The Role of Canonical Transient Receptor Potential Channels in Seizure and Excitotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Group I mGluRs in Seizure and Excitotoxicity
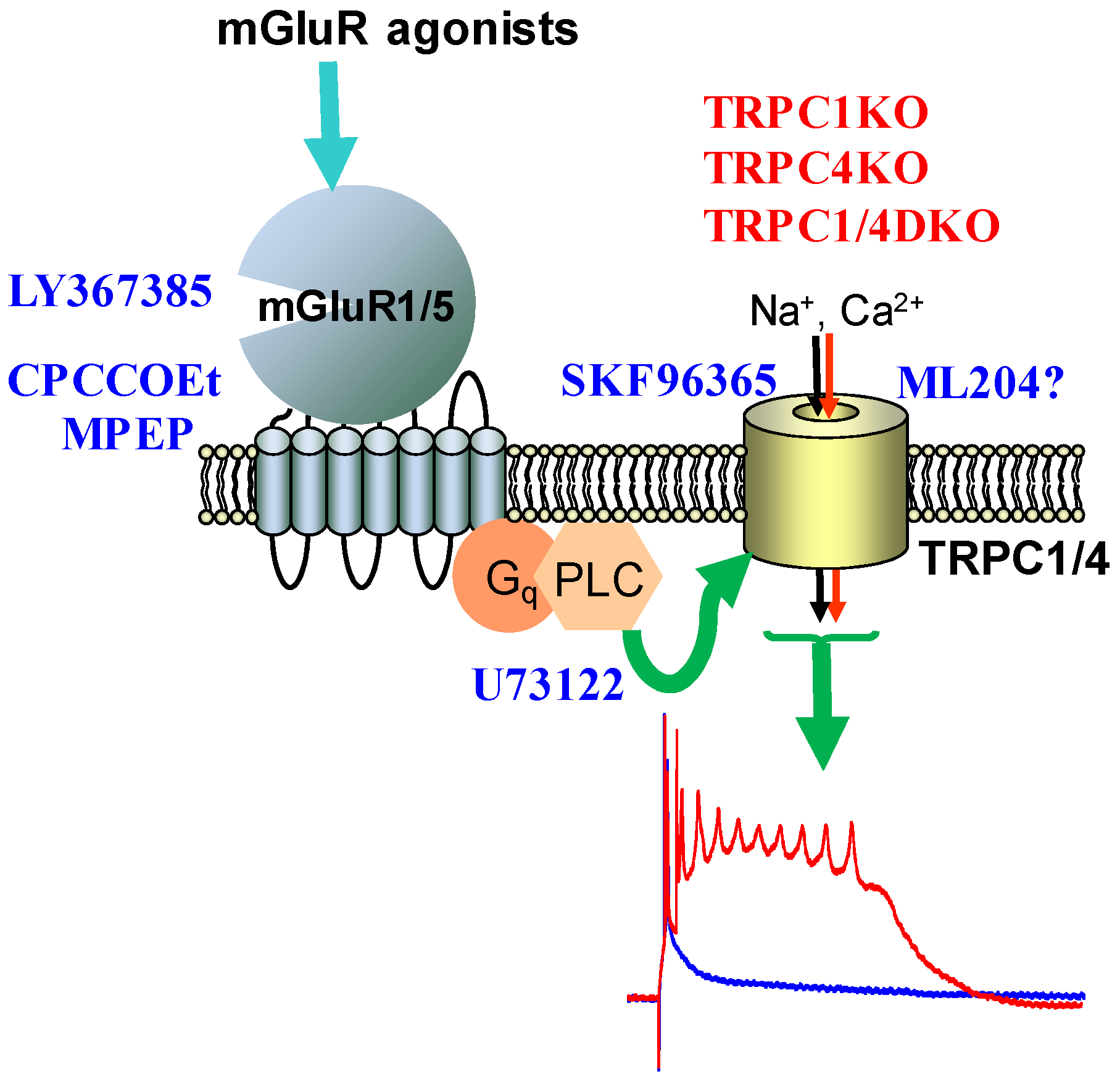
3. Expression, Structure and Pharmacology of TRPC Channels
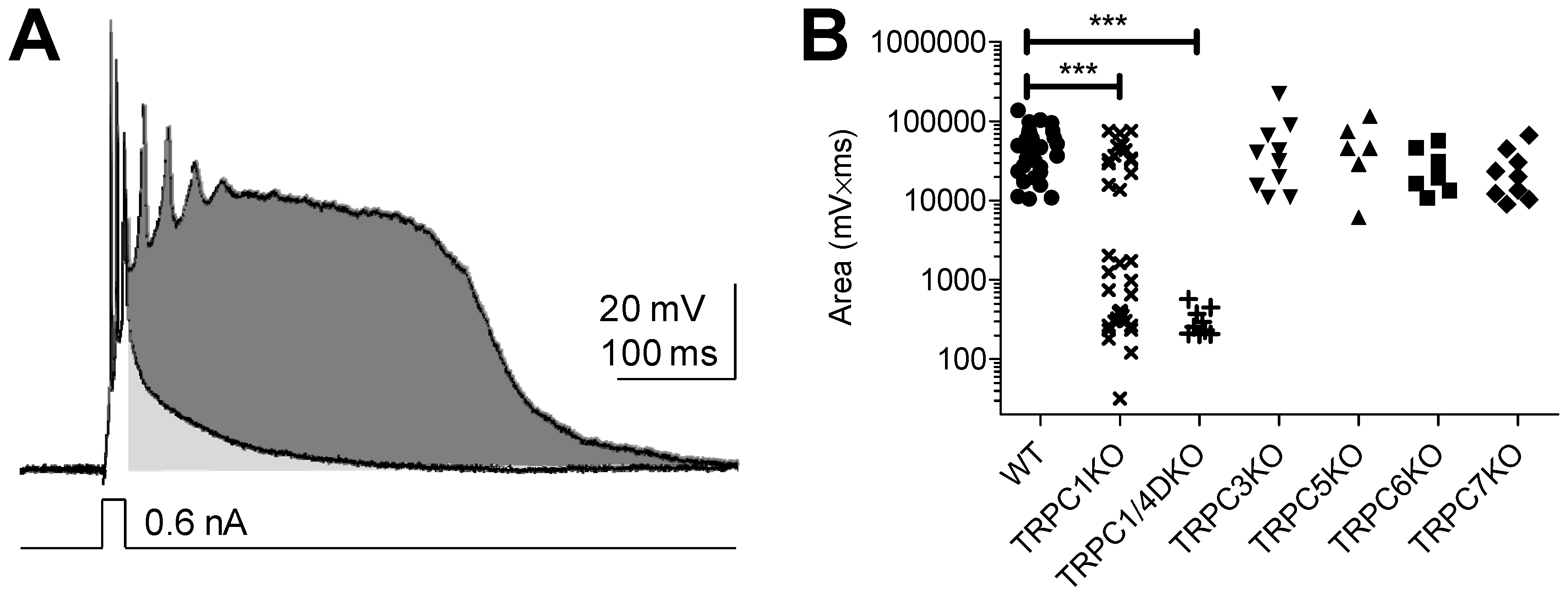
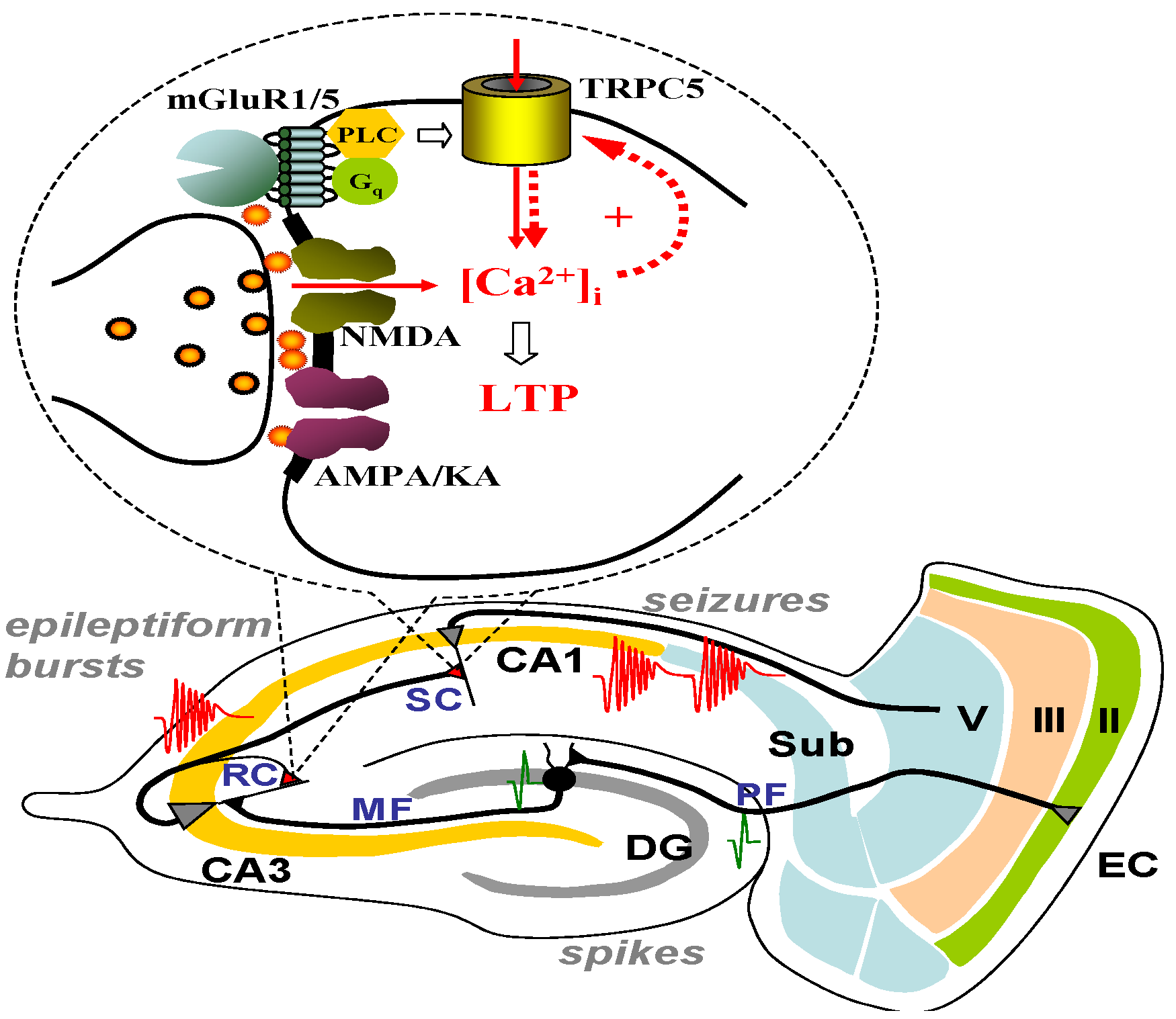
4. TRPC Channels, the Ca2+-Activated Non-Selective (CAN) Current and Epileptiform Burst Firing in the Lateral Septum
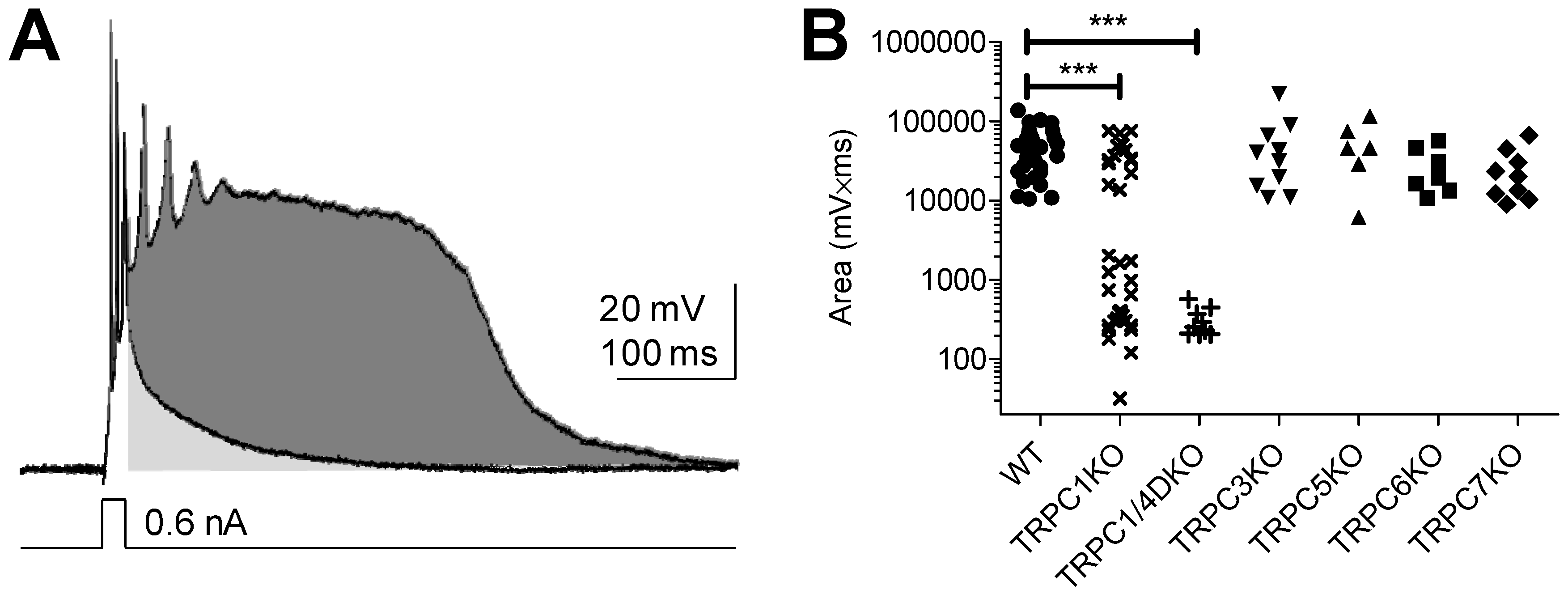
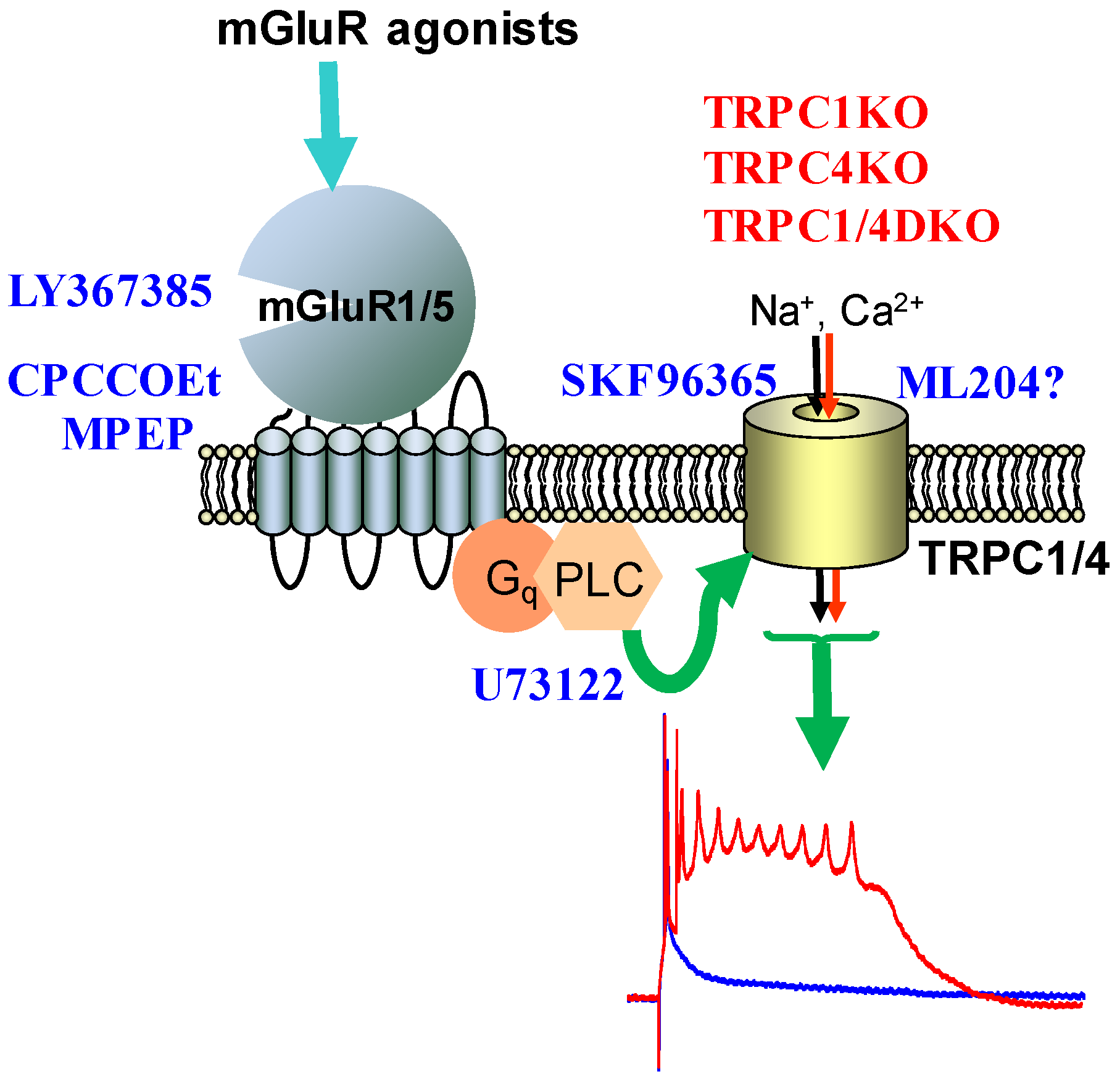
5. TRPC Channels and Epileptiform Burst Firing in the Hippocampus
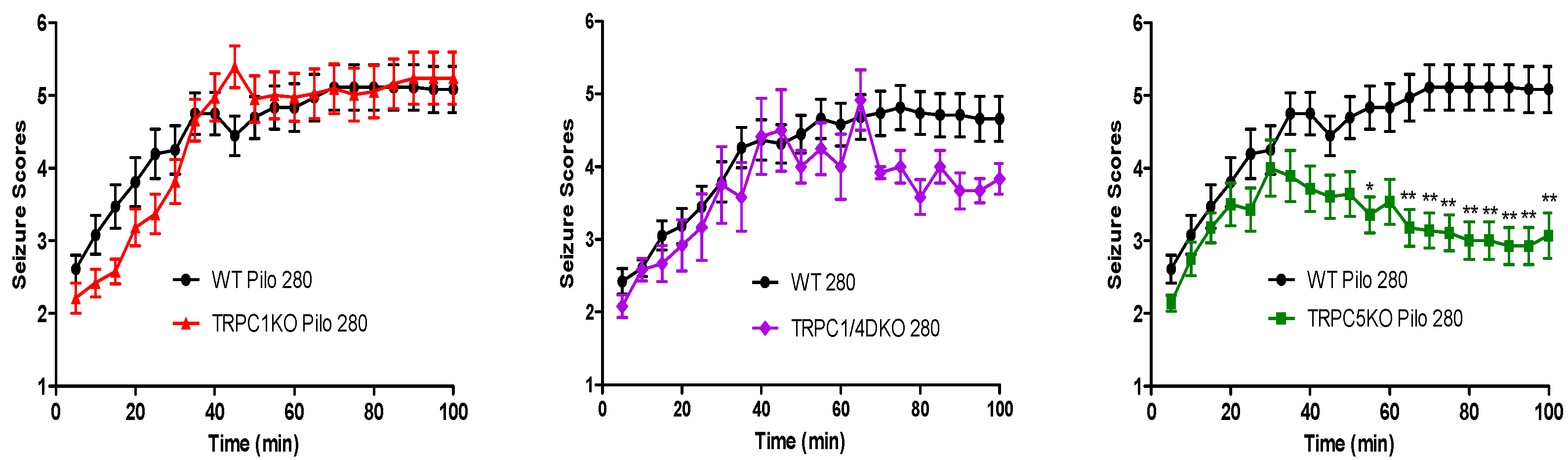
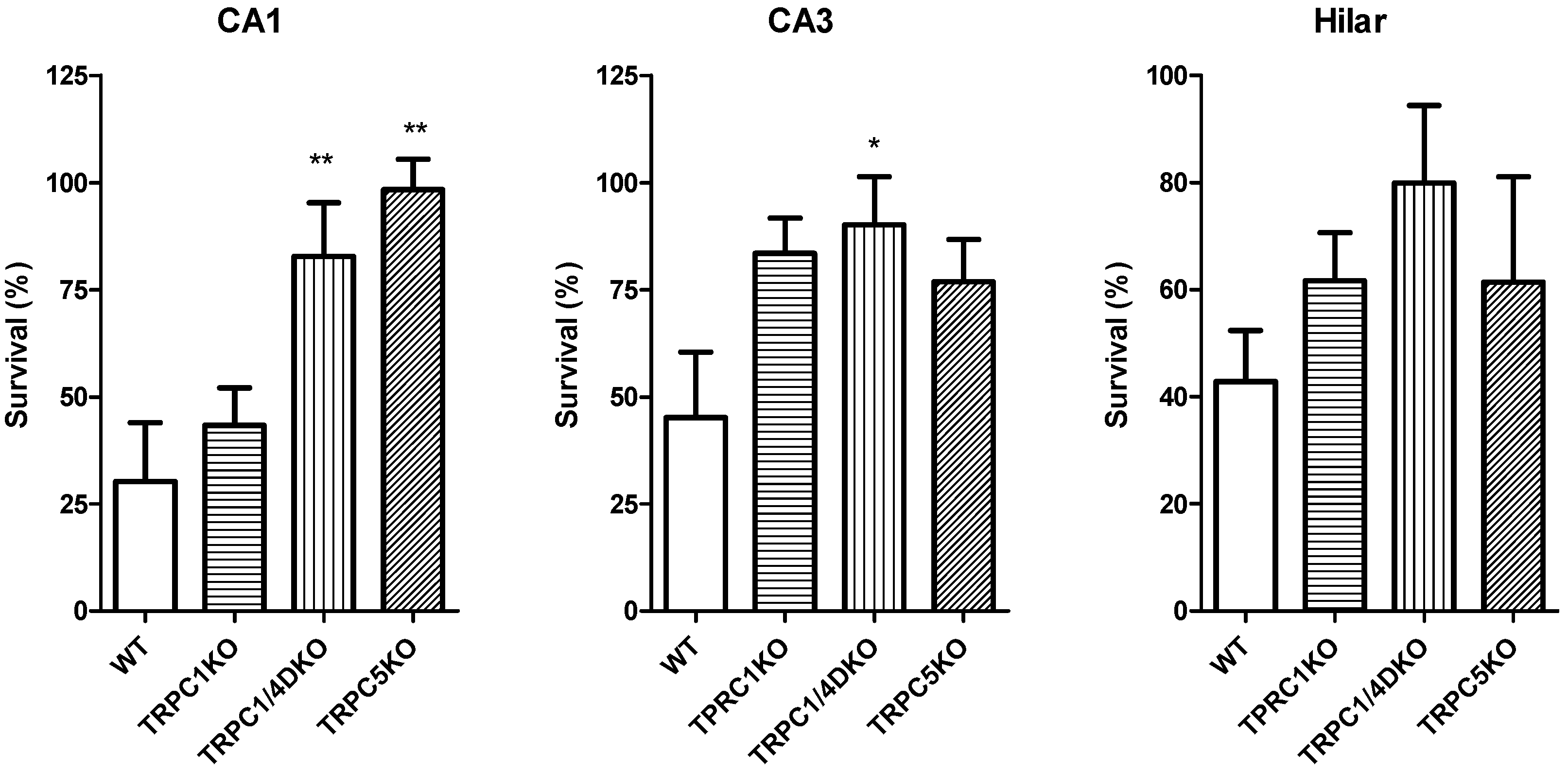
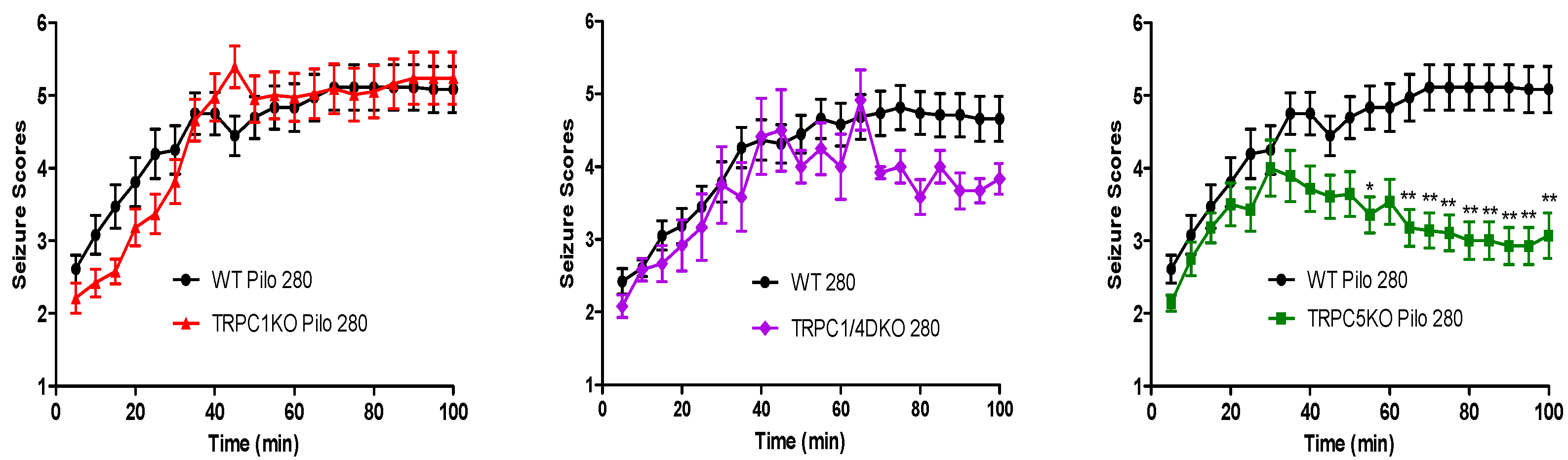
6. The Role of TRPC1, 4, 5 in Seizure and Excitotoxicity in Vivo
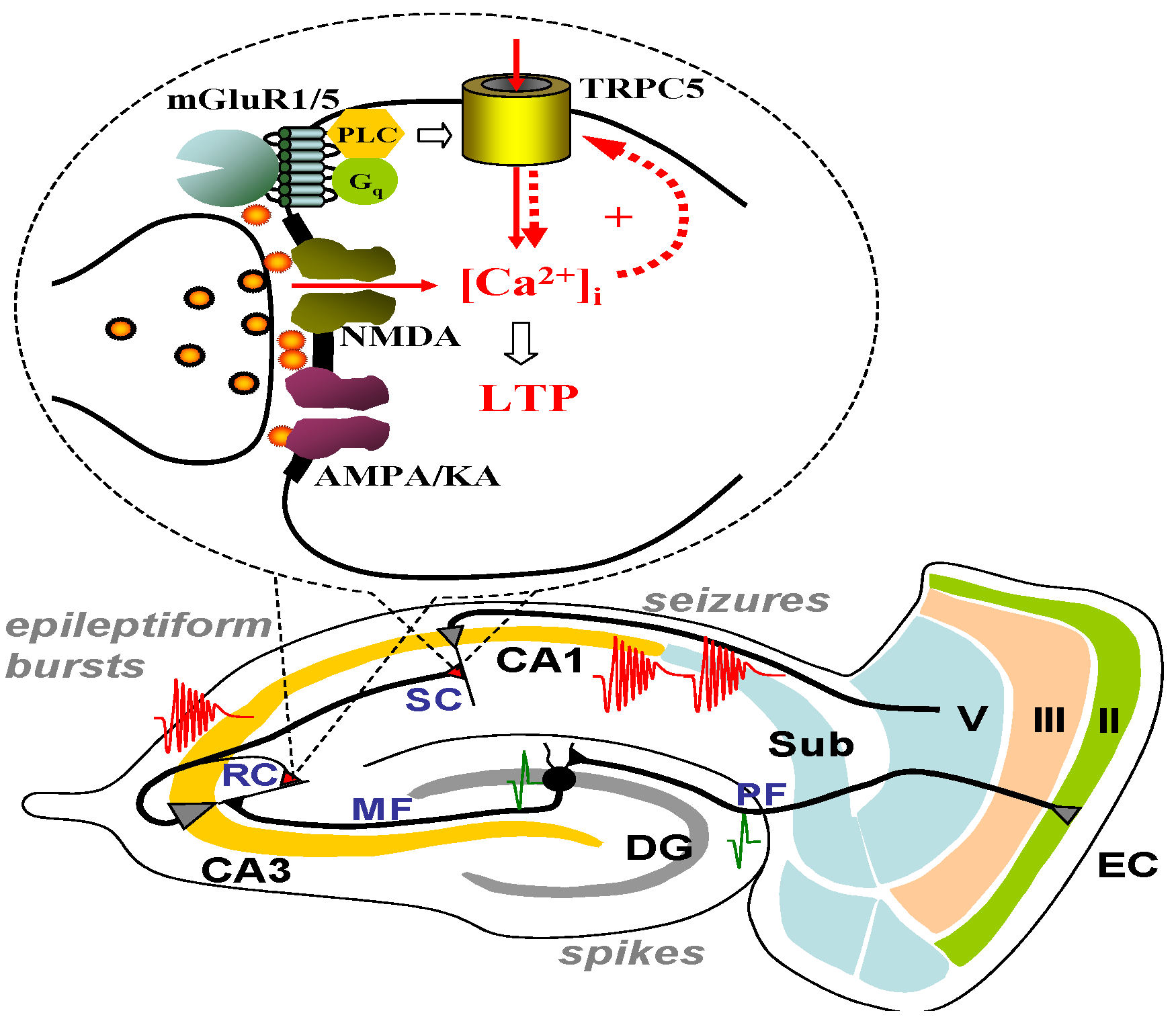
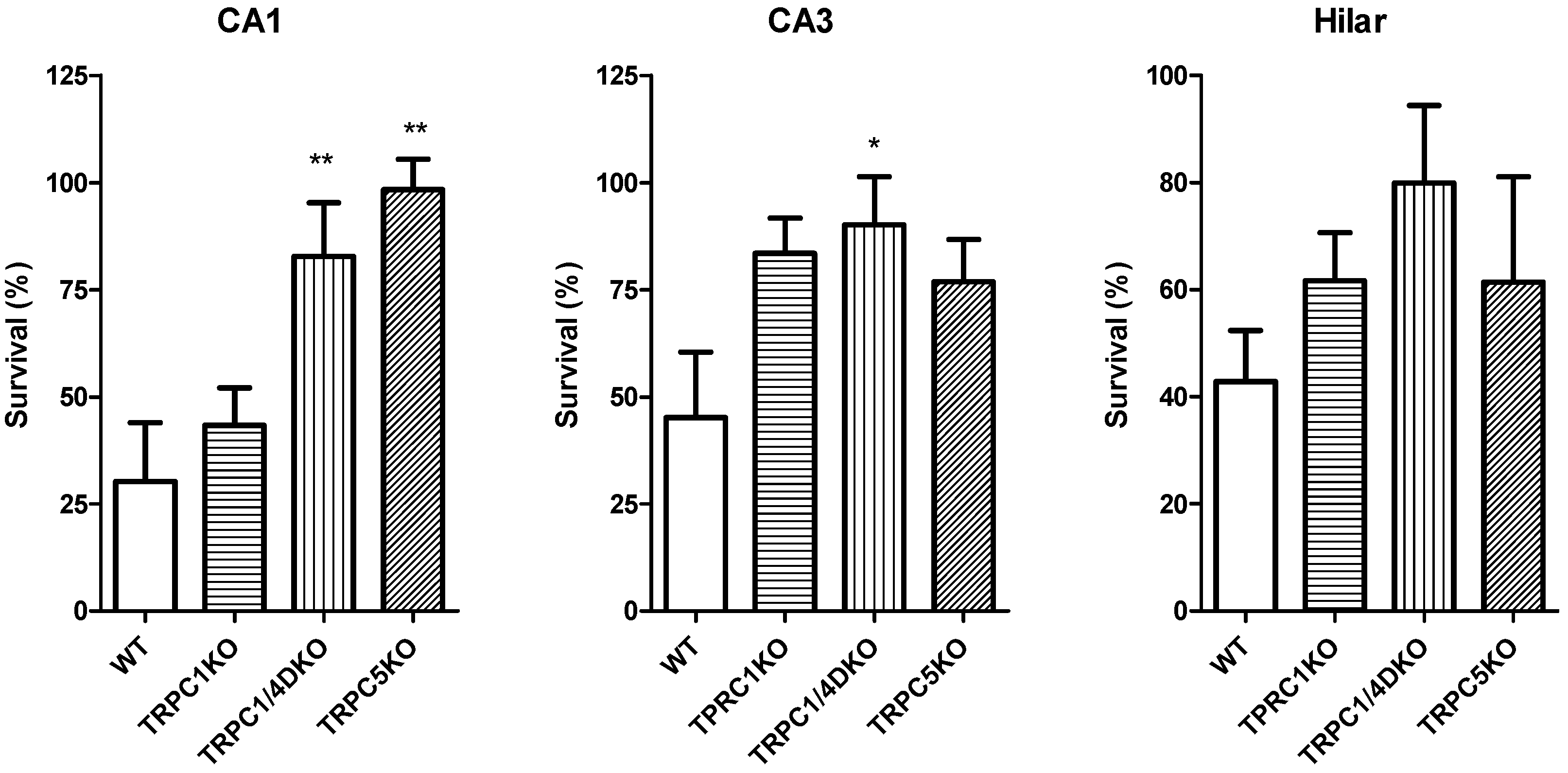
7. Do TRPC3, 6, or 7 Play a Role in Seizure and Excitotoxicity?
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Olney, J.W. Brain lesion, obesity and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, R.A. Mechanisms of disease: Excitatory amino acids as a final common pathway in neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5, 1039–1042. [Google Scholar] [CrossRef]
- Davis, S.M.; Lees, K.R.; Albers, G.W.; Diener, H.C.; Markabi, S.; Karlsson, G.; Norris, J. Selfotel in acute ischemic stroke: Possible neurotoxic effects of an NMDA antagonist. Stroke 2000, 31, 347–354. [Google Scholar] [CrossRef]
- Lees, K.R.; Asplund, K.; Carolei, A.; Davis, S.M.; Diener, H.C.; Kaste, M.; Orgogozo, J.M.; Whitehead, J. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: A randomised controlled trial. GAIN International Investigators. Lancet 2000, 355, 1949–1954. [Google Scholar] [CrossRef]
- Pin, J.P.; Conn, P.J. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef]
- Sacaan, A.I.; Schoepp, D.D. Activation of hippocampal metabotropic excitatory amino acid receptors leads to seizures and neuronal damage. Neurosci. Lett. 1992, 139, 77–82. [Google Scholar] [CrossRef]
- McDonald, J.W.; Fix, A.S.; Tizzano, J.P.; Schoepp, D.D. Seizures and brain injury in neonatal rats induced by 1S, 3R-ACPD, a metabotropic glutamate receptor agonist. J. Neurosci. 1993, 13, 4445–4455. [Google Scholar]
- Barton, M.E.; Shannon, H.E. Behavioral and convulsant effects of the (S) enantiomer of the group I metabotropic glutamate receptor agonist 3,5-DHPG in mice. Neuropharmacology 2005, 48, 779–787. [Google Scholar] [CrossRef]
- Lothman, E.W.; Collins, R.C. Kainic acid induced limbic seizures: Metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981, 218, 299–318. [Google Scholar] [CrossRef]
- Kliot, M.; Poletti, C.E. Hippocampal afterdischarges: differential spread of activity shown by the [14C]deoxyglucose technique. Science 1979, 204, 641–643. [Google Scholar]
- Zheng, F.; Gallagher, J.P. Trans-ACPD (trans-D,L-1-amino-1,3-cyclopentane-dicarboxylic acid) elicited oscillation of membrane potentials in rat dorsolateral septal nucleus neurons recorded intracellularly in vitro. Neurosci. Lett. 1991, 125, 147–150. [Google Scholar] [CrossRef]
- Zheng, F.; Gallagher, J.P. (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid-induced burst firing is mediated by a native pertussis toxin-sensitive metabotropic receptor at rat dorsolateral septal nucleus neurons. Neuroscience 1995, 68, 423–434. [Google Scholar] [CrossRef]
- Zheng, F.; Gallagher, J.P. Burst firing of rat septal neurons induced by 1S,3R-ACPD requires influx of extracellular calcium. Eur. J. Pharmacol. 1992, 211, 281–282. [Google Scholar] [CrossRef]
- Rutecki, P.A.; Yang, Y. Metabotropic glutamate receptor activation modulates epileptiform activity in the hippocampus. Neuroscience 1997, 81, 927–935. [Google Scholar] [CrossRef]
- Chuang, S.C.; Zhao, W.; Young, S.R.; Conquet, F.; Bianchi, R.; Wong, R.K. Activation of group I mGluRs elicits different responses in murine CA1 and CA3 pyramidal cells. J. Physiol. 2002, 541, 113–121. [Google Scholar] [CrossRef]
- Caeser, M.; Brown, D.A.; Gahwiler, B.H.; Knopfel, T. Characterization of a calcium-dependent current generating a slow afterdepolarization of CA3 pyramidal cells in rat hippocampal slice cultures. Eur. J. Neurosci. 1993, 5, 560–569. [Google Scholar] [CrossRef]
- Raggenbass, M.; Pierson, P.; Metzger, D.; Alberi, S. Action of a metabotropic glutamate receptor agonist in rat lateral septum: Induction of a sodium-dependent inward aftercurrent. Brain Res. 1997, 776, 75–87. [Google Scholar] [CrossRef]
- Luthi, A.; Gahwiler, B.H.; Gerber, U. 1S,3R-ACPD induces a region of negative slope conductance in the steady-state current-voltage relationship of hippocampal pyramidal cells. J. Neurophysiol. 1997, 77, 221–228. [Google Scholar]
- Zheng, F.; Gallagher, J.P.; Connor, J.A. Activation of a metabotropic glutamate receptor potentiates spike-driven calcium increases in neurons of the dorsolateral septum. J. Neurosci. 1996, 16, 6079–6088. [Google Scholar]
- Gee, C.E.; Benquet, P.; Gerber, U. Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J. Physiol. 2003, 546, 655–664. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W. The mammalian TRPC cation channels. Biochim. Biophys. Acta 2004, 1742, 21–36. [Google Scholar]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef]
- Yildirim, E.; Birnbaumer, L. TRPC2: Molecular biology and functional importance. Handb. Exp. Pharmacol. 2007, 179, 53–75. [Google Scholar] [CrossRef]
- Riccio, A.; Medhurst, A.D.; Mattei, C.; Kelsell, R.E.; Calver, A.R.; Randall, A.D.; Benham, C.D.; Pangalos, M.N. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res. Mol. Brain Res. 2002, 109, 95–104. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar]
- Mori, Y.; Takada, N.; Okada, T.; Wakamori, M.; Imoto, K.; Wanifuchi, H.; Oka, H.; Oba, A.; Ikenaka, K.; Kurosaki, T. Differential distribution of TRP Ca2+ channel isoforms in mouse brain. Neuroreport 1998, 9, 507–515. [Google Scholar]
- Zechel, S.; Werner, S.; von Bohlen und Halbach, O. Distribution of TRPC4 in developing and adult murine brain. Cell Tissue Res. 2007, 328, 651–656. [Google Scholar] [CrossRef]
- Phelan, K.D.; Mock, M.M.; Kretz, O.; Shwe, U.T.; Kozhemyakin, M.; Greenfield, L.J.; Dietrich, A.; Birnbaumer, L.; Freichel, M.; Flockerzi, V.; et al. Heteromeric canonical transient receptor potential 1 and 4 channels play a critical role in epileptiform burst firing and seizure-induced neurodegeneration. Mol. Pharmacol. 2012, 81, 384–392. [Google Scholar] [CrossRef]
- Philipp, S.; Hambrecht, J.; Braslavski, L.; Schroth, G.; Freichel, M.; Murakami, M.; Cavalie, A.; Flockerzi, V. A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 1998, 17, 4274–4282. [Google Scholar] [CrossRef]
- Strübing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef]
- Bonaventure, P.; Guo, H.; Tian, B.; Liu, X.; Bittner, A.; Roland, B.; Salunga, R.; Ma, X.J.; Kamme, F.; Meurers, B.; et al. Nuclei and subnuclei gene expression profiling in mammalian brain. Brain Res. 2002, 943, 38–47. [Google Scholar] [CrossRef]
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca(2+)-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef]
- Flockerzi, V.; Jung, C.; Aberle, T.; Meissner, M.; Freichel, M.; Philipp, S.E.; Nastainczyk, W.; Maurer, P.; Zimmermann, R. Specific detection and semi-quantitative analysis of TRPC4 protein expression by antibodies. Pflugers Arch. 2005, 451, 81–86. [Google Scholar] [CrossRef]
- Nagamine, K.; Kudoh, J.; Minoshima, S.; Kawasaki, K.; Asakawa, S.; Ito, F.; Shimizu, N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 1998, 54, 124–131. [Google Scholar] [CrossRef]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef]
- Strübing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Boulay, G.; Brown, D.M.; Qin, N.; Jiang, M.; Dietrich, A.; Zhu, M.X.; Chen, Z.; Birnbaumer, M.; Mikoshiba, K.; Birnbaumer, L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): Evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. USA 1999, 96, 14955–14960. [Google Scholar] [CrossRef]
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211. [Google Scholar]
- Kim, S.J.; Kim, Y.S.; Yuan, J.P.; Petralia, R.S.; Worley, P.F.; Linden, D.J. Activation of TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 2003, 426, 285–291. [Google Scholar] [CrossRef]
- Stroh, O.; Freichel, M.; Kretz, O.; Birnbaumer, L.; Hartmann, J.; Egger, V. NMDA receptor-dependent synaptic activation of TRPC channels in olfactory bulb granule cells. J. Neurosci. 2012, 32, 5737–5746. [Google Scholar] [CrossRef]
- Trebak, M.; Lemonnier, L.; Smyth, J.T.; Vazquez, G.; Putney, J.W., Jr. Phospholipase C-coupled receptors and activation of TRPC channels. Handb. Exp. Pharmacol. 2007, 179, 593–614. [Google Scholar] [CrossRef]
- Putney, J. Physiological mechanisms of TRPC activation. Pflugers Arch. 2005, 451, 29–34. [Google Scholar] [CrossRef]
- Singh, A.; Hildebrand, M.E.; Garcia, E.; Snutch, T.P. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharm. 2010, 160, 1464–1475. [Google Scholar] [CrossRef]
- Jung, S.; Mühle, A.; Schaefer, M.; Strotmann, R.; Schultz, G.; Plant, T.D. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J. Biol. Chem. 2003, 278, 3562–3571. [Google Scholar]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.-B.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef]
- Tian, J.; Thakur, D.P.; Lu, Y.; Zhu, Y.; Freichel, M.; Flockerzi, V.; Zhu, M.X. Dual depolarization responses generated within the same lateral septal neurons by TRPC4-containing channels. Pflugers Arch. 2013, in press. [Google Scholar]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Nat. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef]
- Leuner, K.; Heiser, J.H.; Derksen, S.; Mladenov, M.I.; Fehske, C.J.; Schubert, R.; Gollasch, M.; Schneider, G.; Harteneck, C.; Chatterjee, S.S.; et al. Simple 2,4-Diacylphloroglucinols as classic transient receptor potential-6 activators-Identification of a novel pharmacophore. Mol. Pharmacol. 2010, 77, 368–377. [Google Scholar] [CrossRef]
- Urban, N.; Hill, K.; Wang, L.; Kuebler, W.M.; Schaefer, M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium 2012, 51, 194–206. [Google Scholar] [CrossRef]
- Washburn, D.G.; Holt, D.A.; Dodson, J.; McAtee, J.J.; Terrell, L.R.; Barton, L.; Manns, S.; Waszkiewicz, A.; Pritchard, C.; Gillie, D.J.; et al. The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg. Med. Chem. Lett. 2013, 23, 4979–4984. [Google Scholar] [CrossRef]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.-H.; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef]
- Chen, W.; Oberwinkler, H.; Werner, F.; Gaßner, B.; Nakagawa, H.; Feil, R.; Hofmann, F.; Schlossmann, J.; Dietrich, A.; Gudermann, T.; et al. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase I and TRPC6 channels. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2121–2129. [Google Scholar]
- Shwe, U.T.; Phelan, K.D.; Birnbaumer, L.; Zheng, F. The effects of putative TRPC channel blockers on the neuronal TRPC channels with defined molecular identity. (to be submitted).
- Gasparini, F.; Kuhn, R.; Pin, J.-P. Allosteric modulators of group I metabotropic glutamate receptors: Novel subtype-selective ligands and therapeutic perspectives. Curr. Opin. Pharmacol. 2002, 2, 43–49. [Google Scholar] [CrossRef]
- Bleasdale, J.E.; Thakur, N.R.; Gremban, R.S.; Bundy, G.L.; Fitzpatrick, F.A.; Smith, R.J.; Bunting, S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J. Pharmacol. Exp. Ther. 1990, 255, 756–768. [Google Scholar]
- Kandel, E.R.; Spencer, W.A. Electrophysiology of hippocampal neurons. II. After-potentials and repetitive firing. J. Neurophysiol. 1961, 24, 243–259. [Google Scholar]
- McCormick, D.A.; Contreras, D. On the cellular and network bases of epileptic seizures. Ann. Rev. Physiol. 2001, 63, 815–846. [Google Scholar] [CrossRef]
- Azouz, R.; Jensen, M.S.; Yaari, Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J. Physiol. (Lond.) 1996, 492, 211–223. [Google Scholar]
- Hildebrand, M.E.; Isope, P.; Miyazaki, T.; Nakaya, T.; Garcia, E.; Feltz, A.; Schneider, T.; Hescheler, J.; Kano, M.; Sakimura, K.; et al. Functional coupling between mGluR1 and Cav3.1 T-type calcium channels contributes to parallel fiber-induced fast calcium signaling within Purkinje cell dendritic spines. J. Neurosci. 2009, 29, 9668–9682. [Google Scholar] [CrossRef]
- Park, J.-Y.; Remy, S.; Varela, J.; Cooper, D.C.; Chung, S.; Kang, H.W.; Lee, J.-H.; Spruston, N. A post-burst afterdepolarization is mediated by Group I metabotropic glutamate receptor-dependent upregulation of Cav2.3 R-Type calcium channels in CA1 Pyramidal Neurons. PLoS Biol. 2010, 8, e1000534. [Google Scholar] [CrossRef]
- Wong, R.K.S.; Prince, D.A. Afterpotential generation in hippocampal pyramidal neurons. J. Neurophysiol. 1981, 45, 86–97. [Google Scholar]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [CrossRef]
- Yan, H.D.; Villalobos, C.; Andrade, R. TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J. Neurosci. 2009, 29, 10038–10046. [Google Scholar]
- Tai, C.; Hines, D.J.; Choi, H.B.; MacVicar, B.A. Plasma membrane insertion of TRPC5 channels contributes to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. Hippocampus 2011, 21, 958–967. [Google Scholar]
- Zhang, Z.; Reboreda, A.; Alonso, A.; Barker, P.A.; Séguéla, P. TRPC channels underlie cholinergic plateau potentials and persistent activity in entorhinal cortex. Hippocampus 2011, 21, 386–397. [Google Scholar] [CrossRef]
- Stoop, R.; Conquet, F.; Zuber, B.; Voronin, L.L.; Pralong, E. Activation of metabotropic glutamate 5 and NMDA receptors underlies the induction of persistent bursting and associated long-lasting changes in CA3 recurrent connections. J. Neurosci. 2003, 23, 5634–5644. [Google Scholar]
- Bains, J.S.; Longacher, J.M.; Staley, K.J. Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat. Neurosci. 1999, 2, 720–726. [Google Scholar] [CrossRef]
- He, Z.; Jia, C.; Feng, S.; Zhou, K.; Tai, Y.; Bai, X.; Wang, Y. TRPC5 channel is the mediator of neurotrophin-3 in regulating dendritic growth via CaMKIIα in rat hippocampal neurons. J. Neurosci. 2012, 32, 9383–9395. [Google Scholar] [CrossRef]
- Narayanan, K.L.; Irmady, K.; Subramaniam, S.; Unsicker, K.; von Bohlen und Halbach, O. Evidence that TRPC1 is involved in hippocampal glutamate-induced cell death. Neurosci. Lett. 2008, 446, 117–122. [Google Scholar] [CrossRef]
- Amaral, M.D.; Pozzo-Miller, L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 2007, 27, 5179–5189. [Google Scholar] [CrossRef]
- Kim, D.S.; Ryu, H.J.; Kim, J.E.; Kang, T.C. The reverse roles of transient receptor potential canonical channel-3 and -6 in neuronal death following pilocarpine-induced status epilepticus. Cell Mol. Neurobiol. 2013, 33, 99–109. [Google Scholar] [CrossRef]
- Li, H.; Huang, J.; Du, W.; Jia, C.; Yao, H.; Wang, Y. TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity. J. Neurochem. 2012, 123, 1010–1018. [Google Scholar] [CrossRef]
- Lin, Y.; Zhang, J.C.; Fu, J.; Chen, F.; Wang, J.; Wu, Z.L.; Yuan, S.Y. Hyperforin attenuates brain damage induced by transient middle cerebral artery occlusion (MCAO) in rats via inhibition of TRPC6 channels degradation. J. Cereb. Blood Flow Metable 2013, 33, 253–262. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zheng, F.; Phelan, K.D. The Role of Canonical Transient Receptor Potential Channels in Seizure and Excitotoxicity. Cells 2014, 3, 288-303. https://doi.org/10.3390/cells3020288
Zheng F, Phelan KD. The Role of Canonical Transient Receptor Potential Channels in Seizure and Excitotoxicity. Cells. 2014; 3(2):288-303. https://doi.org/10.3390/cells3020288
Chicago/Turabian StyleZheng, Fang, and Kevin D. Phelan. 2014. "The Role of Canonical Transient Receptor Potential Channels in Seizure and Excitotoxicity" Cells 3, no. 2: 288-303. https://doi.org/10.3390/cells3020288