Redox Mechanisms in Regulation of Adipocyte Differentiation: Beyond a General Stress Response

Abstract
:
1. Introduction
2. ROS and Redox-Dependent Signaling Mechanisms

3. Molecular Mechanisms Governing Adipocyte Differentiation
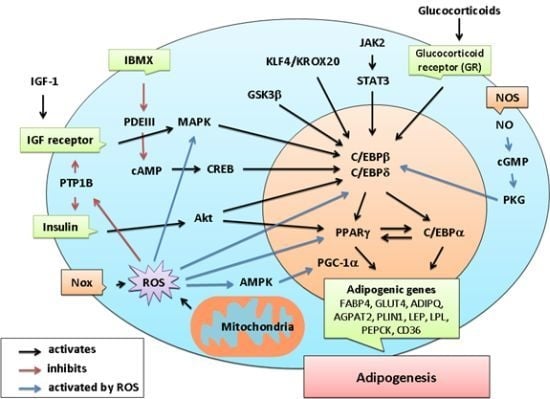
4. Emerging Evidence of Redox-Dependent Regulation of Adipogenesis

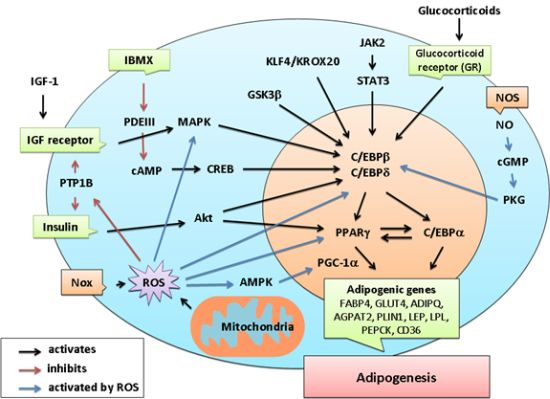{kind=link}
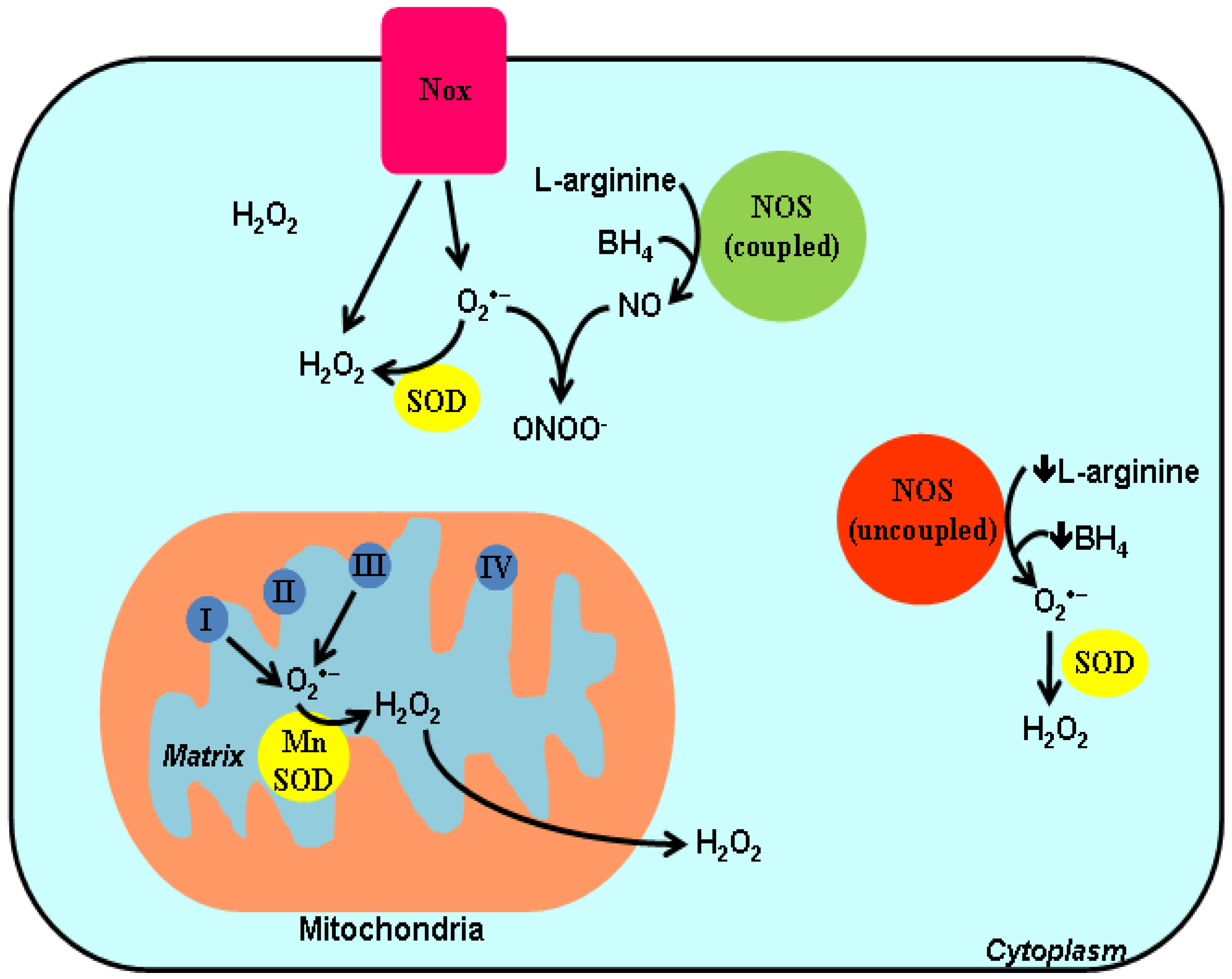{kind=link}
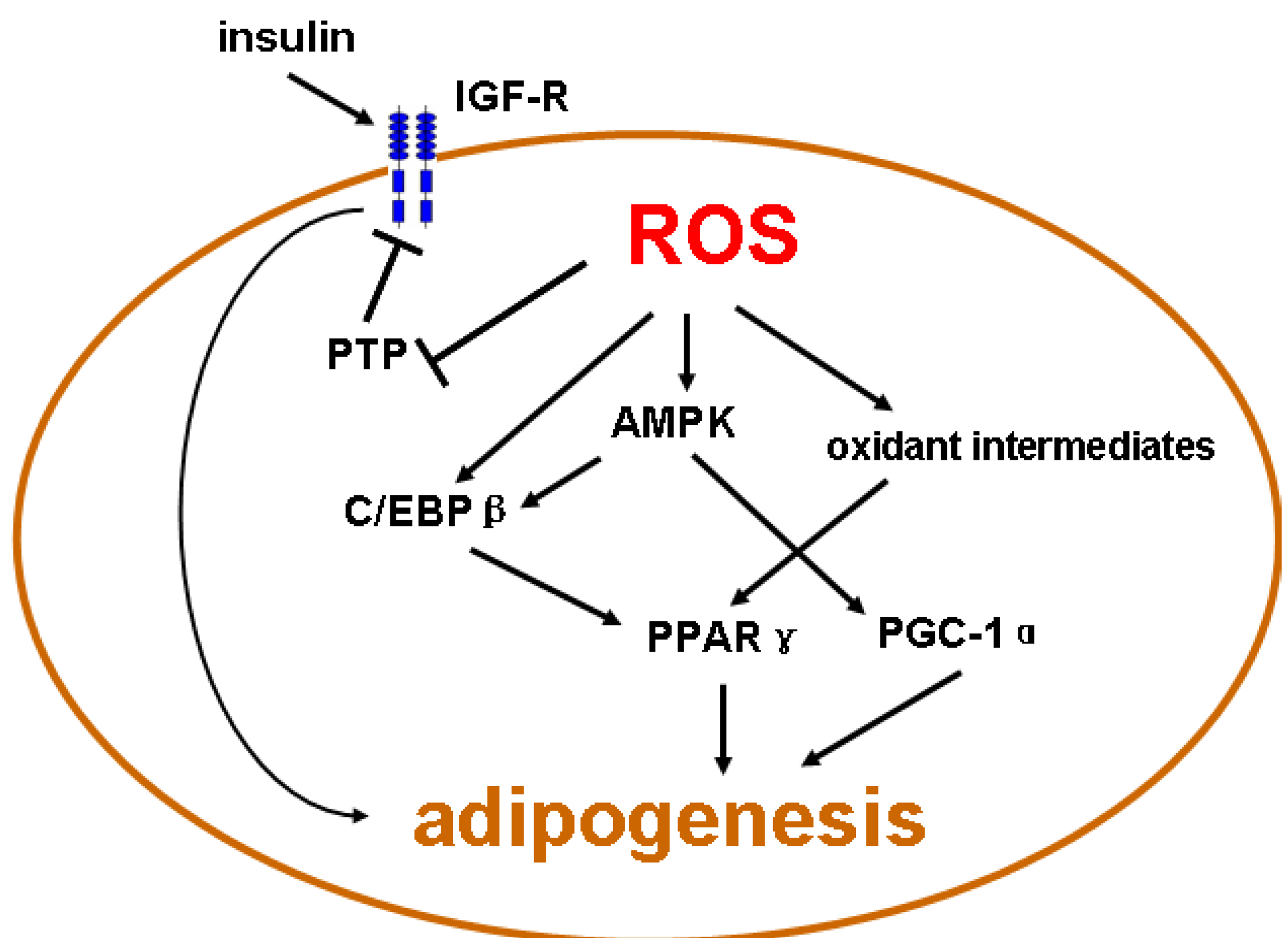{kind=link}
| Source of ROS | Experimental models | Primary findings | References |
|---|---|---|---|
| NADPH oxidase | 3T3-L1 adipocytes | Increased ROS production in accumulated fat contributes to metabolic syndrome. | [64] |
| NADPH oxidase | 3T3-L1 adipocytes, human preadipocytes | Nox4 acts as a switch from insulin-induced proliferation to differentiation by controlling MKP-1 expression, which limits ERK1/2 signaling. | [16] |
| NADPH oxidase | Mouse MSCs | Increase in the intracellular ROS level via Nox4 mediates adipocyte differentiation through CREB in MSC. | [15] |
| Mitochondria | 3T3-L1 adipocytes | Increase in mitochondrial ROS production caused by inhibition of the electron transport chain (complex I and V) prevented preadipocyte proliferation. | [69] |
| Mitochondria | Human MSCs | Mitochondrial metabolism and ROS generation are not simply a consequence of differentiation but are a causal factor in promoting adipocyte differentiation. | [70] |
| NOS | Rat preadipocytes | NO is involved in the positive modulation of preadipocyte differentiation and eNOS rather than iNOS may be the major isoform involved in modulating adipogenesis. | [71] |
5. Redox-Sensitive Mechanisms Related to Adipogenesis
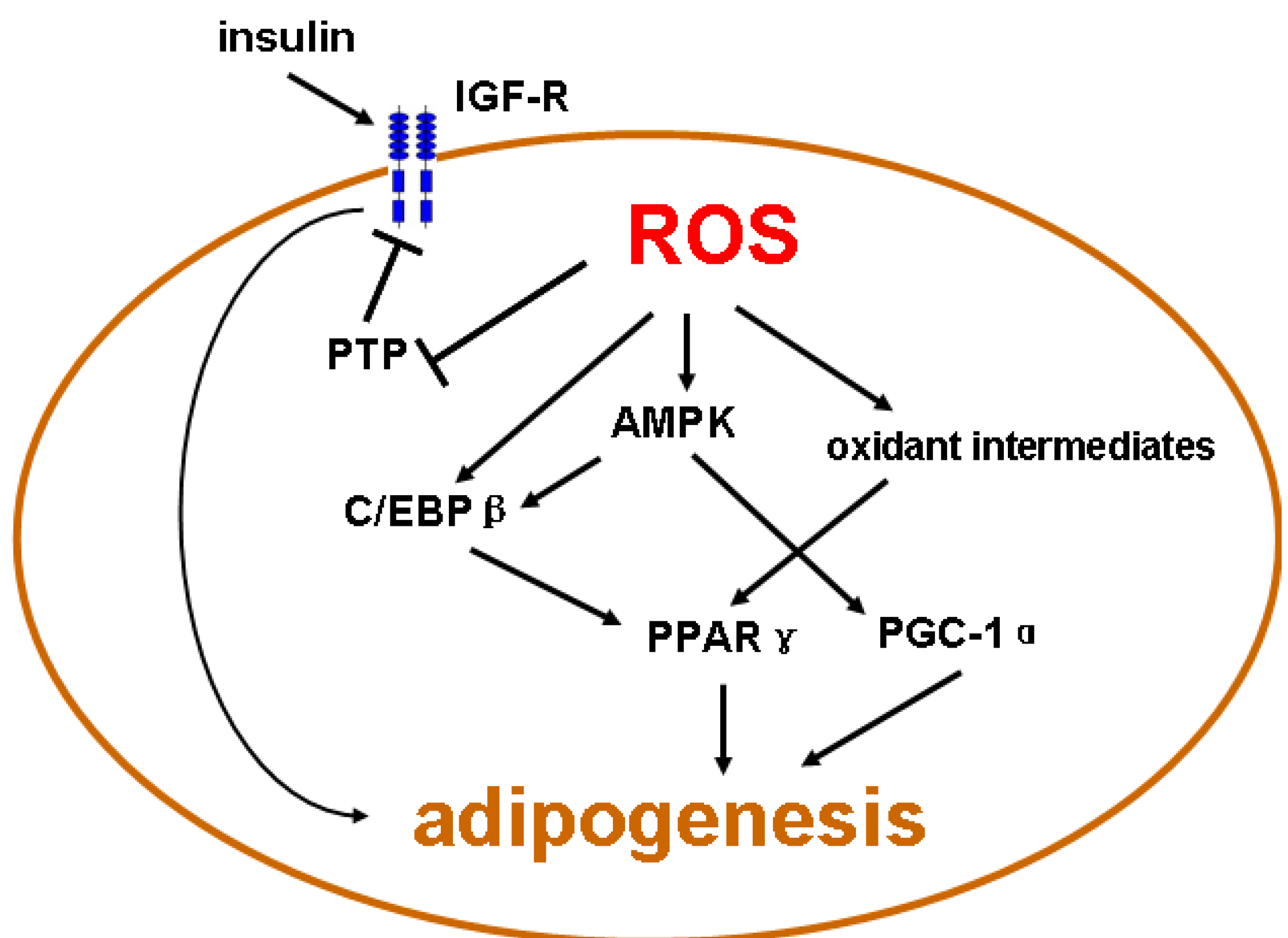
6. Conclusion Remarks
Acknowledgments
Conflict of Interest
References and Notes
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect. 1994, 102 (Suppl. 10), 5–12. [Google Scholar]
- Thannickal, V.; Fanburg, B. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar]
- Jiang, F.; Zhang, Y.; Dusting, G. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Rains, J.J. SK Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef]
- Abe, J.; Berk, B. Reactive oxygen species as mediators of signal transduction in cardiovascular disease. Trends Cardiovasc. Med. 1998, 8, 59–64. [Google Scholar] [CrossRef]
- Chiarugi, P. PTPs versus PTKs: The redox side of the coin. Free Radic. Res. 2005, 39, 353–364. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; Loehneysen, K.V.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Mouche, S.; Mkaddem, S.B.; Wang, W.; Katic, M.; Tseng, Y.H.; Carnesecchi, S.; Steger, K.; Foti, M.; Meier, C.A.; Muzzin, P.; et al. Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochim. Biophys. Acta 2007, 1773, 1015–1027. [Google Scholar] [CrossRef]
- Kanda, Y.; Hinata, T.; Kang, S.W.; Watanabe, Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011, 89, 250–258. [Google Scholar] [CrossRef]
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.; Shah, A.; Morel, F.; Brandes, R. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; Rasmo, D.D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–35. [Google Scholar] [CrossRef]
- Murphy, M. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Jr, K.A.P. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell. Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Lazar, M.A. Transcriptional control of adipogenesis. Annu. Rev. Nutr. 2000, 20, 535–559. [Google Scholar]
- Huang, H.; Song, T.J.; Li, X.; Hu, L.; He, Q.; Liu, M.; Lane, M.D.; Tang, Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2009, 106, 12670–12675. [Google Scholar]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. USA 2004, 101, 9607–9611. [Google Scholar] [CrossRef]
- Zhou, H.; Mak, W.; Zheng, Y.; Dunstan, C.R.; Seibel, M.J. Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J. Biol. Chem. 2008, 283, 1936–1945. [Google Scholar]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad.Sci. USA 2005, 102, 3324–3329. [Google Scholar]
- Shao, D.; Lazar, M.A. Peroxisome proliferator activated receptor γ, CCAAT/enhancer-binding protein α, and cell cycle status regulate the commitment to adipocyte differentiation. J. Biol. Chem. 1997, 272, 21473–21478. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef]
- Siersbæk, R.; Nielsen, R.; Mandrup, S. Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol. Metab. 2012, 23, 56–64. [Google Scholar] [CrossRef]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell. Metab. 2008, 7, 339–347. [Google Scholar] [CrossRef]
- Zhang, J.W.; Klemm, D.J.; Vinson, C.; Lane, M.D. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein β gene during adipogenesis. J. Biol. Chem. 2004, 279, 4471–4478. [Google Scholar]
- Chen, Z.; Torrens, J.I.; Anand, A.; Spiegelman, B.M.; Friedman, J.M. Krox20 stimulates adipogenesis via C/EBP[beta]-dependent and-independent mechanisms. Cell. Metab. 2005, 1, 93–106. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, W.; Yang, Y.; Wu, J. JAK2/STAT3 pathway is involved in the early stage of adipogenesis through regulating C/EBPβ transcription. J. Biol. Chem. 2011, 112, 488–497. [Google Scholar]
- Tang, Q.Q.; Grønborg, M.; Huang, H.; Kim, J.W.; Otto, T.C.; Pandey, A.; Lane, M.D. Sequential phosphorylation of CCAAT enhancer-binding protein β by MAPK and glycogen synthase kinase 3β is required for adipogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 9766–9771. [Google Scholar]
- Yeh, W.C.; Cao, Z.; Classon, M.; McKnight, S.L. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995, 9, 168–181. [Google Scholar] [CrossRef]
- Wiper-Bergeron, N.; Salem, H.A.; Tomlinson, J.J.; Wu, D.; Haché, R.J.G. Glucocorticoid-stimulated preadipocyte differentiation is mediated through acetylation of C/EBPβ by GCN5. Proc. Natl. Acad. Sci. USA 2007, 104, 2703–2708. [Google Scholar]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell. Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef]
- Floyd, Z.E.; Stephens, J.M. STAT5A promotes adipogenesis in nonprecursor cells and associates with the glucocorticoid receptor during adipocyte differentiation. Diabetes 2003, 52, 308–314. [Google Scholar]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef]
- Nielsen, R.; Pedersen, T.Å.; Hagenbeek, D.; Moulos, P.; Siersbæk, R.; Megens, E.; Denissov, S.; Børgesen, M.; Francoijs, K.J.; Mandrup, S. Genome-wide profiling of PPARγ: RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008, 22, 2953–2967. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Lowe, C.E.; O'Rahilly, S.; Rochford, J.J. Adipogenesis at a glance. J. Cell Sci. 2011, 124, 2681–2686. [Google Scholar] [CrossRef]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef]
- Bakan, I.; Laplante, M. Connecting mTORC1 signaling to SREBP-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARγ through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar]
- Moldes, M.; Boizard, M.; Liepvre, X.; Fève, B.; Dugail, I.; Pairault, J. Functional antagonism between inhibitor of DNA binding (Id) and adipocyte determination and differentiation factor 1/sterol regulatory element-binding protein-1c (ADD1/SREBP-1c) trans-factors for the regulation of fatty acid synthase promoter in adipocytes. Biochem. J. 1999, 344, 873–880. [Google Scholar] [CrossRef]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 2009, 4, e6189. [Google Scholar]
- Wang, F.; Tong, Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARγ. Mol. Biol. Cell. 2009, 20, 801–808. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I.; Tobe, K.; Tsushima, K.; Shindo, T.; Fujiu, K.; Nishimura, G.; Maemura, K.; Yamauchi, T.; Kubota, N. Krüppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell. Metab. 2005, 1, 27–39. [Google Scholar] [CrossRef]
- Mori, T.; Sakaue, H.; Iguchi, H.; Gomi, H.; Okada, Y.; Takashima, Y.; Nakamura, K.; Nakamura, T.; Yamauchi, T.; Kubota, N. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J. Biol. Chem. 2005, 280, 12867–12875. [Google Scholar]
- Banerjee, S.S.; Feinberg, M.W.; Watanabe, M.; Gray, S.; Haspel, R.L.; Denkinger, D.J.; Kawahara, R.; Hauner, H.; Jain, M.K. The Krüppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-γ expression and adipogenesis. J. Biol. Chem. 2003, 278, 2581–2584. [Google Scholar]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell. Biol. 2012, 13, 225–238. [Google Scholar]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; De Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell. Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef]
- Liu, C.; Lin, J.D. PGC-1 coactivators in the control of energy metabolism. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 248–257. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, S884–S890. [Google Scholar] [CrossRef]
- Bonen, A. PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity. Appl. Physiol. Nutr. Metab. 2009, 34, 307–314. [Google Scholar] [CrossRef]
- Huang, P.I.; Chen, Y.C.; Chen, L.H.; Juan, C.C.; Ku, H.H.; Wang, S.T.; Chiou, S.H.; Chiou, G.Y.; Chi, C.W.; Hsu, C.C.; et al. PGC-1alpha mediates differentiation of mesenchymal stem cells to brown adipose cells. J. Atheroscler. Thromb. 2011, 18, 966–980. [Google Scholar] [CrossRef]
- Spiegelman, B.M.; Puigserver, P.; Wu, Z. Regulation of adipogenesis and energy balance by PPARgamma and PGC-1. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. 4), S8–S10. [Google Scholar]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar]
- Park, H.S.; Jin, D.K.; Shin, S.M.; Jang, M.K.; Longo, N.; Park, J.W.; Bae, D.S.; Bae, Y.S. Impaired generation of reactive oxygen species in leprechaunism through downregulation of Nox4. Diabetes 2005, 54, 3175–3181. [Google Scholar] [CrossRef]
- Jiang, F.; Lim, H.K.; Morris, M.J.; Prior, L.; Velkoska, E.; Wu, X.; Dusting, G.J. Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Rep. 2011, 16, 223–229. [Google Scholar] [CrossRef]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef]
- Li, Y.; Mouche, S.; Sajic, T.; Veyrat-Durebex, C.; Supale, R.; Pierroz, D.; Ferrari, S.; Negro, F.; Hasler, U.; Feraille, E.; et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int. J. Obes. (Lond) 2012, in press. [Google Scholar]
- Carriere, A.; Fernandez, Y.; Rigoulet, M.; Penicaud, L.; Casteilla, L. Inhibition of preadipocyte proliferation by mitochondrial reactive oxygen species. FEBS Lett. 2003, 550, 163–167. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell. Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef]
- Yan, H.; Aziz, E.; Shillabeer, G.; Wong, A.; Shanghavi, D.; Kermouni, A.; Abdel-Hafez, M.; Lau, D.C. Nitric oxide promotes differentiation of rat white preadipocytes in culture. J. Lipid. Res. 2002, 43, 2123–2129. [Google Scholar] [CrossRef]
- Ribiere, C.; Jaubert, A.M.; Gaudiot, N.; Sabourault, D.; Marcus, M.L.; Boucher, J.L.; Denis-Henriot, D.; Giudicelli, Y. White adipose tissue nitric oxide synthase: A potential source for NO production. Biochem. Biophys. Res. Commun. 1996, 222, 706–712. [Google Scholar] [CrossRef]
- Engeli, S.; Janke, J.; Gorzelniak, K.; Bohnke, J.; Ghose, N.; Lindschau, C.; Luft, F.C.; Sharma, A.M. Regulation of the nitric oxide system in human adipose tissue. J. Lipid. Res. 2004, 45, 1640–1648. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, J.; Yan, G.; Wu, J.; Sun, X.; Shen, J.; Jiang, H.; Wang, H. Sildenafil promotes adipogenesis through a PKG pathway. Biochem. Biophys. Res. Commun. 2010, 396, 1054–1059. [Google Scholar] [CrossRef]
- Choi, J.W.; Pai, S.H.; Kim, S.K.; Ito, M.; Park, C.S.; Cha, Y.N. Increases in nitric oxide concentrations correlate strongly with body fat in obese humans. Clin. Chem. 2001, 47, 1106–1109. [Google Scholar]
- Elizalde, M.; Ryden, M.; van Harmelen, V.; Eneroth, P.; Gyllenhammar, H.; Holm, C.; Ramel, S.; Olund, A.; Arner, P.; Andersson, K. Expression of nitric oxide synthases in subcutaneous adipose tissue of nonobese and obese humans. J. Lipid. Res. 2000, 41, 1244–1251. [Google Scholar]
- Ryden, M.; Elizalde, M.; van Harmelen, V.; Ohlund, A.; Hoffstedt, J.; Bringman, S.; Andersson, K. Increased expression of eNOS protein in omental versus subcutaneous adipose tissue in obese human subjects. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 811–815. [Google Scholar]
- Younce, C.; Kolattukudy, P. MCP-1 induced protein promotes adipogenesis via oxidative stress, endoplasmic reticulum stress and autophagy. Cell Physiol. Biochem. 2012, 30, 307–320. [Google Scholar] [CrossRef]
- Liu, H.; Yang, X.; Zhang, Y.; Dighe, A.; Li, X.; Cui, Q. Fullerol antagonizes dexamethasone-induced oxidative stress and adipogenesis while enhancing osteogenesis in a cloned bone marrow mesenchymal stem cell. J. Orthop. Res. 2012, 30, 1051–1057. [Google Scholar] [CrossRef]
- Nam, W.S.; Park, K.M.; Park, J.W. RNA interference targeting cytosolic NADP(+)-dependent isocitrate dehydrogenase exerts anti-obesity effect in vitro and in vivo. Biochim. Biophys. Acta 2012, 1822, 1181–1188. [Google Scholar] [CrossRef]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S.; et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein beta during adipogenesis. Free Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef]
- Imhoff, B.R.; Hansen, J.M. Differential redox potential profiles during adipogenesis and osteogenesis. Cell. Mol. Biol. Lett. 2011, 16, 149–161. [Google Scholar] [CrossRef]
- Saitoh, Y.; Xiao, L.; Mizuno, H.; Kato, S.; Aoshima, H.; Taira, H.; Kokubo, K.; Miwa, N. Novel polyhydroxylated fullerene suppresses intracellular oxidative stress together with repression of intracellular lipid accumulation during the differentiation of OP9 preadipocytes into adipocytes. Free Radic. Res. 2010, 44, 1072–1081. [Google Scholar] [CrossRef]
- Samuni, Y.; Cook, J.A.; Choudhuri, R.; Degraff, W.; Sowers, A.L.; Krishna, M.C.; Mitchell, J.B. Inhibition of adipogenesis by Tempol in 3T3-L1 cells. Free Radic. Biol. Med. 2010, 49, 667–673. [Google Scholar] [CrossRef]
- Vigilanza, P.; Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Modulation of intracellular glutathione affects adipogenesis in 3T3-L1 cells. J. Cell Physiol. 2011, 226, 2016–2024. [Google Scholar] [CrossRef]
- Calzadilla, P.; Sapochnik, D.; Cosentino, S.; Diz, V.; Dicelio, L.; Calvo, J.C.; Guerra, L.N. N-Acetylcysteine Reduces Markers of Differentiation in 3T3-L1 Adipocytes. Int. J. Mol. Sci. 2011, 12, 6936–6951. [Google Scholar] [CrossRef]
- Imhoff, B.R.; Hansen, J.M. Extracellular redox environments regulate adipocyte differentiation. Differentiation 2010, 80, 31–39. [Google Scholar] [CrossRef]
- Higuchi, M.; Dusting, G.J.; Peshavariya, H.; Jiang, F.; Hsiao, S.T.-F.; Chan, E.C.; Liu, G.-S. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead Box O1 mediated upregulation of antioxidant enzymes. Stem. Cells Dev. 2012, in press. [Google Scholar]
- Mitchell, J.B.; Xavier, S.; DeLuca, A.M.; Sowers, A.L.; Cook, J.A.; Krishna, M.C.; Hahn, S.M.; Russo, A. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radic. Biol. Med. 2003, 34, 93–102. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, E.J.; Kim, S.H.; Choi, I.; Lee, D.M.; Lee, H.J.; Yoon, D.; Chun, T. IL-17A promotes transdifferentiation of mouse myoblast cells (C2C12) into adipocytes by increasing the expression of peroxisome proliferator-activated receptor gamma through CAAT/enhancer binding protein beta signaling. Biotechnol. Lett. 2011, 33, 229–235. [Google Scholar] [CrossRef]
- Itoigawa, Y.; Kishimoto, K.N.; Okuno, H.; Sano, H.; Kaneko, K.; Itoi, E. Hypoxia induces adipogenic differentitation of myoblastic cell lines. Biochem. Biophys. Res. Commun. 2010, 399, 721–726. [Google Scholar] [CrossRef]
- Yamanouchi, K.; Yada, E.; Ishiguro, N.; Nishihara, M. 18alpha-glycyrrhetinic acid induces phenotypic changes of skeletal muscle cells to enter adipogenesis. Cell. Physiol. Biochem. 2007, 20, 781–790. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Wang, L.; Wang, N.; Li, Y.; Li, H. Transdifferentiation of fibroblasts into adipocyte-like cells by chicken adipogenic transcription factors. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2010, 156, 502–508. [Google Scholar] [CrossRef]
- Smith, P.J.; Wise, L.S.; Berkowitz, R.; Wan, C.; Rubin, C.S. Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J. Biol. Chem. 1988, 263, 9402–9408. [Google Scholar]
- Prusty, D.; Park, B.H.; Davis, K.E.; Farmer, S.R. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2002, 277, 46226–46232. [Google Scholar]
- Lawrence, J.C., Jr.; Larner, J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J. Biol. Chem. 1978, 253, 2104–2113. [Google Scholar]
- May, J.M.; de Haen, C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 1979, 254, 2214–2220. [Google Scholar]
- Meng, T.C.; Buckley, D.A.; Galic, S.; Tiganis, T.; Tonks, N.K. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 2004, 279, 37716–37725. [Google Scholar]
- Goldstein, B.J.; Mahadev, K.; Wu, X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 2005, 54, 311–321. [Google Scholar]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.H.; Park, S.G.; Choi, J.S.; Xia, Y.; Sung, J.H. The pivotal role of reactive oxygen species generation in the hypoxia-induced stimulation of adipose-derived stem cells. Stem. Cells. Dev. 2011, 20, 1753–1761. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Scheller, E.L.; MacDougald, O.A. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J. Lipid. Res. 2012, 53, 227–246. [Google Scholar] [CrossRef]
- Bost, F.; Aouadi, M.; Caron, L.; Binetruy, B. The role of MAPKs in adipocyte differentiation and obesity. Biochimie 2005, 87, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Seltmann, H.; Zouboulis, C.C.; Konger, R.L. Involvement of PPARgamma in oxidative stress-mediated prostaglandin E(2) production in SZ95 human sebaceous gland cells. J. Invest. Dermatol. 2006, 126, 42–48. [Google Scholar] [CrossRef]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Schopfer, F.J.; Cole, M.P.; Groeger, A.L.; Chen, C.S.; Khoo, N.K.; Woodcock, S.R.; Golin-Bisello, F.; Motanya, U.N.; Li, Y.; Zhang, J.; et al. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: Selective ligand activity and anti-diabetic signaling actions. J. Biol. Chem. 2010, 285, 12321–12333. [Google Scholar]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; O'Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef]
- Kim, J.W.; Tang, Q.Q.; Li, X.; Lane, M.D. Effect of phosphorylation and S–S bond-induced dimerization on DNA binding and transcriptional activation by C/EBPbeta. Proc. Natl. Acad. Sci. USA 2007, 104, 1800–1804. [Google Scholar]
- Lee, H.; Lee, Y.J.; Choi, H.; Ko, E.H.; Kim, J.W. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 2009, 284, 10601–10609. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, T. Metabolic syndrome and oxidative stress. Free Radic. Biol. Med. 2009, 47, 213–218. [Google Scholar] [CrossRef]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J.; et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012, 936486. [Google Scholar]
- Fujita, K.; Nishizawa, H.; Funahashi, T.; Shimomura, I.; Shimabukuro, M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ. J. 2006, 70, 1437–1442. [Google Scholar] [CrossRef]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. (Lond) 2006, 30, 400–418. [Google Scholar] [CrossRef]
- Galinier, A.; Carriere, A.; Fernandez, Y.; Carpene, C.; Andre, M.; Caspar-Bauguil, S.; Thouvenot, J.P.; Periquet, B.; Penicaud, L.; Casteilla, L. Adipose tissue proadipogenic redox changes in obesity. J. Biol. Chem. 2006, 281, 12682–12687. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, G.-S.; Chan, E.C.; Higuchi, M.; Dusting, G.J.; Jiang, F. Redox Mechanisms in Regulation of Adipocyte Differentiation: Beyond a General Stress Response. Cells 2012, 1, 976-993. https://doi.org/10.3390/cells1040976
Liu G-S, Chan EC, Higuchi M, Dusting GJ, Jiang F. Redox Mechanisms in Regulation of Adipocyte Differentiation: Beyond a General Stress Response. Cells. 2012; 1(4):976-993. https://doi.org/10.3390/cells1040976
Chicago/Turabian StyleLiu, Guei-Sheung, Elsa C. Chan, Masayoshi Higuchi, Gregory J. Dusting, and Fan Jiang. 2012. "Redox Mechanisms in Regulation of Adipocyte Differentiation: Beyond a General Stress Response" Cells 1, no. 4: 976-993. https://doi.org/10.3390/cells1040976