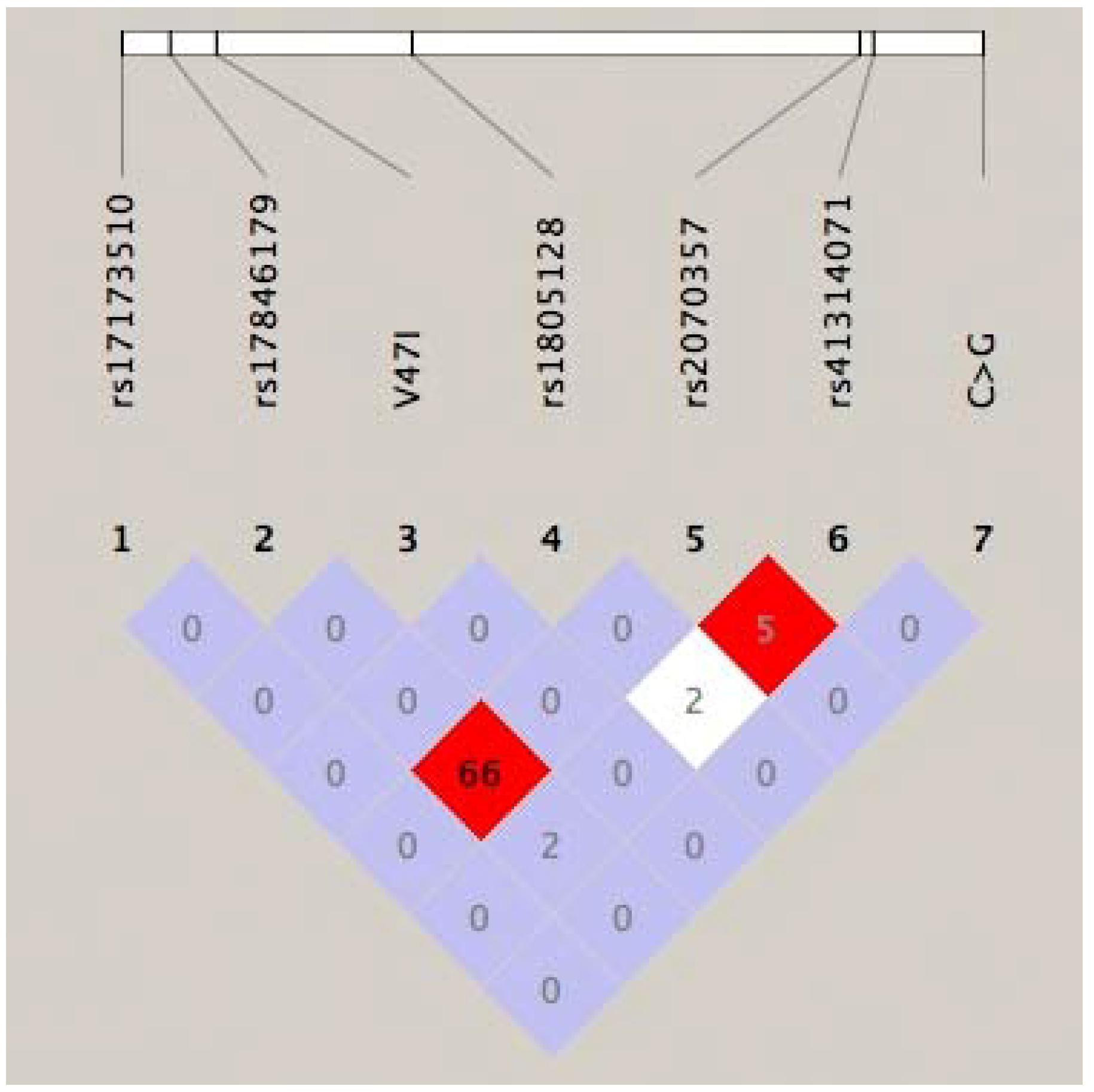{kind=link}
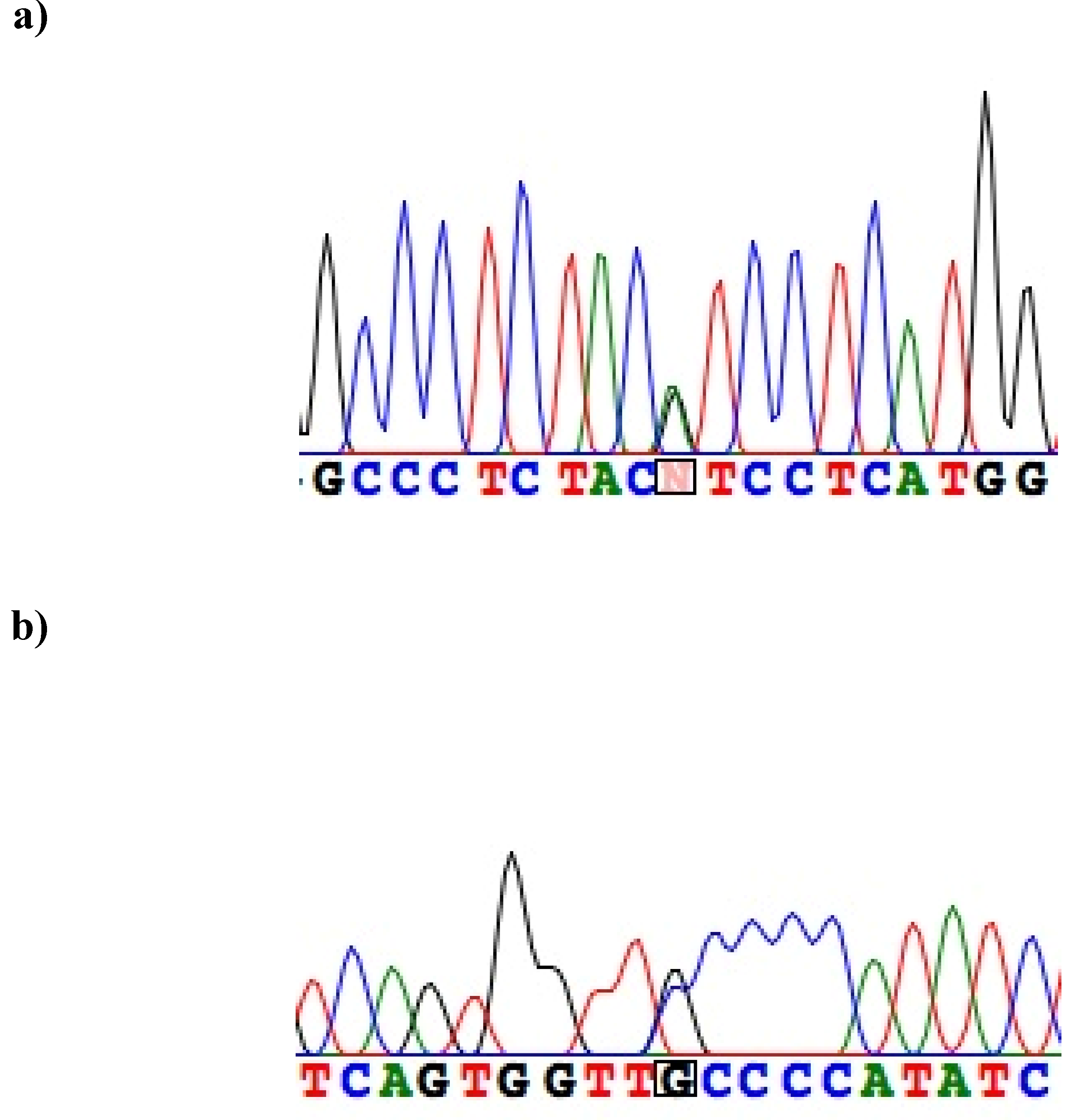{kind=link}
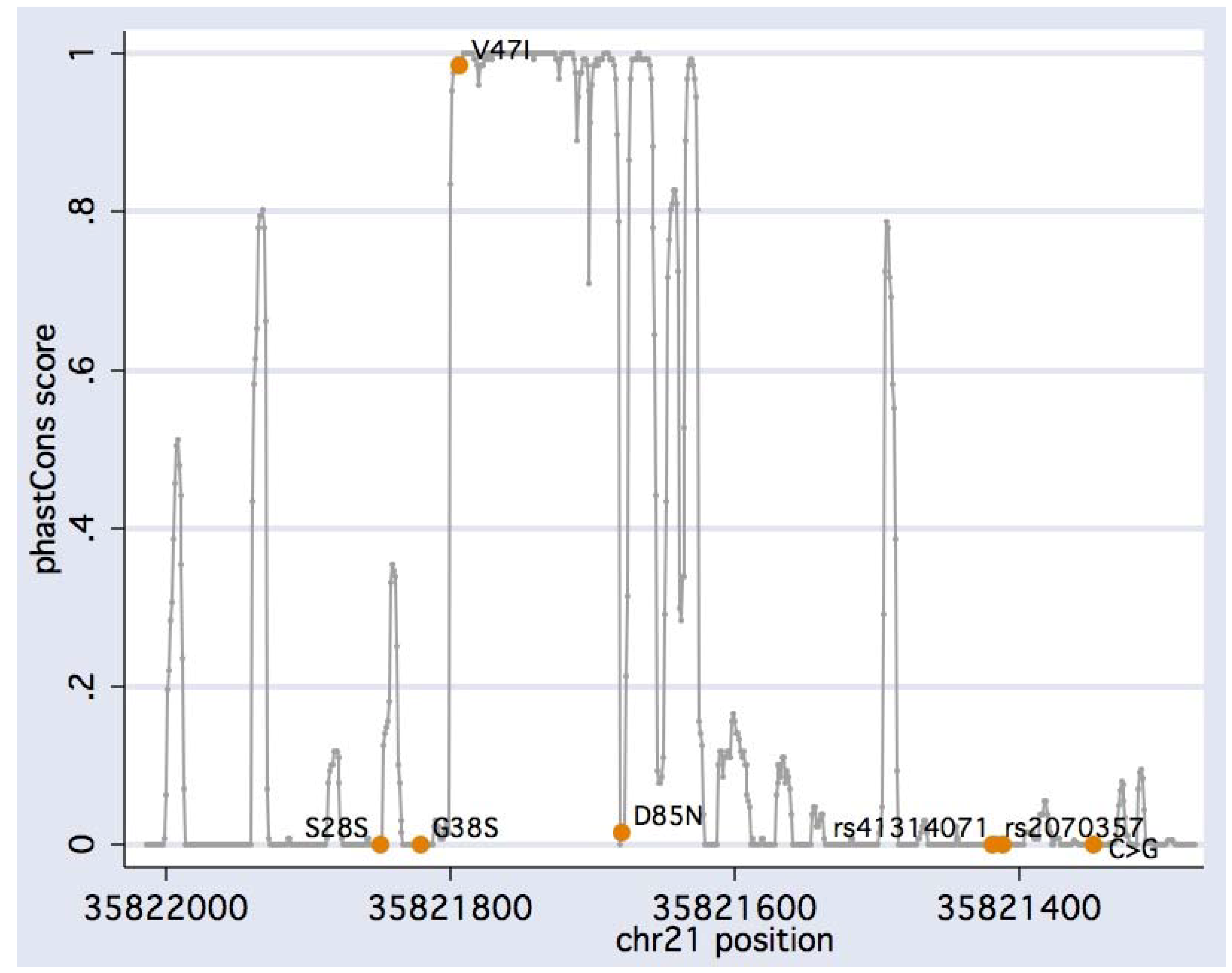{kind=link}
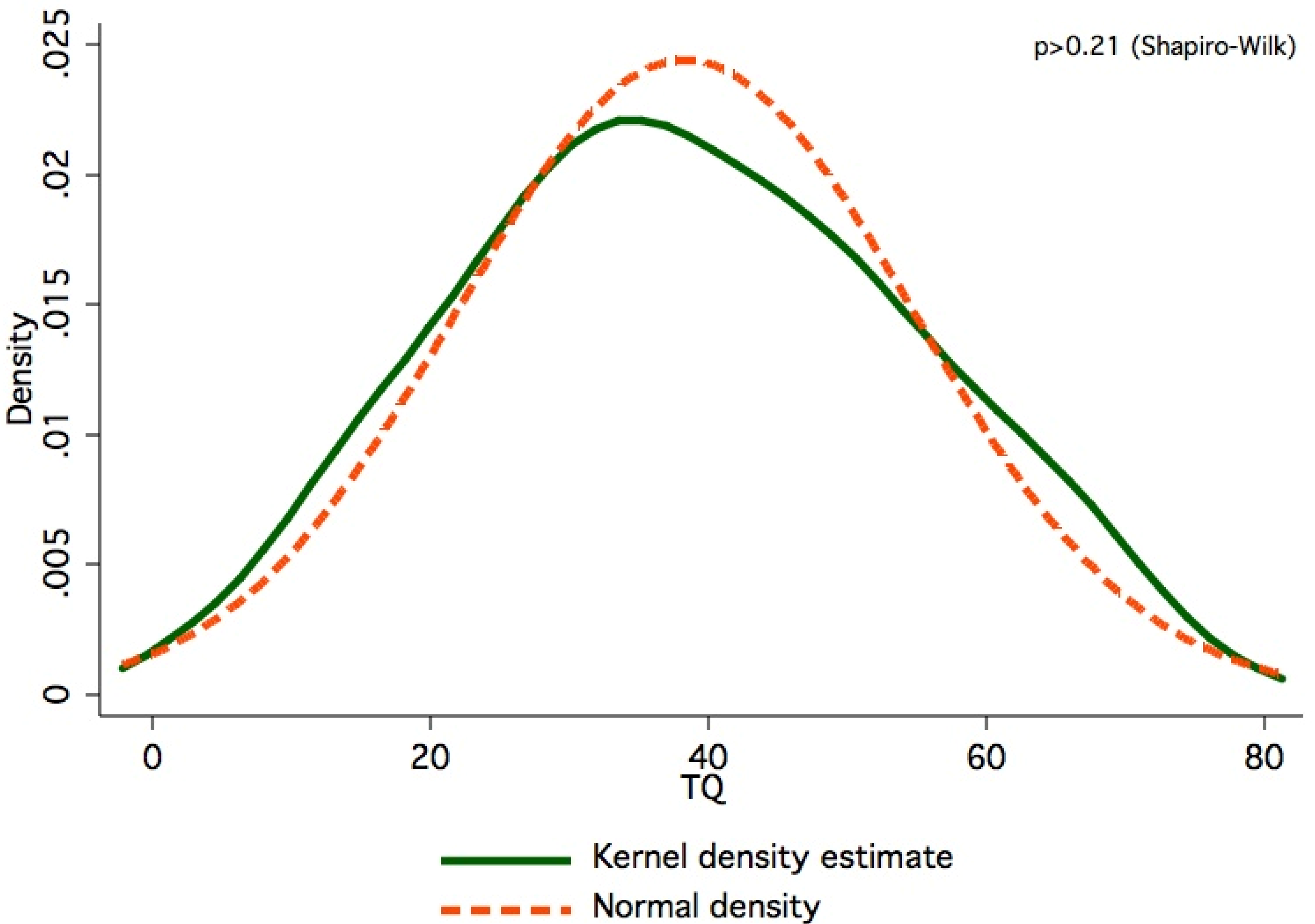{kind=link}
| dbSNP ID | chr21 position | major>minor allelesa | variant amino acid | minor allele frequency in chronic tinnitus | homozygous/heterozygous carriers of the minor allele (pHWE) |
| rs28933384 | 35,821,913 | C>T | T7I | 0.000 | - |
| - | 35,821,910 | C>T | A8V | 0.000 | - |
| - | 35,821,904 | C>T | T10M | 0.000 | - |
| - | 35,821,903 | G>A | T10T | 0.000 | - |
| - | 35,821,883 | G>A | W17X | 0.000 | - |
| - | 35,821,874 | C>T | T20I | 0.000 | - |
| - | 35,821,850 | C>T | S28L | 0.000 | - |
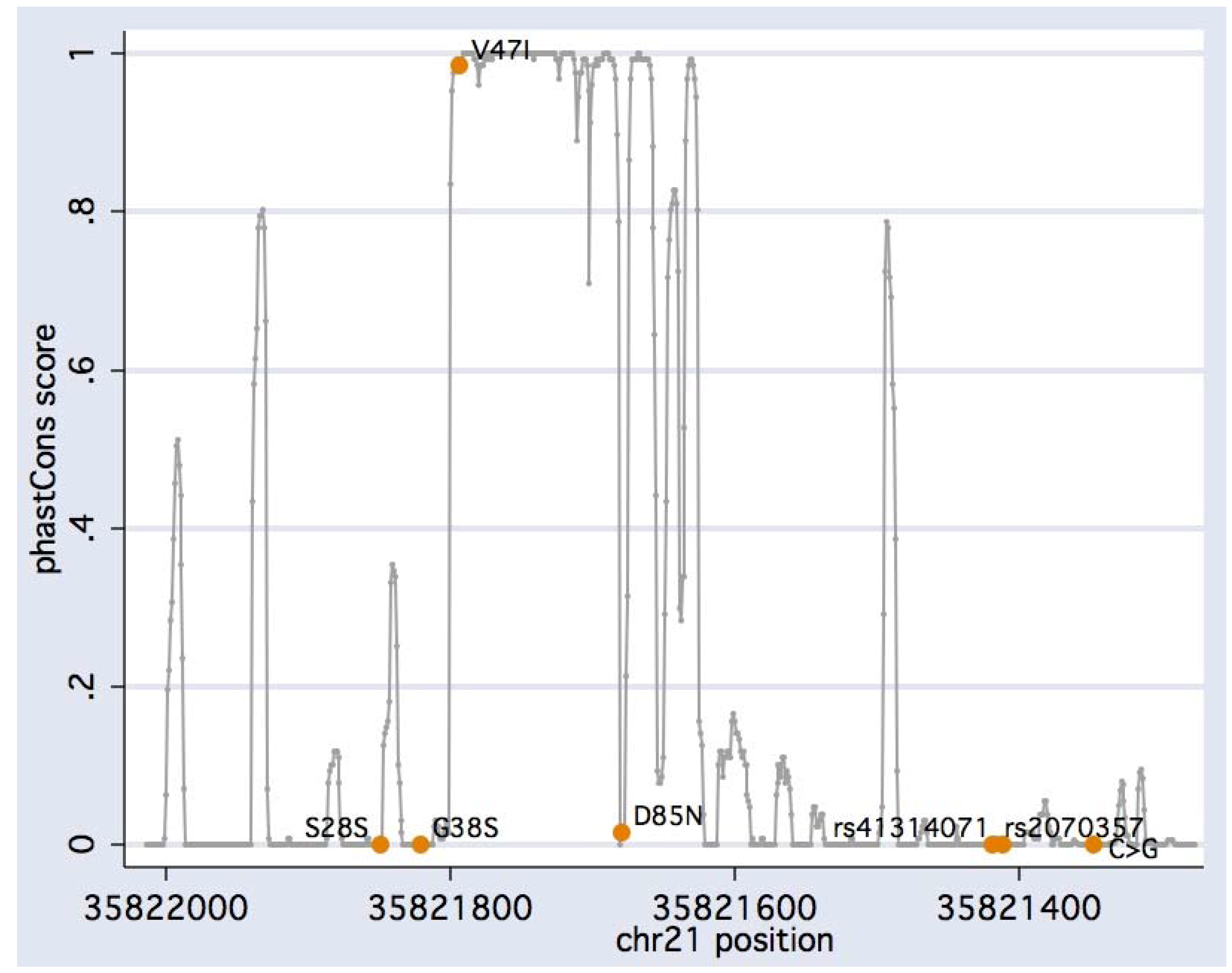
| rs17173510 | 35,821,849 | G>A | S28S | 0.002 | 0/1 (0.972) |
| rs17857111 | 35,821,838 | G>A | R32H | 0.000 | - |
| - | 35,821,826 | G>A | R36H | 0.000 | - |
| rs1805127 | 35,821,821 | G>A | G38S | 0.359 | 28/88 (0.498) |
| - | 35,821,794 | G>T | V47F | 0.000 | - |
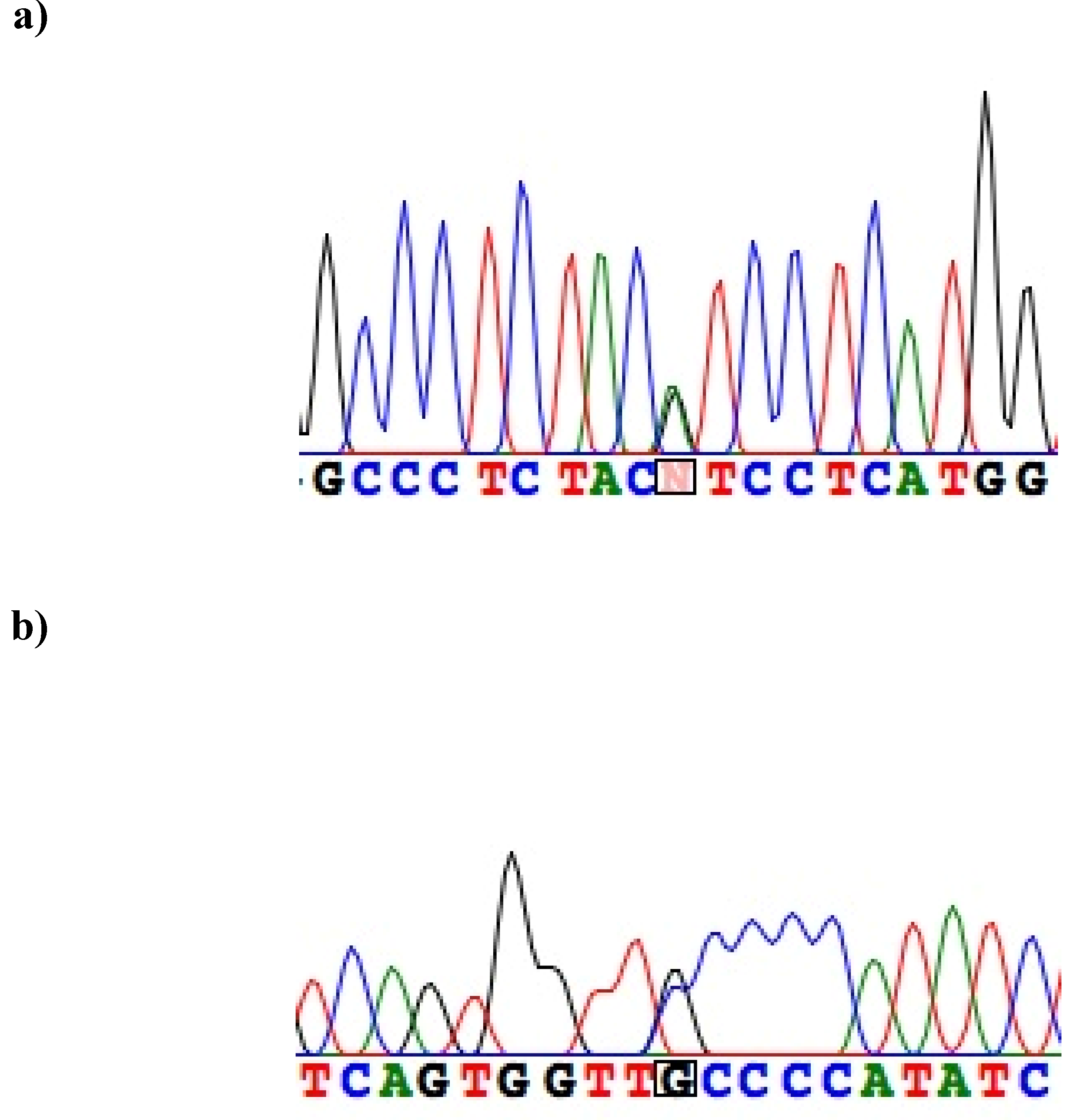
| (novel) | 35,821,794 | G>A | V47I | 0.002 | 0/1 (0.972) |
| - | 35,821,780-1 | TG>AC | L51H | 0.000 | - |
| rs17173509 | 35,821,778 | G>C | G52A | 0.000 | - |
| - | 35,821,779 | G>A | G52R | 0.000 | - |
| - | 35,821,775 | T>C | F53S | 0.000 | - |
| - | 35,821,774 | C>T | F53F | 0.000 | - |
| rs17173508 | 35,821,771 | C>T | F54F | 0.000 | - |
| - | 35,821,770 | G>A | G55S | 0.000 | - |
| - | 35,821,761 | A>C | T58P | 0.000 | - |
| - | 35,821,757 | T>C | L59P | 0.000 | - |
| - | 35,821,734 | C>T | R67C | 0.000 | - |
| - | 35,821,733 | G>A | R67H | 0.000 | - |
| - | 35,821,727 | A>G | K69R | 0.000 | - |
| - | 35,821,724 | A>T | K70M | 0.000 | - |
| - | 35,821,723 | G>C | K70N | 0.000 | - |
| - | 35,821,712 | C>T | S74L | 0.000 | - |
| - | 35,821,708 | C>T | N75N | 0.000 | - |
| - | 35,821,707 | G>A | D76N | 0.000 | - |
| - | 35,821,693 | C>G | V80V | 0.000 | - |
| - | 35,821,693 | C>T | V80V | 0.000 | - |
| - | 35,821,691 | A>G | Y81C | 0.000 | - |
| - | 35,821,686 | G>A | E83K | 0.000 | - |
| rs1805128 | 35,821,680 | G>A | D85N | 0.007 | 0/3 (0.915) |
| - | 35,821,674 | T>C | W87R | 0.000 | - |
| - | 35,821,641 | C>T | R98W | 0.000 | - |
| rs17853625 | 35,821,615 | C>A | C106X | 0.000 | - |
| - | 35,821,608 | G>A | V109I | 0.000 | - |
| - | 35,821,584 | C>T | Q117X | 0.000 | - |
| - | 35,821,559 | C>T | T125M | 0.000 | - |
| - | 35,821,554 | C>A | P127T | 0.000 | - |
| rs2070357 | 35,821,419 | G>A | - | 0.455 | 42/98 (0.865) |
| rs41314071 | 35,821,411 | A>G | - | 0.045 | 1/16 (0.328) |
| rs41314069 | 35,821,376 | C>A | - | 0.000 | - |
| (novel) | 35,821,347 | C>G | - | 0.003 | 0/1 (0.972) |
| rs41312371 | 35,821,283 | A>C | - | 0.000 | - |
| rs41314807 | 35,821,275 | C>T | - | 0.000 | - |

| healthy controls (Nunrelated) | source | fSer28(TCA) | vs. fSer28(TCA) in present study (p) | fSer38 | vs. fSer38 in present study (p) | fIle47 | vs. fIle47 in present study (p) | fAsn85 | vs. fAsn85 in present study (p) |
|---|---|---|---|---|---|---|---|---|---|
| U.S., European descent (187) | [36] | 0.000 | n.s. | - | - | 0.000 | n.s. | - | - |
| Dutch (32) | [58] | 0.000 | n.s. | 0.33 | n.s. | 0.000 | n.s. | 0.000 | n.s. |
| German (141) | [59] | - | - | - | - | 0.000 | n.s. | - | - |
| French (398) | [60,62] | 0.000 | n.s. | 0.372 | n.s. | 0.000 | n.s. | 0.018 | n.s. |
| Polish (129) | [26] | - | - | (0.582) | (<0.0001) | - | - | - | - |
| German (3,916) | [63] | - | - | 0.368 | n.s. | - | - | - | - |
| Finnish (5,043) | [64] | - | - | - | - | - | - | 0.014 | n.s. |
| U.S., European descent (180) | [51] | 0.006 | n.s. | 0.378 | n.s. | 0.000 | n.s. | 0.008 | n.s. |
| Central Europeans (59) | [27] HapMap CEU | - | - | 0.381 | n.s. | - | - | 0.008 | n.s. |
| Caucasian panel (47) | [27] Coriell Cell Repository R31 CAU | - | - | 0.394 | n.s. | - | - | 0.021 | n.s. |
3. Experimental Section
4. Conclusions
References and Notes
- Sanchez, L. The epidemiology of tinnitus. Audiol. Med. 2004, 2, 8–17. [Google Scholar] [CrossRef]
- Hoffman, H.J.; Reed, G.W. Epidemiology of Tinnitus. In Tinnitus: Theory and Management; Snow, J.B., Ed.; BC Decker: London, UK, 2004; pp. 16–41. [Google Scholar]
- Eggermont, J.J. Pathophysiology of tinnitus. Prog. Brain Res. 2007, 166, 19–35. [Google Scholar] [CrossRef]
- Attias, J.; Furman, V.; Shemesh, Z.; Bresloff, I. Impaired brain processing in noise-induced tinnitus patients as measured by auditory and visual event-related potentials. Ear Hear. 1996, 17, 327–333. [Google Scholar]
- Sand, P.G.; Langguth, B.; Kleinjung, T.; Eichhammer, P. Genetics of chronic tinnitus. Prog. Brain Res. 2007, 166, 159–168. [Google Scholar] [CrossRef]
- Wangemann, P. K+ cycling and the endocochlear potential. Hear Res. 2002, 165, 1–9. [Google Scholar] [CrossRef]
- Hibino, H.; Nin, F.; Tsuzuki, C.; Kurachi, Y. How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 2010, 459, 521–533. [Google Scholar] [CrossRef]
- Coucke, P.J.; Van Hauwe, P.; Kelley, P.M.; Kunst, H.; Schatteman, I.; Van Velzen, D.; Meyers, J.; Ensink, R.J.; Verstreken, M.; Declau, F.; Marres, H.; Kastury, K.; Bhasin, S.; McGuirt, W.T.; Smith, R.J.H.; Cremers, C.W.R.J.; Van de Heyning, P.; Willems, P.J.; Smith, S.D.; Van Camp, G. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Molec. Genet. 1999, 8, 1321–1328. [Google Scholar] [CrossRef]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Neyroud, N.; Tesson, F.; Denjoy, I.; Leibovici, M.; Donger, C.; Barhanin, J.; Faure, S.; Gary, F.; Coumel, P.; Petit, C.; Schwartz, K.; Guicheney, P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nature Genet. 1997, 15, 186–189. [Google Scholar] [CrossRef]
- Tyson, J.; Tranebjaerg, L.; Bellman, S.; Wren, C.; Taylor, J.F.; Bathen, J.; Aslaksen, B.; Sørland, S.J.; Lund, O.; Malcolm, S.; Pembrey, M.; Bhattacharya, S.; Bitner-Glindzicz, M. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet. 1997, 6, 2179–2185. [Google Scholar] [CrossRef]
- Vetter, D.E.; Mann, J.R.; Wangemann, P.; Liu, J.; McLaughlin, K.J.; Lesage, F.; Marcus, D.C.; Lazdunski, M.; Heinemann, S.F.; Barhanin, J. Inner ear defects induced by null mutation of the isk gene. Neuron 1996, 17, 1251–1264. [Google Scholar] [CrossRef]
- Letts, V.A.; Valenzuela, A.; Dunbar, C.; Zheng, Q.Y.; Johnson, K.R.; Frankel, W.N. A new spontaneous mouse mutation in the Kcne1 gene. Mamm. Genome 2000, 11, 831–835. [Google Scholar] [CrossRef]
- Clancy, S.M.; Chen, B.; Bertaso, F.; Mamet, J.; Jegla, T. KCNE1 and KCNE3 beta-subunits regulate membrane surface expression of Kv12.2 K(+) channels in vitro and form a tripartite complex in vivo. PLoS One 2009, 4, e6330. [Google Scholar]
- Doi, K.; Sato, T.; Kuramasu, T.; Hibino, H.; Kitahara, T.; Horii, A.; Matsushiro, N.; Fuse, Y.; Kubo, T. Ménière's disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J. Otorhinolaryngol. Relat. Spec. 2005, 67, 289–293. [Google Scholar] [CrossRef]
- Van Eyken, E.; Van Laer, L.; Fransen, E.; Topsakal, V.; Lemkens, N.; Laureys, W.; Nelissen, N.; Vandevelde, A.; Wienker, T.; Van De Heyning, P.; Van Camp, G. KCNQ4: a gene for age-related hearing impairment? Hum. Mutat. 2006, 27, 1007–1016. [Google Scholar] [CrossRef]
- Van Laer, L.; Carlsson, P.I.; Ottschytsch, N.; Bondeson, M.L.; Konings, A.; Vandevelde, A.; Dieltjens, N.; Fransen, E.; Snyders, D.; Borg, E.; Raes, A.; Van Camp, G. The contribution of genes involved in potassium-recycling in the inner ear to noise-induced hearing loss. Hum. Mutat. 2006, 27, 786–795. [Google Scholar] [CrossRef]
- Pawelczyk, M; Van Laer, L; Fransen, E. Analysis of gene polymorphisms associated with K ion circulation in the inner ear of patients susceptible and resistant to noise-induced hearing loss. Ann. Hum. Genet. 2009, 73, 411–421. [Google Scholar] [CrossRef]
- Axelsson, A.; Sandh, A. Tinnitus in noise-induced hearing loss. Br. J. Audiol. 1985, 19, 271–276. [Google Scholar] [CrossRef]
- Goebel, G.; Hiller, W. The tinnitus questionnaire.A standard instrument for grading the degree of tinnitus. Results of a multicenter study with the tinnitus questionnaire. HNO 1994, 42, 166–172. [Google Scholar]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; Weinstock, G.M.; Wilson, R.K.; Gibbs, R.A.; Kent, W.J.; Miller, W.; Haussler, D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef]
- Blanchette, M.; Kent, W.J.; Riemer, C.; Elnitski, L.; Smit, A.F.; Roskin, K.M.; Baertsch, R.; Rosenbloom, K.; Clawson, H.; Green, E.D.; Haussler, D.; Miller, W. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004, 14, 708–715. [Google Scholar] [CrossRef]
- Rhead, B.; Karolchik, D.; Kuhn, R.M.; Hinrichs, A.S.; Zweig, A.S.; Fujita, P.A.; Diekhans, M.; Smith, K.E.; Rosenbloom, K.R.; Raney, B.J.; Pohl, A.; Pheasant, M.; Meyer, L.R.; Learned, K.; Hsu, F.; Hillman-Jackson, J.; Harte, R.A.; Giardine, B.; Dreszer, T.R.; Clawson, H.; Barber, G.P.; Haussler, D.; Kent, W.J. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010, 38, D613–619. [Google Scholar] [CrossRef]
- Dupont, W.D.; Plummer, W.D., Jr. Power and sample size calculations: a review and computer program. Control Clin. Trials 1990, 11, 116–128. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Prystupa, A.; Dzida, G.; Myśliński, W.; Małaj, G.; Lorenc, T. MinK gene polymorphism in the pathogenesis of lone atrial fibrillation. Kardiol. Pol. 2006, 64, 1205–1211. [Google Scholar]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; Feolo, M.; Geer, L.Y.; Helmberg, W.; Kapustin, Y.; Landsman, D.; Lipman, D.J.; Lu, Z.; Madden, T.L.; Madej, T.; Maglott, D.R.; Marchler-Bauer, A.; Miller, V.; Mizrachi, I.; Ostell, J.; Panchenko, A.; Pruitt, K.D.; Schuler, G.D.; Sequeira, E.; Sherry, S.T.; Shumway, M.; Sirotkin, K.; Slotta, D.; Souvorov, A.; Starchenko, G.; Tatusova, T.A.; Wagner, L.; Wang, Y.; John Wilbur, W.; Yaschenko, E.; Ye, J. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38, D5-16. [Google Scholar] [CrossRef]
- Genome Variation Server. Available online: http://gvs.gs.washington.edu/GVS (accessed 4 March 2010).
- Ohno, S.; Zankov, D.P.; Yoshida, H.; Tsuji, K.; Makiyama, T.; Itoh, H.; Akao, M.; Hancox, J.C.; Kita, T.; Horie, M. N- and C-terminal KCNE1 mutations cause distinct phenotypes of long QT syndrome. Heart Rhythm 2007, 4, 332–340. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef]
- Koo, S.H.; Ho, W.F.; Lee, E.J. Genetic polymorphisms in KCNQ1, HERG, KCNE1 and KCNE2 genes in the Chinese, Malay and Indian populations of Singapore. Br. J. Clin. Pharmacol. 2006, 61, 301–308. [Google Scholar] [CrossRef]
- Millat, G.; Kugener, B.; Chevalier, P.; Chahine, M.; Huang, H.; Malicier, D.; Rodriguez-Lafrasse, C.; Rousson, R. Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatr. Cardiol. 2009, 30, 502–509. [Google Scholar] [CrossRef]
- Shim, S.H.; Ito, M.; Maher, T.; Milunsky, A. Gene sequencing in neonates and infants with the long QT syndrome. Genet. Test 2005, 9, 281–284. [Google Scholar]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. J.A.M.A. 2005, 294, 2975–2980. [Google Scholar] [CrossRef]
- Bianchi, L.; Shen, Z.; Dennis, A.T.; Priori, S.G.; Napolitano, C.; Ronchetti, E.; Bryskin, R.; Schwartz, P.J.; Brown, A.M. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 1999, 8, 1499–1507. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Tester, D.J.; Jones, G.S.; Will, M.L.; Burrow, C.R.; Curran, M.E. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin. Proc. 2003, 78, 1479–1487. [Google Scholar] [CrossRef]
- Ma, L.; Lin, C.; Teng, S.; Chai, Y.; Bähring, R.; Vardanyan, V.; Li, L.; Pongs, O.; Hui, R. Characterization of a novel Long QT syndrome mutation G52R-KCNE1 in a Chinese family. Cardiovasc. Res. 2003, 59, 612–619. [Google Scholar] [CrossRef]
- Lai, L.P.; Su, Y.N.; Hsieh, F.J.; Chiang, F.T.; Juang, J.M.; Liu, Y.B.; Ho, Y.L.; Chen, W.J.; Yeh, S.J.; Wang, C.C.; Ko, Y.L.; Wu, T.J.; Ueng, K.C.; Lei, M.H.; Tsao, H.M.; Chen, S.A.; Lin, T.K.; Wu, M.H.; Lo, H.M.; Huang, S.K.; Lin, J.L. Denaturing high-performance liquid chromatography screening of the long QT syndrome-related cardiac sodium and potassium channel genes and identification of novel mutations and single nucleotide polymorphisms. J. Hum. Genet. 2005, 50, 490–496. [Google Scholar] [CrossRef]
- Splawski, I.; Tristani-Firouzi, M.; Lehmann, M.H.; Sanguinetti, M.C.; Keating, M.T. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 1997, 17, 338–340. [Google Scholar] [CrossRef]
- Westenskow, P.; Splawski, I.; Timothy, K.W.; Keating, M.T.; Sanguinetti, M.C. Compound mutations: a common cause of severe long-QT syndrome. Circulation 2004, 109, 1834–1841. [Google Scholar] [CrossRef]
- Berge, K.E.; Haugaa, K.H.; Früh, A.; Anfinsen, O.G.; Gjesdal, K.; Siem, G.; Oyen, N.; Greve, G.; Carlsson, A.; Rognum, T.O.; Hallerud, M.; Kongsgård, E.; Amlie, J.P.; Leren, T.P. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand. J. Clin. Lab. Invest. 2008, 68, 362–368. [Google Scholar] [CrossRef]
- Schulze-Bahr, E.; Wang, Q.; Wedekind, H.; Haverkamp, W.; Chen, Q.; Sun, Y.; Rubie, C.; Hördt, M.; Towbin, J.A.; Borggrefe, M.; Assmann, G.; Qu, X.; Somberg, J.C.; Breithardt, G.; Oberti, C.; Funke, H. KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat. Genet. 1997, 17, 267–268. [Google Scholar] [CrossRef]
- Duggal, P.; Vesely, M.R.; Wattanasirichaigoon, D.; Villafane, J.; Kaushik, V.; Beggs, A.H. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation 1998, 97, 142–146. [Google Scholar] [CrossRef]
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 507–517. [Google Scholar] [CrossRef]
- Tester, D.J.; Arya, P.; Will, M.; Haglund, C.M.; Farley, A.L.; Makielski, J.C.; Ackerman, M.J. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm 2006, 3, 800–805. [Google Scholar] [CrossRef]
- Friedlander, Y.; Vatta, M.; Sotoodehnia, N.; Sinnreich, R.; Li, H.; Manor, O.; Towbin, J.A.; Siscovick, D.S.; Kark, J.D. Possible association of the human KCNE1 (minK) gene and QT interval in healthy subjects: evidence from association and linkage analyses in Israeli families. Ann. Hum. Genet. 2005, 69, 645–656. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Petrov-Kondratov, V.I.; Zakharova, E.; Nam, E.G.; MacRae, C.A. Potassium channel gene mutations rarely cause atrial fibrillation. BMC Med. Genet. 2006, 7, 70. [Google Scholar] [CrossRef]
- Wu, DM; Lai, LP; Zhang, M. Characterization of an LQT5-related mutation in KCNE1, Y81C: implications for a role of KCNE1 cytoplasmic domain in IKs channel function. Heart Rhythm 2006, 3, 1031–1040. [Google Scholar] [CrossRef]
- Millat, G.; Chevalier, P.; Restier-Miron, L.; Da Costa, A.; Bouvagnet, P.; Kugener, B.; Fayol, L.; Gonzàlez Armengod, C.; Oddou, B.; Chanavat, V.; Froidefond, E.; Perraudin, R.; Rousson, R.; Rodriguez-Lafrasse, C. Spectrum of pathogenic mutations and associated polymorphisms in a cohort of 44 unrelated patients with long QT syndrome. Clin. Genet. 2006, 70, 214–227. [Google Scholar] [CrossRef]
- Schulze-Bahr, E.; Schwarz, M.; Hauenschild, S.; Wedekind, H.; Funke, H.; Haverkamp, W.; Breithardt, G.; Pongs, O.; Isbrandt, D. A novel long-QT 5 gene mutation in the C-terminus (V109I) is associated with a mild phenotype. J. Mol. Med. 2001, 79, 504–509. [Google Scholar] [CrossRef]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; Keating, M.T. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef]
- Campbell, C.A.; Della Santina, C.C.; Meyer, N.C.; Smith, N.B.; Myrie, O.A.; Stone, E.M.; Fukushima, K.; Califano, J.; Carey, J.P.; Hansen, M.R.; Gantz, B.J.; Minor, L.B.; Smith, R.J. Polymorphisms in KCNE1 or KCNE3 are not associated with Ménière disease in the Caucasian population. Am. J. Med. Genet. A 2010, 152A, 67–74. [Google Scholar] [CrossRef]
- Tesson, F.; Donger, C.; Denjoy, I.; Berthet, M.; Bennaceur, M.; Petit, C.; Coumel, P.; Schwarts, K.; Guicheney, P. Exclusion of KCNE1 (IsK) as a candidate gene for Jervell and Lange-Nielsen syndrome. J. Mol. Cell. Cardiol. 1996, 28, 2051–205. [Google Scholar] [CrossRef]
- Jongbloed, R.; Marcelis, C.; Velter, C.; Doevendans, P.; Geraedts, J.; Smeets, H. DHPLC analysis of potassium ion channel genes in congenital long QT syndrome. Hum. Mutat. 2002, 20, 382–391. [Google Scholar] [CrossRef]
- Herlyn, H.; Zechner, U.; Oswald, F.; Pfeufer, A.; Zischler, H.; Haaf, T. Positive selection at codon 38 of the human KCNE1 (= minK) gene and sporadic absence of 38Ser-coding mRNAs in Gly38Ser heterozygotes. BMC Evol. Biol. 2009, 9, 188. [Google Scholar] [CrossRef]
- Nishio, Y.; Makiyama, T.; Itoh, H.; Sakaguchi, T.; Ohno, S.; Gong, Y.Z.; Yamamoto, S.; Ozawa, T.; Ding, W.G; Toyoda, F.; Kawamura, M.; Akao, M.; Matsuura, H.; Kimura, T.; Kita, T.; Horie, M. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J. Am. Coll. Cardiol. 2009, 54, 812–819. [Google Scholar] [CrossRef]
- Jones, M. A discussion on the etiology of tinnitus aurium. Br. Med. J. 1890, 2, 667–672. [Google Scholar] [CrossRef]
- Paulussen, A.D.; Gilissen, R.A.; Armstrong, M.; Doevendans, P.A.; Verhasselt, P.; Smeets, H.J.; Schulze-Bahr, E.; Haverkamp, W.; Breithardt, G.; Cohen, N.; Aerssens, J. Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J. Mol. Med. 2004, 82, 182–188. [Google Scholar] [CrossRef]
- Aydin, A.; Bähring, S.; Dahm, S.; Guenther, U.P.; Uhlmann, R.; Busjahn, A.; Luft, F.C. Single nucleotide polymorphism map of five long-QT genes. J. Mol. Med. 2005, 83, 159–165. [Google Scholar] [CrossRef]
- Gouas, L.; Nicaud, V.; Chaouch, S.; Berthet, M.; Forhan, A.; Tichet, J.; Tiret, L.; Balkau, B.; Guicheney, P. Confirmation of associations between ion channel gene SNPs and QTc interval duration in healthy subjects. Eur. J. Hum. Genet. 2007, 15, 974–979. [Google Scholar] [CrossRef]
- Tian, C.; Vanoye, C.G.; Kang, C.; Welch, R.C.; Kim, H.J.; George, A.L., Jr.; Sanders, C.R. Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry 2007, 46, 11459–11472. [Google Scholar]
- Gouas, L.; Nicaud, V.; Berthet, M.; Forhan, A.; Tiret, L.; Balkau, B.; Guicheney, P.; D.E.S.I.R. Study Group. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur. J. Hum. Genet. 2005, 13, 1213–1222. [Google Scholar] [CrossRef]
- Akyol, M.; Jalilzadeh, S.; Sinner, M.F.; Perz, S.; Beckmann, B.M.; Gieger, C.; Illig, T.; Wichmann, H.E.; Meitinger, T.; Kääb, S.; Pfeufer, A. The common non-synonymous variant G38S of the KCNE1-(minK)-gene is not associated to QT interval in Central European Caucasians: results from the KORA study. Eur. Heart J. 2007, 28, 305–309. [Google Scholar] [CrossRef]
- Marjamaa, A.; Newton-Cheh, C.; Porthan, K.; Reunanen, A.; Lahermo, P.; Väänänen, H.; Jula, A.; Karanko, H.; Swan, H.; Toivonen, L.; Nieminen, M.S.; Viitasalo, M.; Peltonen, L.; Oikarinen, L.; Palotie, A.; Kontula, K.; Salomaa, V. Common candidate gene variants are associated with QT interval duration in the general population. J. Intern. Med. 2009, 265, 448–458. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sand, P.G.; Luettich, A.; Kleinjung, T.; Hajak, G.; Langguth, B. An Examination of KCNE1 Mutations and Common Variants in Chronic Tinnitus. Genes 2010, 1, 23-37. https://doi.org/10.3390/genes1010023
Sand PG, Luettich A, Kleinjung T, Hajak G, Langguth B. An Examination of KCNE1 Mutations and Common Variants in Chronic Tinnitus. Genes. 2010; 1(1):23-37. https://doi.org/10.3390/genes1010023
Chicago/Turabian StyleSand, Philipp G., Alexander Luettich, Tobias Kleinjung, Goeran Hajak, and Berthold Langguth. 2010. "An Examination of KCNE1 Mutations and Common Variants in Chronic Tinnitus" Genes 1, no. 1: 23-37. https://doi.org/10.3390/genes1010023