Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation

 ,
,  and
and 
Abstract
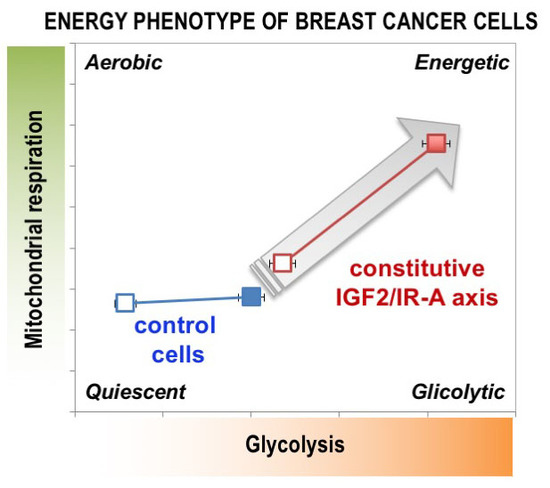
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. Generation of Lentiviral Vector pLEXG418–EV and pLEXG418–hIRA–FLAG
2.4. Lentiviral Particle Production and Lentiviral Transduction and Generation of MCF7-KOIGF1R-IR-A-FLAG
2.5. Western Blot Analysis
2.6. Real-Time PCR and Mitochondrial DNA Copy Number
2.7. IR Isoform mRNA Expression
2.8. Cell Proliferation
2.9. Migration Assay
2.10. Invasion Assay
2.11. Soft-Agar Colony Formation
2.12. Glucose Uptake and Lactate Production
2.13. Mitochondrial Staining and Flow Cytometry
2.14. Glycolytic Function and Mitochondrial Respiration Assay
2.15. Densitometric and Statistical Analysis
3. Results
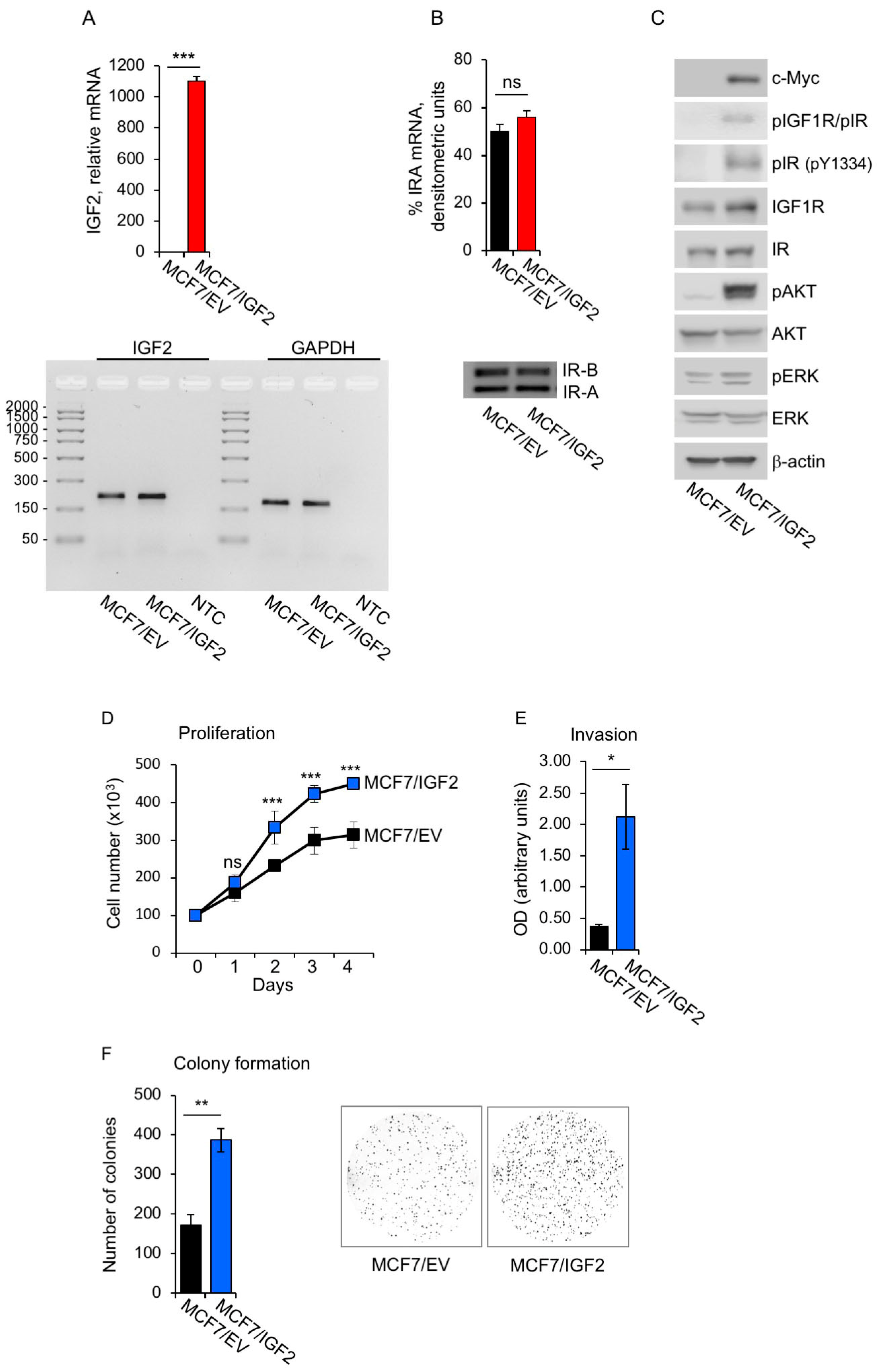
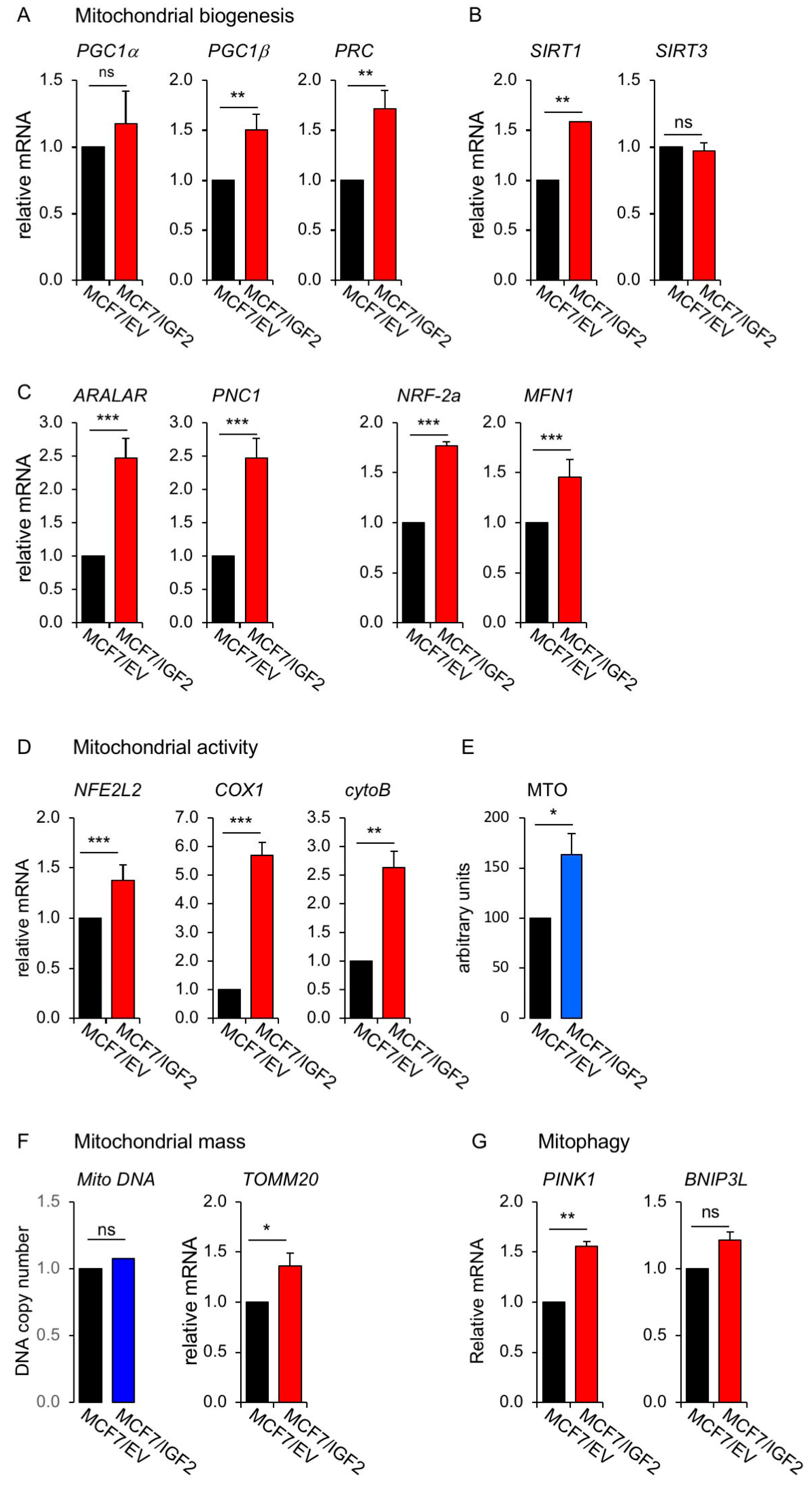
3.1. Establishment of Human BC Cells with Constitutive Autocrine IGF2 Secretion
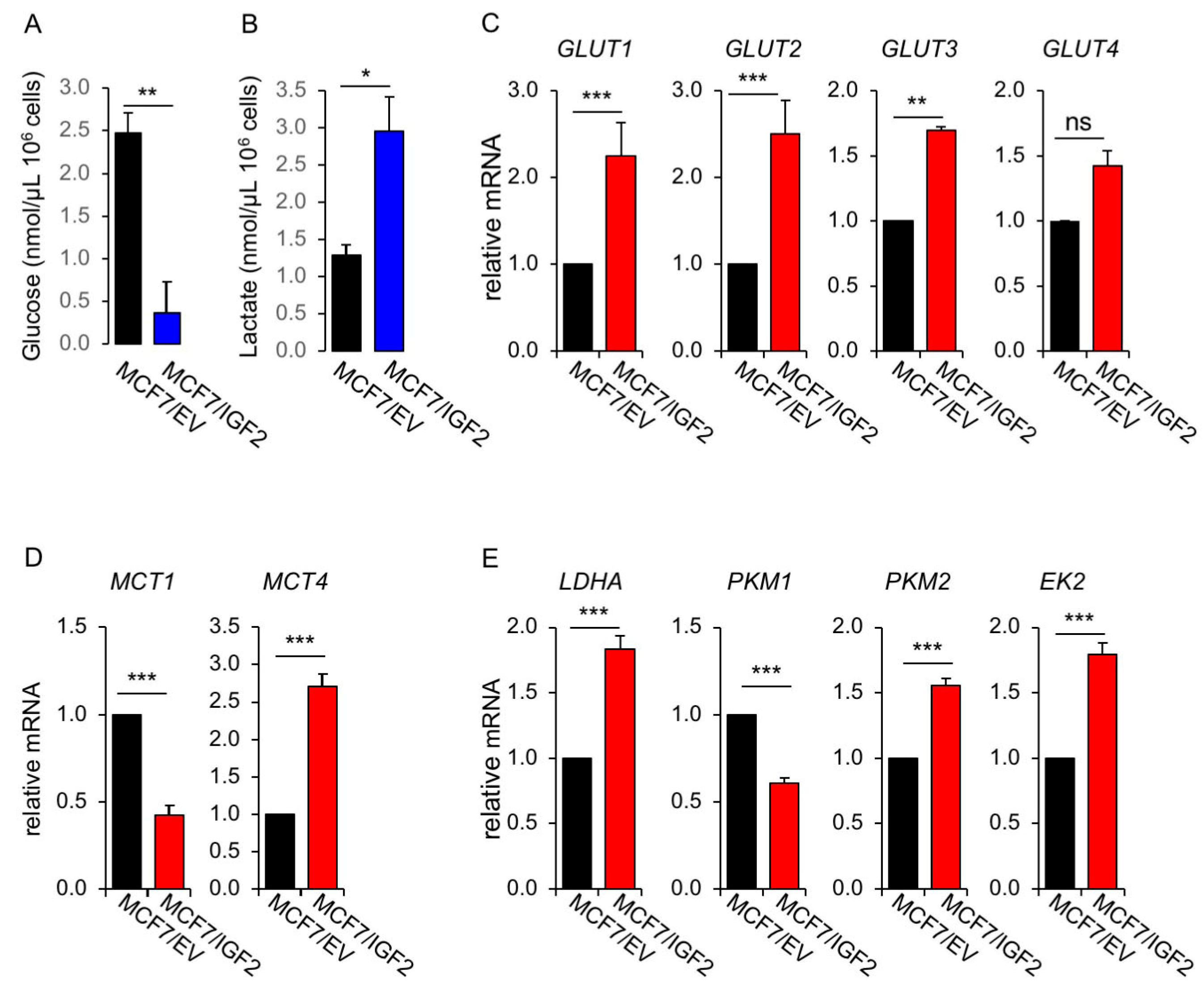
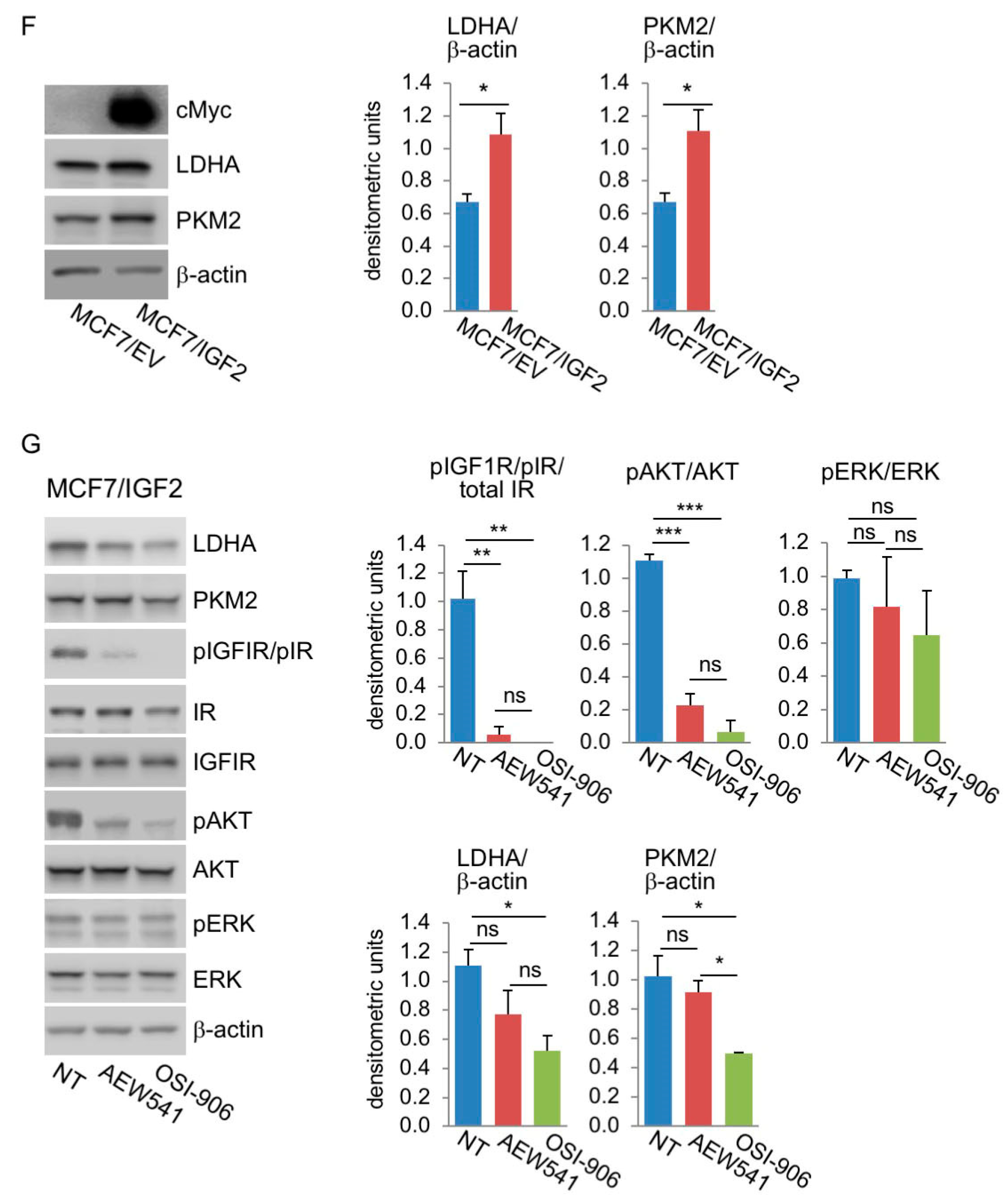
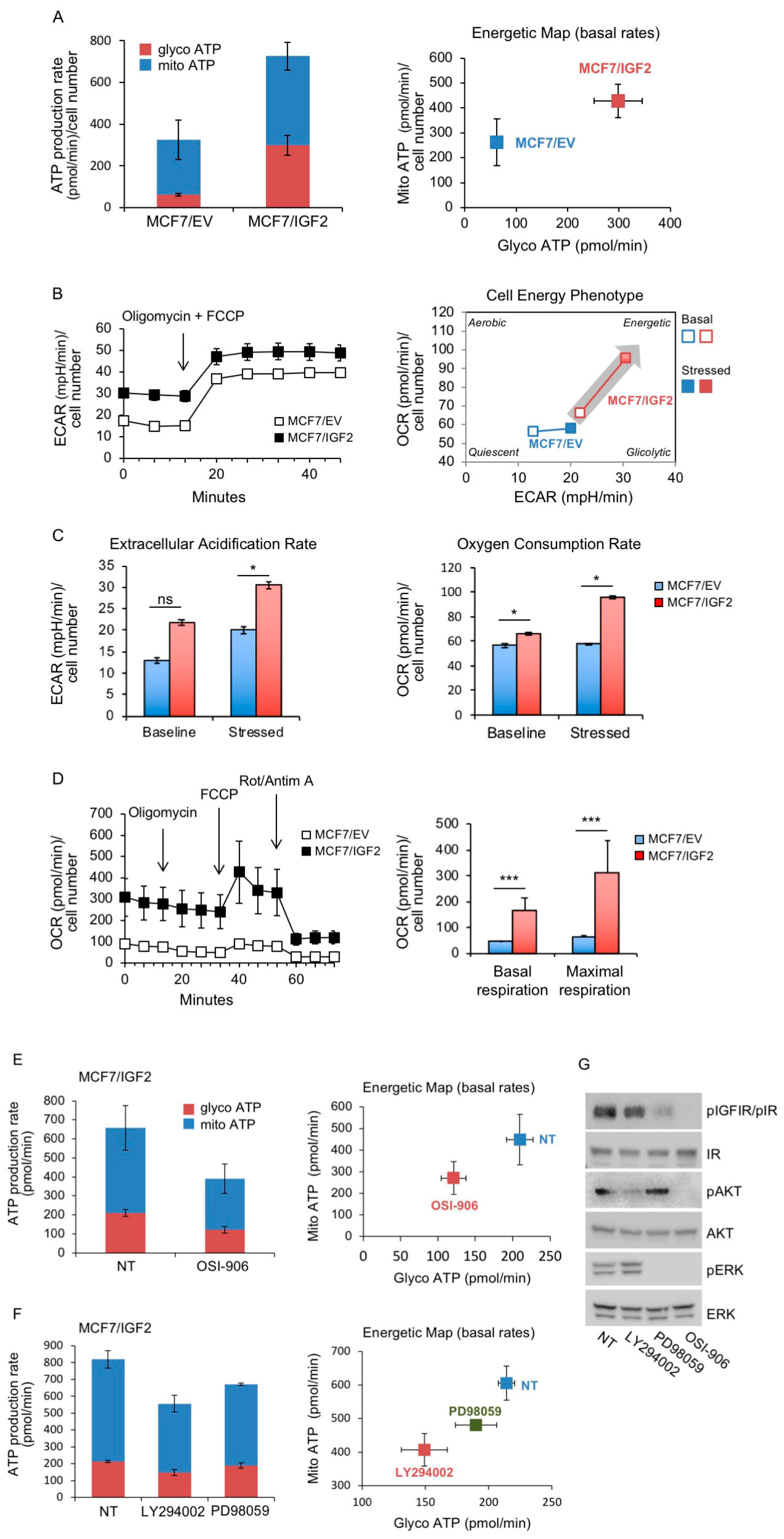
MCF7/IGF2 Cells Showed Increased Glycolytic and Mitochondrial Activities
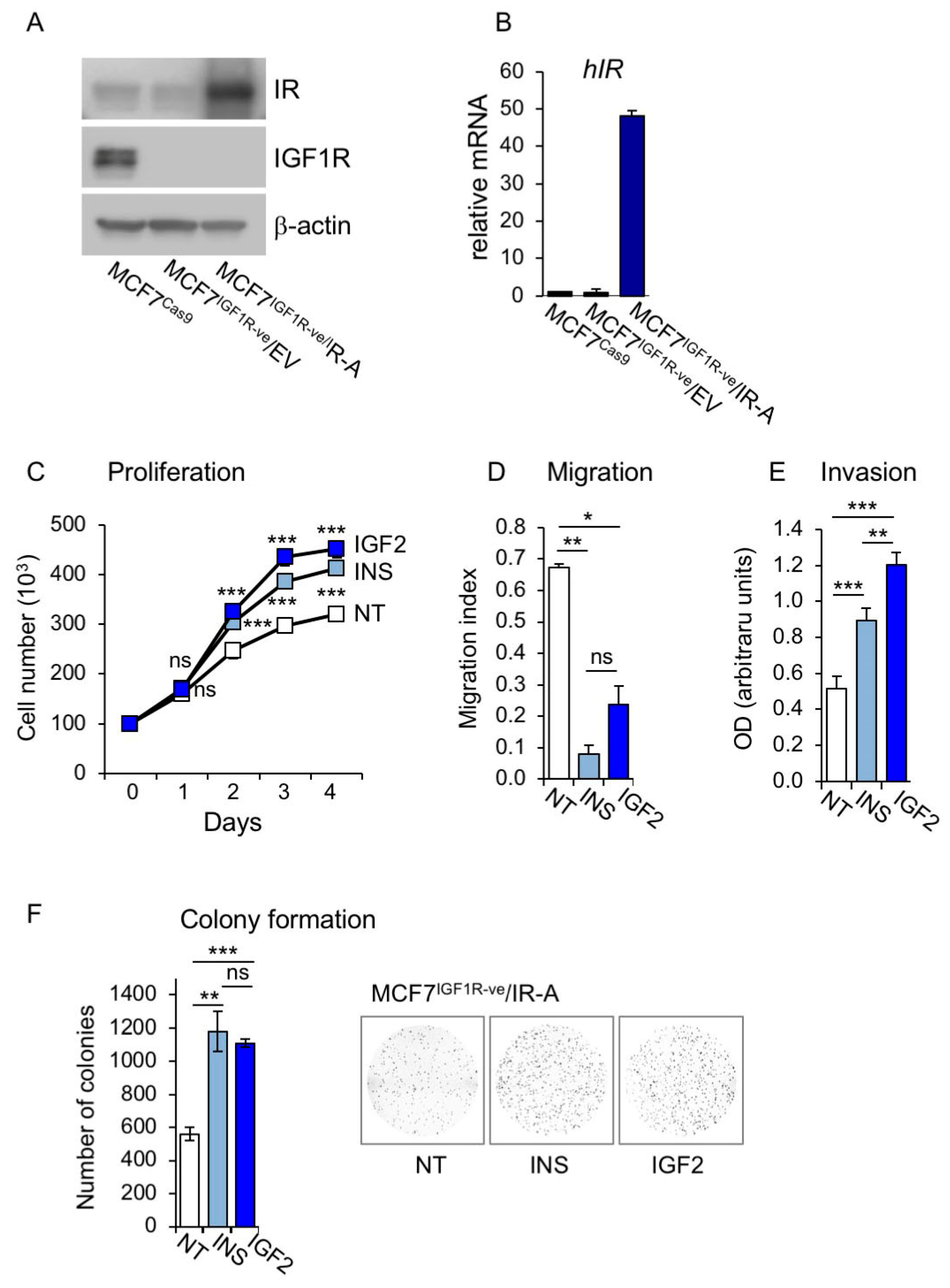
3.2. Establishment of MCF7 Cells Lacking IGF1R and Overexpressing IR-A
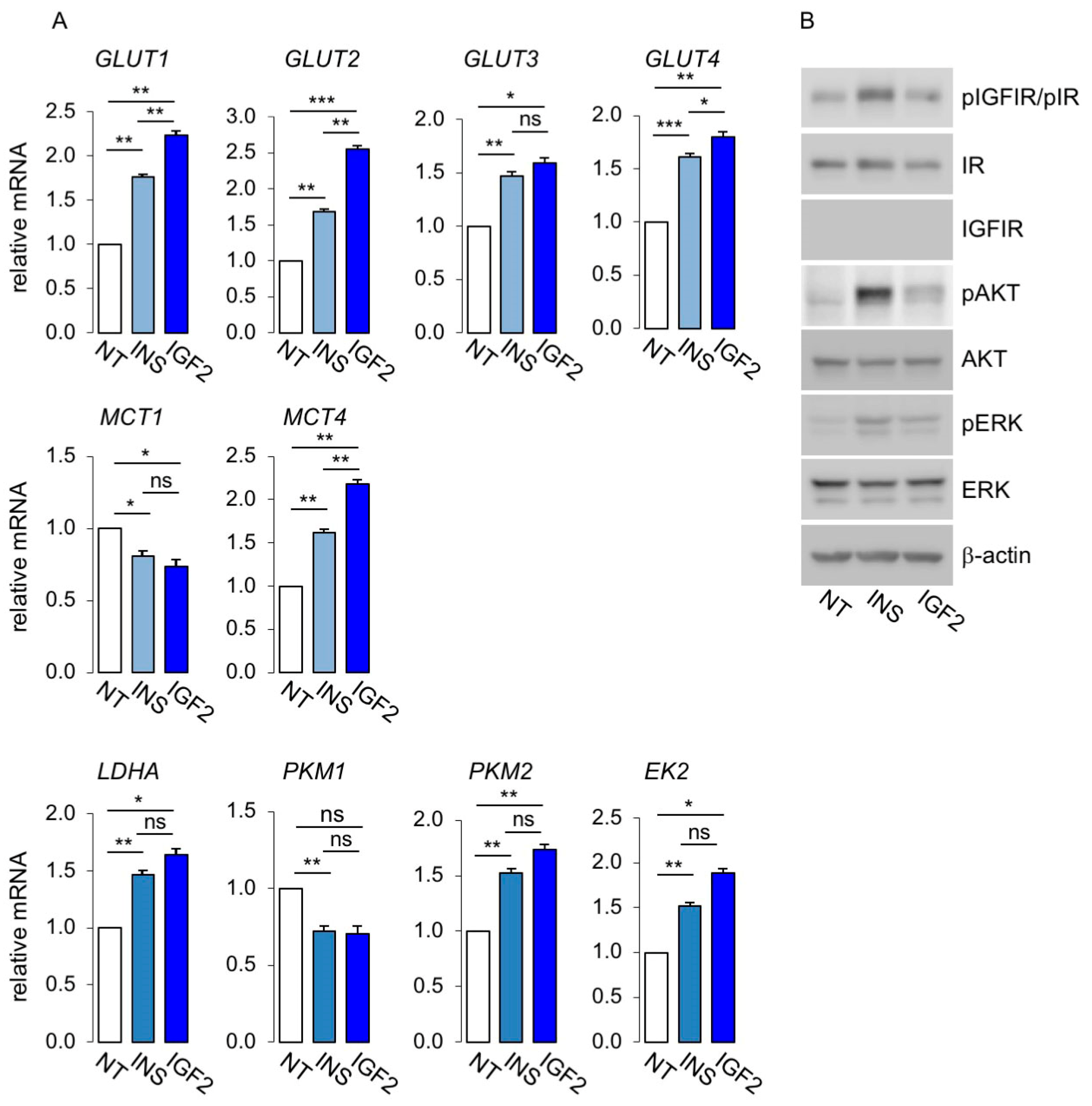
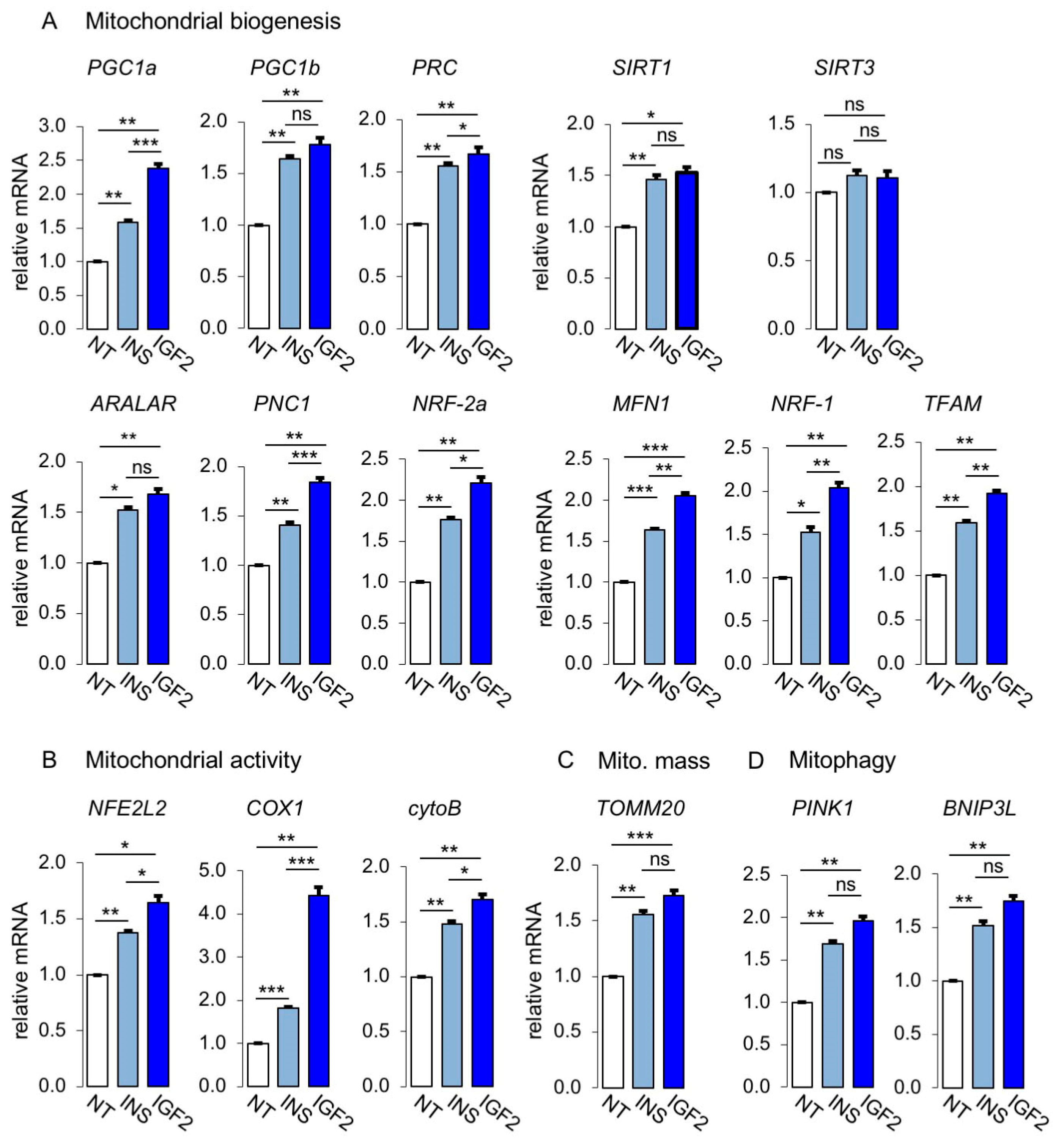
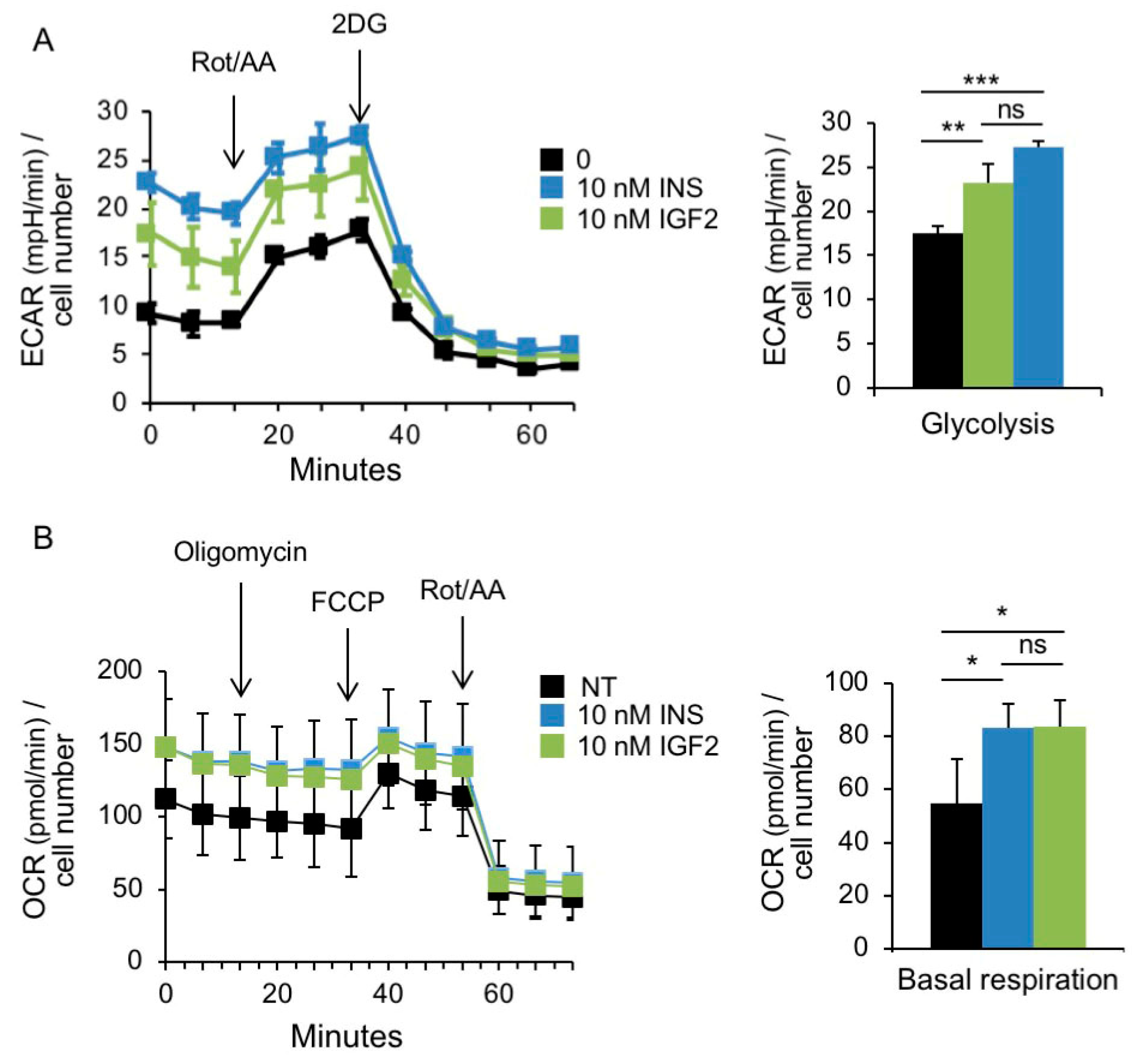
3.3. Metabolic Effects of Insulin and IGF2 in MCF7IGF1R-ve/IR-A Cells
3.4. In BC Cells, Combination Treatment with Inhibitors of Glycolysis and Mitochondrial Respiration Has Additive Inhibitory Effects on IR-A Driven Migration and Colony Formation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R. The Emerging Role of Insulin Receptor Isoforms in Thyroid Cancer: Clinical Implications and New Perspectives. Int. J. Mol. Sci. 2018, 19, 3814. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Palladino, C.; Spoleti, C.; Massimino, M.; Vigneri, P.; Purrello, M.; Ragusa, M.; Morrione, A.; et al. Discoidin domain receptor 1 modulates insulin receptor signaling and biological responses in breast cancer cells. Oncotarget 2017, 8, 43248–43270. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Cantafio, P.; Massimino, M.; Lappano, R.; Vigneri, P.; Ciuni, R.; Gangemi, P.; Morrione, A.; Malaguarnera, R.; et al. DDR1 regulates thyroid cancer cell differentiation via IGF-2/IR-A autocrine signaling loop. Endocr.-Relat. Cancer 2019, 26, 197–214. [Google Scholar] [CrossRef]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Frasca, F.; Garozzo, A.; Giani, F.; Pandini, G.; Vella, V.; Vigneri, R.; Belfiore, A. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J. Clin. Endocrinol. Metab. 2011, 96, 766–774. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Blackburn, O.A.; Marchildon, F.; Cohen, P. Insights into the Link Between Obesity and Cancer. Curr. Obes. Rep. 2017, 6, 195–203. [Google Scholar] [CrossRef]
- Godsland, I.F. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin. Sci. 2009, 118, 315–332. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Vella, V.; Nicolosi, M.L.; Belfiore, A. Insulin Resistance: Any Role in the Changing Epidemiology of Thyroid Cancer? Front. Endocrinol. 2017, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Pothiwala, P.; Jain, S.K.; Yaturu, S. Metabolic syndrome and cancer. Metab. Syndr. Relat. Disord. 2009, 7, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-gamma Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.E.; Sorensen, P.H. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin. Ther. Targets 2012, 16, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin receptor activation by IGF-II in breast cancers: Evidence for a new autocrine/paracrine mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Conte, E.; Medico, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann. N. Y. Acad. Sci. 2004, 1028, 450–456. [Google Scholar] [CrossRef]
- Belfiore, A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007, 13, 671–686. [Google Scholar] [CrossRef]
- Belfiore, A.; Pandini, G.; Vella, V.; Squatrito, S.; Vigneri, R. Insulin/IGF-I hybrid receptors play a major role in IGF-I signaling in thyroid cancer. Biochimie 1999, 81, 403–407. [Google Scholar] [CrossRef]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial Metabolism: Yin and Yang for Tumor Progression. Trends Endocrinol. Metab. TEM 2017, 28, 748–757. [Google Scholar] [CrossRef]
- Belfiore, A.; Frittitta, L.; Costantino, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Goldfine, I.D.; Vigneri, R. Insulin receptors in breast cancer. Ann. N. Y. Acad. Sci. 1996, 784, 173–188. [Google Scholar] [CrossRef]
- Frittitta, L.; Cerrato, A.; Sacco, M.G.; Weidner, N.; Goldfine, I.D.; Vigneri, R. The insulin receptor content is increased in breast cancers initiated by three different oncogenes in transgenic mice. Breast Cancer Res. Treat. 1997, 45, 141–147. [Google Scholar] [CrossRef]
- Papa, V.; Pezzino, V.; Costantino, A.; Belfiore, A.; Giuffrida, D.; Frittitta, L.; Vannelli, G.B.; Brand, R.; Goldfine, I.D.; Vigneri, R. Elevated insulin receptor content in human breast cancer. J. Clin. Investig. 1990, 86, 1503–1510. [Google Scholar] [CrossRef]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; Alikhani, N.; Tobin-Hess, A.; Blank, J.; Buffin, N.J.; Zelenko, Z.; Tennagels, N.; Werner, U.; LeRoith, D. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes 2013, 62, 3553–3560. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frittitta, L.; Vigneri, R.; Papa, V.; Goldfine, I.D.; Grasso, G.; Trischitta, V. Structural and functional studies of insulin receptors in human breast cancer. Breast Cancer Res. Treat. 1993, 25, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Happerfield, L.C.; Miles, D.W.; Barnes, D.M.; Thomsen, L.L.; Smith, P.; Hanby, A. The localization of the insulin-like growth factor receptor 1 (IGFR-1) in benign and malignant breast tissue. J. Pathol. 1997, 183, 412–417. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, Y.H.; Park, Y.L.; Sohn, J.H.; Kim, H.S. Prognostic significance of insulin growth factor-I receptor and insulin growth factor binding protein-3 expression in primary breast cancer. Oncol. Rep. 2010, 23, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Peiro, G.; Adrover, E.; Sanchez-Tejada, L.; Lerma, E.; Planelles, M.; Sanchez-Paya, J.; Aranda, F.I.; Giner, D.; Gutierrez-Avino, F.J. Increased insulin-like growth factor-1 receptor mRNA expression predicts poor survival in immunophenotypes of early breast carcinoma. Mod. Pathol. 2011, 24, 201–208. [Google Scholar] [CrossRef][Green Version]
- Jiang, W.; Hua, R.; Wei, M.; Li, C.; Qiu, Z.; Yang, X.; Zhang, C. An optimized method for high-titer lentivirus preparations without ultracentrifugation. Sci. Rep. 2015, 5, 13875. [Google Scholar] [CrossRef]
- Chang, C.C.; Hsu, W.H.; Wang, C.C.; Chou, C.H.; Kuo, M.Y.; Lin, B.R.; Chen, S.T.; Tai, S.K.; Kuo, M.L.; Yang, M.H. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013, 73, 4147–4157. [Google Scholar] [CrossRef]
- Frasca, F.; Vella, V.; Nicolosi, M.L.; Messina, R.L.; Giani, F.; Lotta, S.; Vigneri, P.; Regalbuto, C.; Vigneri, R. Thyroid cancer cell resistance to gefitinib depends on the constitutive oncogenic activation of the ERK pathway. J. Clin. Endocrinol. Metab. 2013, 98, 2502–2512. [Google Scholar] [CrossRef]
- Pacher, M.; Seewald, M.J.; Mikula, M.; Oehler, S.; Mogg, M.; Vinatzer, U.; Eger, A.; Schweifer, N.; Varecka, R.; Sommergruber, W.; et al. Impact of constitutive IGF1/IGF2 stimulation on the transcriptional program of human breast cancer cells. Carcinogenesis 2007, 28, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Nicolosi, M.L.; Sacco, A.; Morcavallo, A.; Vella, V.; Voci, C.; Spatuzza, M.; Xu, S.Q.; Iozzo, R.V.; Vigneri, R.; et al. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget 2015, 6, 16084–16105. [Google Scholar] [CrossRef] [PubMed]
- Slaaby, R. Specific insulin/IGF1 hybrid receptor activation assay reveals IGF1 as a more potent ligand than insulin. Sci. Rep. 2015, 5, 7911. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed]
- van Roozendaal, C.E.; Gillis, A.J.; Klijn, J.G.; van Ooijen, B.; Claassen, C.J.; Eggermont, A.M.; Henzen-Logmans, S.C.; Oosterhuis, J.W.; Foekens, J.A.; Looijenga, L.H. Loss of imprinting of IGF2 and not H19 in breast cancer, adjacent normal tissue and derived fibroblast cultures. FEBS Lett. 1998, 437, 107–111. [Google Scholar] [CrossRef]
- Ito, Y.; Koessler, T.; Ibrahim, A.E.; Rai, S.; Vowler, S.L.; Abu-Amero, S.; Silva, A.L.; Maia, A.T.; Huddleston, J.E.; Uribe-Lewis, S.; et al. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum. Mol. Genet. 2008, 17, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Yballe, C.M.; Vu, T.H.; Hoffman, A.R. Imprinting and expression of insulin-like growth factor-II and H19 in normal breast tissue and breast tumor. J. Clin. Endocrinol. Metab. 1996, 81, 1607–1612. [Google Scholar] [CrossRef][Green Version]
- Lee, J.S.; Kang, J.H.; Boo, H.J.; Hwang, S.J.; Hong, S.; Lee, S.C.; Park, Y.J.; Chung, T.M.; Youn, H.; Mi Lee, S.; et al. STAT3-mediated IGF-2 secretion in the tumour microenvironment elicits innate resistance to anti-IGF-1R antibody. Nat. Commun. 2015, 6, 8499. [Google Scholar] [CrossRef]
- Lee, S.C.; Min, H.Y.; Jung, H.J.; Park, K.H.; Hyun, S.Y.; Cho, J.; Woo, J.K.; Kwon, S.J.; Lee, H.J.; Johnson, F.M.; et al. Essential role of insulin-like growth factor 2 in resistance to histone deacetylase inhibitors. Oncogene 2016, 35, 5515–5526. [Google Scholar] [CrossRef]
- Unger, C.; Kramer, N.; Unterleuthner, D.; Scherzer, M.; Burian, A.; Rudisch, A.; Stadler, M.; Schlederer, M.; Lenhardt, D.; Riedl, A.; et al. Stromal-derived IGF2 promotes colon cancer progression via paracrine and autocrine mechanisms. Oncogene 2017, 36, 5341–5355. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.Y.; Yang, J.T.; Huang, T.H.; Liu, H.W. Crosstalk with cancer-associated fibroblasts increases the growth and radiation survival of cervical cancer cells. Radiat. Res. 2014, 181, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Mutgan, A.C.; Besikcioglu, H.E.; Wang, S.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Insulin/IGF-driven cancer cell-stroma crosstalk as a novel therapeutic target in pancreatic cancer. Mol. Cancer 2018, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.A.; Cullen, K.J. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. Breast Cancer Res. Treat. 1998, 47, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Saleh, A.M.; Al-Aqeel, S.M.; Shamsa, H.B.; Al-Bawab, A.; Al Dubayee, M.; Ahmed, A.A. Quantification of insulin receptor mRNA splice variants as a diagnostic tumor marker in breast cancer. Cancer Biomark. 2015, 15, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Morehouse, C.; Streicher, K.; Higgs, B.W.; Gao, J.; Czapiga, M.; Boutrin, A.; Zhu, W.; Brohawn, P.; Chang, Y.; et al. Altered expression of insulin receptor isoforms in breast cancer. PLoS ONE 2011, 6, e26177. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Sciacca, L.; Pezzino, V.; Squatrito, S.; Belfiore, A.; Vigneri, R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch. Physiol. Biochem. 2008, 114, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bjorner, S.; Rosendahl, A.H.; Simonsson, M.; Markkula, A.; Jirstrom, K.; Borgquist, S.; Rose, C.; Ingvar, C.; Jernstrom, H. Combined and individual tumor-specific expression of insulin-like growth factor-I receptor, insulin receptor and phospho-insulin-like growth factor-I receptor/insulin receptor in primary breast cancer: Implications for prognosis in different treatment groups. Oncotarget 2017, 8, 9093–9107. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Sapio, L.; Spina, A.; Naviglio, D.; Calogero, A.; Naviglio, S. Lactic dehydrogenase and cancer: An overview. Front. Biosci. 2015, 20, 1234–1249. [Google Scholar]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic Acid: No Longer an Inert and End-Product of Glycolysis. Physiology 2017, 32, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Ojo, D.; Yan, J.; Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 2015, 356, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Siddiqui, F.A.; Gupta, V.; Chattopadhyay, S.; Gopinath, P.; Kumar, B.; Manvati, S.; Chaman, N.; Bamezai, R.N. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol. Cancer 2013, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.Y.; Liu, W.T.; Sun, H.R.; Ge, X.; Shi, Z.M.; Wang, M.; Li, W.; Zhang, J.Y.; Liu, L.Z.; Jiang, B.H. IGF-1-mediated PKM2/beta-catenin/miR-152 regulatory circuit in breast cancer. Sci. Rep. 2017, 7, 15897. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Hofmann, M.H.; Garin-Chesa, P.; Schweifer, N.; Wernitznig, A.; Fischer, S.; Jeschko, A.; Meyer, R.; Moll, J.; Pecina, T.; et al. The IGF1R/INSR Inhibitor BI 885578 Selectively Inhibits Growth of IGF2-Overexpressing Colorectal Cancer Tumors and Potentiates the Efficacy of Anti-VEGF Therapy. Mol. Cancer Ther. 2017, 16, 2223–2233. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Belfiore, A. The insulin receptor: A new target for cancer therapy. Front. Endocrinol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Blanquart, C.; Achi, J.; Issad, T. Characterization of IRA/IRB hybrid insulin receptors using bioluminescence resonance energy transfer. Biochem. Pharmacol. 2008, 76, 873–883. [Google Scholar] [CrossRef]
- Morcavallo, A.; Gaspari, M.; Pandini, G.; Palummo, A.; Cuda, G.; Larsen, M.R.; Vigneri, R.; Belfiore, A. Research resource: New and diverse substrates for the insulin receptor isoform A revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol. Endocrinol. 2011, 25, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Morcavallo, A.; Pandini, G.; Vigneri, R.; Belfiore, A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 2009, 150, 3594–3602. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Milazzo, G.; Giorgino, F.; Damante, G.; Sung, C.; Stampfer, M.R.; Vigneri, R.; Goldfine, I.D.; Belfiore, A. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992, 52, 3924–3930. [Google Scholar] [PubMed]
- Liu, S.; Tang, Y.; Yan, M.; Jiang, W. PIK3CA mutation sensitizes breast cancer cells to synergistic therapy of PI3K inhibition and AMPK activation. Investig. New Drugs 2018, 36, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Costantino, A.; Frasca, F.; Pandini, G.; Mineo, R.; Vigneri, P.; Maddux, B.; Goldfine, I.D.; Vigneri, R. Overexpression of membrane glycoprotein PC-1 in MDA-MB231 breast cancer cells is associated with inhibition of insulin receptor tyrosine kinase activity. Mol. Endocrinol. 1996, 10, 1318–1326. [Google Scholar] [CrossRef][Green Version]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers |
|---|---|
| β-actin | Fw 5′-GACAGGATGCAGAAGGAGATCACT-3′ Rv 5′-TGATCCACATCTGCTGGAACCT-3′ |
| ARALAR | Fw 5′-CGAGACATTCCCTTCTCTGC-3′ Rv 5′-GTCCCTTTCCAAAATGCTGA-3′ |
| GAPDH | Fw 5′-ACCCACTCCTCCACCTTTG-3′ Rv 5′-CTCTTGTGCTCTTGCTGGG-3′ |
| GLUT1 | Fw 5′-ATCGTGGCCATCTTTGGCTTTGTG-3′ Rv 5′-CTGGAAGCACATGCCCACAATGAA-3′ |
| GLUT2 | Fw 5′-AGCTGCATTCAGCAATTGGACCTG-3′ Rv 5′-ATGTGAACAGGGTAAAGGCCAGGA-3′ |
| GLUT3 | Fw 5′-AGCTCTCTGGGATCAATGCTGTGT-3′ Rv 5′-ATGGTGGCATAGATGGGCTCTTGA-3′ |
| GLUT4 | Fw 5′-TCGTGGCCATATTTGGCTTTGTGG-3′ Rv 5′-TAAGGACCCATAGCATCCGCAACA-3′ |
| Hexokinase2 | Fw 5′-TTGGCTTTTGCTTGGCAGAG-3′ Rv 5′-TCTTAATAGGGCCAAGCTCAGC-3′ |
| IGF2 | Fw 5′-GACCGCGGCTTCTACTTCAG-3′ Rv 5′-AGAACTTGCCCACGGGGTAT-3′ |
| IR isoform | Fw 5′-CCAAAGACAGACTCTTCAGAT-3′ Rv 5′-AACATCGCCAAGGGACCTGC-3′ |
| IR total | Fw 5′-CGTGGAGGATAATTACATCGTGTT-3′ Rv 5′-TGGTCGGGCAAACTTTCTG-3′ |
| LDHA | Fw 5′-CATGGCAGCCTTTTCCTTAG-3′ Rv 5′-ATGACCAGCTTGGAGTTTGC -3′ |
| MCT1 | Fw 5′-GTGGAATGCTGTCCTGTCCTC-3′ Rv 5′-TCGATAATTGATGCCCATGCC-3′ |
| MCT4 | Fw 5′-ATTGGCCTGGTGCTGCTGATG-3′ Rv 5′-CGAGTCTGCAGGAGGCTTGTG-3′ |
| MFN1 | Fw 5′-TGTTTTGGTCGCAAACTCTG-3′ Rv 5′-CTGTCTGCGTACGTCTTCCA-3′ |
| b2 microglobulin | Fw 5′-TGCTGTCTCCATGTTTGATGTATCT-3′ Rv 5′-TCTCTGCTCCCCACCTCTAAGT-3′ |
| NRF1 | Fw 5′-CGCTCTGAGAACTTCATGGAGGAACAC-3′ Rv 5′-GCCACATGGACCTGCTGCACTT-3′ |
| NRF2 | Fw 5’-AACAAGAACGCCTTGGGATAC-3′ Rv 5’-GTGAGGTCTATATCGGTCATGCT-3’ |
| PGC1α | Fw 5’-CAAGCCAAACCAACAACTTTATCTCT-3′ Rv 5’-CACACTTAAGGTGCGTTCAATAGTC-3′ |
| PGC1β | Fw 5’-GCTCAAGCTCTGGCTCTTCA-3′ Rv 5’-ATGCTTGGCGTTCTGTCTGA-3’ |
| PKM1 | Fw 5′-TGAAGAACTTGTGCGAGCCT-3′ Rv 5′-GCCAGACTCCGTCAGAACTA-3′ |
| PKM2 | Fw 5′-TTACCAGCGACCCCACAGAA-3′ Rv 5′-GACGATTATGGCCCCACTGC-3′ |
| PNC1 | Fw 5′-GCTCTGCAGCTTTTATCACAAATTC-3′ Rv 5′-AACGTAACGAGCACACTGGAGTG-3′ |
| PRC | Fw 5′-CAAGCAGAAACAGAAGAGAGAAG-3′ Rv 5′-GGTGGGATGACAAGACAAGG-3′ |
| tRNAleu (UUR) mtDNA | Fw 5′-CACCCAAGAACAGGGTTTGT-3′ Rv 5′-TGGCCATGGGTATGTTGTTA-3′ |
| hRP-S9 | Fw 5′-CTGGGTTTGTCGCAAAACTT-3′ Rv 5′-GTGGGTCCTTCTCATCAAGC-3′ |
| TFAM | Fw 5’-ACTGCGCTCCCCCTTCAG-3′ Rv 5’-ACAGATGAAAACCACCTCGGTAA-3′ |
| TOMM20 | Fw 5′-GCTGGGCTTTCCAAGTTACC-3′ Rv 5′-TGTCAGATGGTCTACGCCCT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vella, V.; Nicolosi, M.L.; Giuliano, M.; Morrione, A.; Malaguarnera, R.; Belfiore, A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells 2019, 8, 1017. https://doi.org/10.3390/cells8091017
Vella V, Nicolosi ML, Giuliano M, Morrione A, Malaguarnera R, Belfiore A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells. 2019; 8(9):1017. https://doi.org/10.3390/cells8091017
Chicago/Turabian StyleVella, Veronica, Maria Luisa Nicolosi, Marika Giuliano, Andrea Morrione, Roberta Malaguarnera, and Antonino Belfiore. 2019. "Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation" Cells 8, no. 9: 1017. https://doi.org/10.3390/cells8091017
APA StyleVella, V., Nicolosi, M. L., Giuliano, M., Morrione, A., Malaguarnera, R., & Belfiore, A. (2019). Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells, 8(9), 1017. https://doi.org/10.3390/cells8091017