Approaches for Studying Autophagy in Caenorhabditis elegans

Abstract
:1. C. elegans, an Animal Model to Study Autophagy
1.1. General Experimental Advantages
1.2. Physiological Processes Involving Autophagy in C. elegans
2. Methods to Monitor Autophagy in C. elegans
2.1. Imaging Autophagy
2.1.1. In Vivo Imaging
2.1.2. Immunostaining of Autophagy Proteins
2.1.3. Electron Microscopy
2.2. Molecular Approaches
2.2.1. Monitoring Autophagic Flux by Western Blotting
2.2.2. Monitoring Autophagy Genes Expression by RT-qPCR
2.3. Modifying Autophagy
2.3.1. Genetic Approaches
2.3.2. Pharmacological Treatments
3. Tools to Monitor Selective Autophagy
3.1. Aggrephagy
3.1.1. P Granules Degradation through Aggrephagy
3.1.2. Poly-Q Aggregates
3.2. Selective Degradation of Mitochondria by Autophagy
3.2.1. Mitophagy of Paternal Mitochondria
3.2.2. Mitophagy in Stress Conditions
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Paix, A.; Folkmann, A.; Rasoloson, D.; Seydoux, G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics 2015, 201, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, Y.; Wang, F.; Noda, N.N.; Zhang, H. Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 29457–29467. [Google Scholar] [CrossRef] [PubMed]
- Jenzer, C.; Simionato, E.; Legouis, R. Tools and methods to analyze autophagy in C. elegans. Methods 2015, 75, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, J.T.; Guo, B.; Hansen, M.; Jia, K.; Kovács, A.L.; Kumsta, C.; Lapierre, L.R.; Legouis, R.; Lin, L.; et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 2015, 11, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, M.E.; Tavernarakis, N. Monitoring autophagic responses in Caenorhabditis elegans. Methods Enzymol. 2017, 588, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, P.; Huang, X.; Hu, W.; Guo, B.; Wu, F.; Lin, L.; Kovács, A.L.; Yu, L.; Zhang, H. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell 2011, 21, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, L.; Zhou, Z.; Yang, P.; Tian, E.; Zhang, K.; Zhao, Y.; Li, Z.; Song, B.; Han, J.; et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell 2009, 136, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, F.; Wang, X.; Du, H.; Wang, X.; Zhang, H. The two C. elegans ATG-16 homologs have partially redundant functions in the basal autophagy pathway. Autophagy 2013, 9, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Z.; Hu, W.; Ren, H.; Tian, E.; Zhao, Y.; Lu, Q.; Huang, X.; Yang, P.; Li, X.; et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010, 141, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Huang, X.; Zhang, P.; Qi, L.; Liang, Q.; Zhang, X.; Huang, J.; Fang, B.; Hou, W.; Han, J.; et al. Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO Rep. 2014, 15, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Ruck, A.; Attonito, J.; Garces, K.T.; Nuñez, L.; Palmisano, N.J.; Rubel, Z.; Bai, Z.; Nguyen, K.C.Q.; Sun, L.; Grant, B.D.; et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy 2011, 7, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.; Wang, F.; Han, J.; Zhang, H. epg-1 functions in autophagy-regulated processes and may encode a highly divergent Atg13 homolog in C. elegans. Autophagy 2009, 5, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yang, P.; Huang, X.; Zhang, H.; Lu, Q.; Zhang, H. The scaffold protein EPG-7 links cargo–receptor complexes with the autophagic assembly machinery. J. Cell Biol. 2013, 201, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, H. The coiled-coil domain protein EPG-8 plays an essential role in the autophagy pathway in C. elegans. Autophagy 2011, 7, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Yang, P.; Tian, E.; Han, J.; Zhang, H. The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy 2012, 8, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A Role for Autophagy in the Extension of Lifespan by Dietary Restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Gelino, S.; Meléndez, A.; Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 2011, 21, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Tóth, M.L.; Simon, P.; Kovács, A.L.; Vellai, T. Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J. Cell Sci. 2007, 120, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; You, Y.; Avery, L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007, 21, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Scott, B.A.; Crowder, C.M. Autophagy protects against hypoxic injury in C. elegans. Autophagy 2008, 4, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ookuma, S.; Nishida, E. Lifespan extension by suppression of autophagy genes in Caenorhabditis elegans. Genes Cells 2009, 14, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Michelet, X.; Djeddi, A.; Legouis, R. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 2010, 6, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Manil-Ségalen, M.; Lefebvre, C.; Jenzer, C.; Trichet, M.; Boulogne, C.; Satiat-Jeunemaitre, B.; Legouis, R. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev. Cell 2014, 28, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.; Garvis, S.; Bedet, C.; Palladino, F. The Caenorhabditis elegans HP1 family protein HPL-2 maintains ER homeostasis through the UPR and hormesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5956–5961. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Wicky, C.; Magnenat, L.; Tobler, H.; Mori, I.; Müller, F.; Ohshima, Y. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994, 8, 2389–2400. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.M.; Richmond, J.E.; Olsen, J.G.; Hall, D.H.; Bamber, B.A. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 2006, 26, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Roggo, L.; Bernard, V.; Kovacs, A.L.; Rose, A.M.; Savoy, F.; Zetka, M.; Wymann, M.P.; Müller, F. Membrane transport in Caenorhabditis elegans: An essential role for VPS34 at the nuclear membrane. EMBO J. 2002, 21, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Hall, D.H.; Hansen, M. Monitoring the role of autophagy in C. elegans aging. Methods Enzymol. 2008, 451, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.-L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Thomas, C.; Akbar, M.; Sun, Q.; Adams-Huet, B.; Gilpin, C.; Levine, B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc. Natl. Acad. Sci. USA 2009, 106, 14564–14569. [Google Scholar] [CrossRef] [PubMed]
- Curt, A.; Zhang, J.; Minnerly, J.; Jia, K. Intestinal autophagy activity is essential for host defense against Salmonella typhimurium infection in Caenorhabditis elegans. Dev. Comp. Immunol. 2014, 45, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Sigmond, T.; Fehér, J.; Baksa, A.; Pásti, G.; Pálfia, Z.; Takács-Vellai, K.; Kovács, J.; Vellai, T.; Kovács, A.L. Qualitative and quantitative characterization of autophagy in Caenorhabditis elegans by electron microscopy. Methods Enzymol. 2008, 451, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Kumsta, C.; Chang, J.T.; Schmalz, J.; Hansen, M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun. 2017, 8, 14337. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Levine, B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 2007, 3, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Conradt, B.; Xue, D. Programmed Cell Death. Available online: https://www.ncbi.nlm.nih.gov/books/NBK19668/ (accessed on 28 June 2017).
- Takacs-Vellai, K.; Vellai, T.; Puoti, A.; Passannante, M.; Wicky, C.; Streit, A.; Kovacs, A.L.; Müller, F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 2005, 15, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, G.; Trinkwalder, M.; Irmisch, L.; Wehman, A.M. C. elegans midbodies are released, phagocytosed and undergo LC3-dependent degradation independent of macroautophagy. J. Cell Sci. 2016, 129, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Samara, C.; Syntichaki, P.; Tavernarakis, N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008, 15, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Odedra, D.; Dikic, I.; Pohl, C. Autophagy and modular restructuring of metabolism control germline tumor differentiation and proliferation in C. elegans. Autophagy 2016, 12, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Gosai, S.J.; Kwak, J.H.; Luke, C.J.; Long, O.S.; King, D.E.; Kovatch, K.J.; Johnston, P.A.; Shun, T.Y.; Lazo, J.S.; Perlmutter, D.H.; et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS ONE 2010, 5, e15460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padman, B.S.; Nguyen, T.N.; Lazarou, M. Autophagosome formation and cargo sequestration in the absence of LC3/GABARAPs. Autophagy 2017, 13, 772–774. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Erdélyi, P.; Borsos, É.; Takács-Vellai, K.; Kovács, T.; Kovács, A.L.; Sigmond, T.; Hargitai, B.; Pásztor, L.; SenGupta, T.; Dengg, M.; et al. Shared developmental roles and transcriptional control of autophagy and apoptosis in Caenorhabditis elegans. J. Cell Sci. 2011, 124, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pak, S.C.; O’Reilly, L.P.; Benson, J.A.; Wang, Y.; Hidvegi, T.; Hale, P.; Dippold, C.; Ewing, M.; Silverman, G.A.; et al. Fluphenazine reduces proteotoxicity in C. elegans and mammalian models of alpha-1-antitrypsin deficiency. PLoS ONE 2014, 9, e87260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.H.; Hartwieg, E.; Nguyen, K.C.Q. Modern electron microscopy methods for C. elegans. Methods Cell Biol. 2012, 107, 93–149. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, N.J.; Meléndez, A. Detection of Autophagy in Caenorhabditis elegans by western blotting analysis of LGG-1. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot086512. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wu, F.; Zhang, H. Aggrephagy: Lessons from C. elegans. Biochem. J. 2013, 452, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Visvikis, O.; Ihuegbu, N.; Labed, S.A.; Luhachack, L.G.; Alves, A.-M.F.; Wollenberg, A.C.; Stuart, L.M.; Stormo, G.D.; Irazoqui, J.E. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014, 40, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, K.; Le, W. Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol. Sin. 2013, 34, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.-C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wu, Y.; Lu, Q.; Yan, J.; Zhang, H.; Wang, X. Autophagy genes coordinate with the class II PI/PtdIns 3-kinase PIKI-1 to regulate apoptotic cell clearance in C. elegans. Autophagy 2013, 9, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Qadota, H.; Inoue, M.; Hikita, T.; Köppen, M.; Hardin, J.D.; Amano, M.; Moerman, D.G.; Kaibuchi, K. Establishment of a tissue-specific RNAi system in C. elegans. Gene 2007, 400, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.B.; Johnson, C.D. Genetic pharmacology: Interactions between drugs and gene products in Caenorhabditis elegans. Methods Cell Biol. 1995, 48, 187–204. [Google Scholar] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Mariño, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J. Cell Biol. 2011, 192, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Ash, P.E.A.; Gowda, V.; Liu, L.; Shirihai, O.; Wolozin, B. Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol. Neurodegener. 2015, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Hird, S.N.; Paulsen, J.E.; Strome, S. Segregation of germ granules in living Caenorhabditis elegans embryos: Cell-type-specific mechanisms for cytoplasmic localisation. Development (Camb. Engl.) 1996, 122, 1303–1312. [Google Scholar]
- Morfini, G.; Pigino, G.; Brady, S.T. Polyglutamine expansion diseases: Failing to deliver. Trends Mol. Med. 2005, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Satyal, S.H.; Schmidt, E.; Kitagawa, K.; Sondheimer, N.; Lindquist, S.; Kramer, J.M.; Morimoto, R.I. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2000, 97, 5750–5755. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.H.; Lim, Y.; Oh, H.J.; Park, S.M.; Lee, S.-K.; Ahnn, J.; Kim, D.H.; Song, W.K.; Kwak, T.H.; Park, W.J. Pharmacological activation of Sirt1 ameliorates polyglutamine-induced toxicity through the regulation of autophagy. PLoS ONE 2013, 8, e64953. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Khan, L.A.; Yamanaka, T.; Nukina, N. Genetic impairment of autophagy intensifies expanded polyglutamine toxicity in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2008, 368, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.; Roberts, T.M.; Strome, S.; Pavalko, F.M.; Hogan, E. Monoclonal antibodies that recognize a polypeptide antigenic determinant shared by multiple Caenorhabditis elegans sperm-specific proteins. J. Cell Biol. 1986, 102, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Thomson, J.N. Monoclonal antibodies which distinguish certain classes of neuronal and supporting cells in the nervous tissue of the nematode Caenorhabditis elegans. J. Neurosci. 1985, 5, 643–653. [Google Scholar] [PubMed]
- Zhou, Q.; Li, H.; Li, H.; Nakagawa, A.; Lin, J.L.J.; Lee, E.-S.; Harry, B.L.; Skeen-Gaar, R.R.; Suehiro, Y.; William, D.; et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 2016, 353, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Lemire, B.D. Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem. Cell Biol. 2002, 80, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Allophagy: A macroautophagic process degrading spermatozoid-inherited organelles. Autophagy 2012, 8, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 2017, 7, 46208. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.-I.; Kanki, T. How autophagy eats large mitochondria: Autophagosome formation coupled with mitochondrial fragmentation. Autophagy 2017, 13, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Ackema, K.B.; Hench, J.; Böckler, S.; Wang, S.C.; Sauder, U.; Mergentaler, H.; Westermann, B.; Bard, F.; Frank, S.; Spang, A. The small GTPase Arf1 modulates mitochondrial morphology and function. EMBO J. 2014, 33, 2659–2675. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
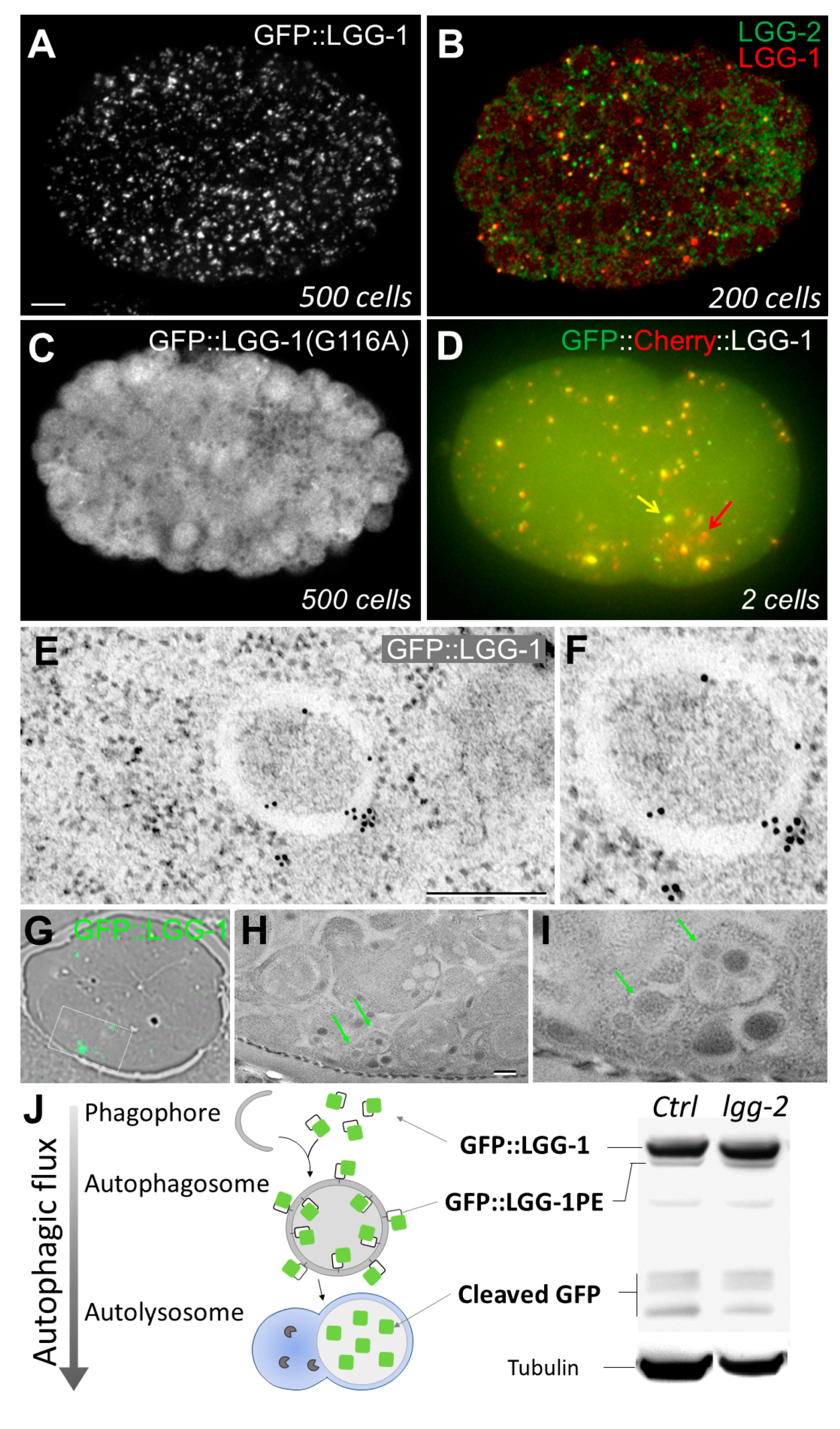


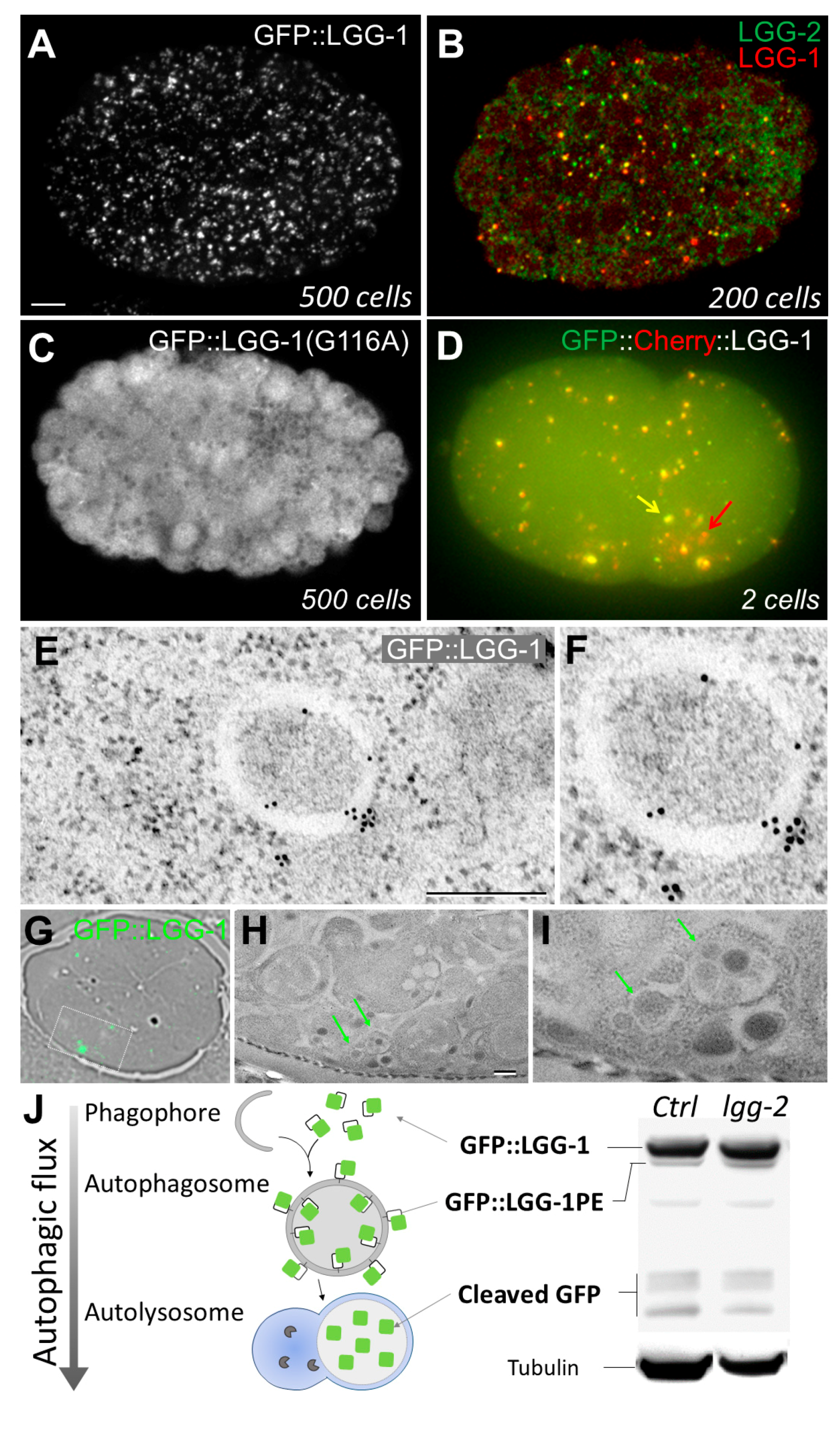{kind=link}
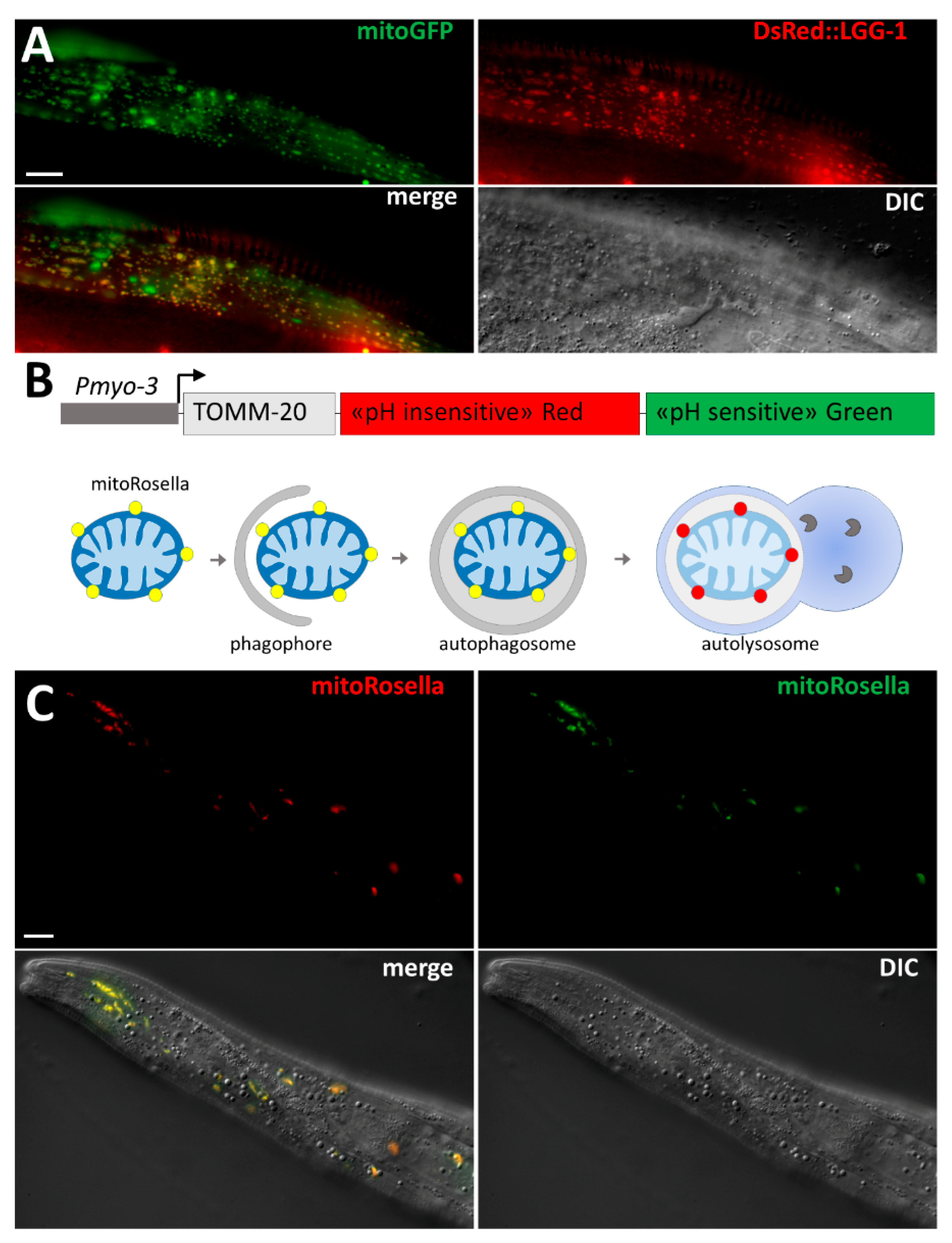{kind=link}
| C. elegans Genes | Mutant Alleles or RNAi | Mammalian Homologs | Yeast Genes | References |
|---|---|---|---|---|
| atg-2 | bp576 | ATG2 | ATG2 | [6] |
| atg-3 | bp412/RNAi | ATG3 | ATG3 | [7] |
| atg-4.1 | bp410 | ATG4A, ATG4B | ATG4 | [2] |
| atg-4.2 | tm3948 | ATG4C, ATG4D | ATG4 | [2] |
| atg-5 | bp546/RNAi | ATG5 | ATG5 | [8] |
| atg-7 | bp422/RNAi | ATG7 | ATG7 | [7] |
| atg-9 | bp564/RNAi | ATG9 | ATG9 | [6] |
| atg-10 | bp421, bp588/RNAi | ATG10 | ATG10 | [7] |
| atg-16.1 | gk668615d | ATG16L1 | ATG16 | [8] |
| atg-16.2 | bp636, ok3224 | ATG16L2 | [8] | |
| atg-18 | gk378/RNAi | WIPI1, WIPI2 | ATG18 | [9,10] |
| bec-1 | ok691, bp613, ok700/RNAi | BECN1 | VPS30/ATG6 | [10,11] |
| epg-1 | bp414 | KIAA0652 | ATG13 | [12] |
| epg-2 | bp444/RNAi | ? | VPS34 | [9] |
| epg-3 | bp405/RNAi | VPM1 | VPS34 | [9] |
| epg-4 | bp425/RNAi | EI24 | VPS34 | [9] |
| epg-5 | tm3425/RNAi | EPG5 | VPS34 | [9] |
| epg-6 | bp242 | WIPI3, WIPI4 | [6] | |
| epg-7 | tm2508 | FIP200 | VPS34 | [13] |
| epg-8 | bp251 | ATG14 | ATG14 | [14] |
| epg-9 | bp320 | ATG101 | VPS34 | [15] |
| let-363 | h98/RNAi | TOR | [16,17,18] | |
| lgg-1 | tm348/RNAi | GABARAP | ATG8 | [7,9,18,19,20,21,22,23,24,25] |
| lgg-2 | tm5755/RNAi | LC3 | ATG8 | [22,24,26] |
| lgg-3 | RNAi | ATG12 | ATG12 | [7,22] |
| pgl-3 | bp439 | ? | [12] | |
| rab-7 | ok511/RNAi | RAB7 | [19,23,24,25] | |
| sepa-1 | bp456 | ? | [9,12] | |
| sqst-1 | ok2892 | SQSTM1/p62 | [13] | |
| unc-51 | e369/RNAi | ULK1 | ATG1 | [18,27,28] |
| vps-34 | h741 | VPS34 | VPS34 | [29] |
| vps-39 | tm2253 | VPS39 | [25] | |
| vps-41 | ep402 | VPS4 | [25] |
| C. elegans Protein | Tools | Localization Pattern | References |
|---|---|---|---|
| LGG-1 | GFP, DsRed, GFP::Cherry, Cherry, mRFP; antibody | Puncta | [9,25,28,31,40,48] |
| GFP::LGG-1(G116A) | Diffuse | [25] | |
| LGG-2 | GFP; antibody | Puncta | [24] |
| GFP::LGG-2(G130A) | Diffuse | [23] | |
| BEC-1 | GFP, mRFP | Puncta/Patch | [28,38] |
| ATG-4.1 | GFP | Diffuse | [2] |
| ATG-9 | GFP | Diffuse | [6] |
| ATG-18 | GFP | Puncta | [47] |
| EPG-1 | GFP | Diffuse | [12] |
| EPG-2 | Antibody | Puncta | [9] |
| EPG-3 | GFP | Diffuse | [9] |
| EPG-4 | GFP | Diffuse | [9] |
| EPG-5 | GFP | Diffuse | [9] |
| EPG-6 | GFP | Diffuse | [6] |
| EPG-7 | GFP | Puncta | [13] |
| EPG-8 | GFP | Diffuse | [14] |
| LMP-1 | GFP | Puncta | [11] |
| SEPA-1 | GFP, RFP; antibody | Puncta/Patch | [7,12] |
| SQST-1 | GFP | Puncta/Patch | [9] |
| PGL-1 | GFP; antibody | Puncta/Patch | [7] |
| PGL-3 | Antibody | Puncta/Patch | [7] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Scarcelli, V.; Legouis, R. Approaches for Studying Autophagy in Caenorhabditis elegans. Cells 2017, 6, 27. https://doi.org/10.3390/cells6030027
Chen Y, Scarcelli V, Legouis R. Approaches for Studying Autophagy in Caenorhabditis elegans. Cells. 2017; 6(3):27. https://doi.org/10.3390/cells6030027
Chicago/Turabian StyleChen, Yanfang, Vincent Scarcelli, and Renaud Legouis. 2017. "Approaches for Studying Autophagy in Caenorhabditis elegans" Cells 6, no. 3: 27. https://doi.org/10.3390/cells6030027