p53 -Dependent and -Independent Nucleolar Stress Responses


{kind=link}
{kind=link}
{kind=link}
Abstract
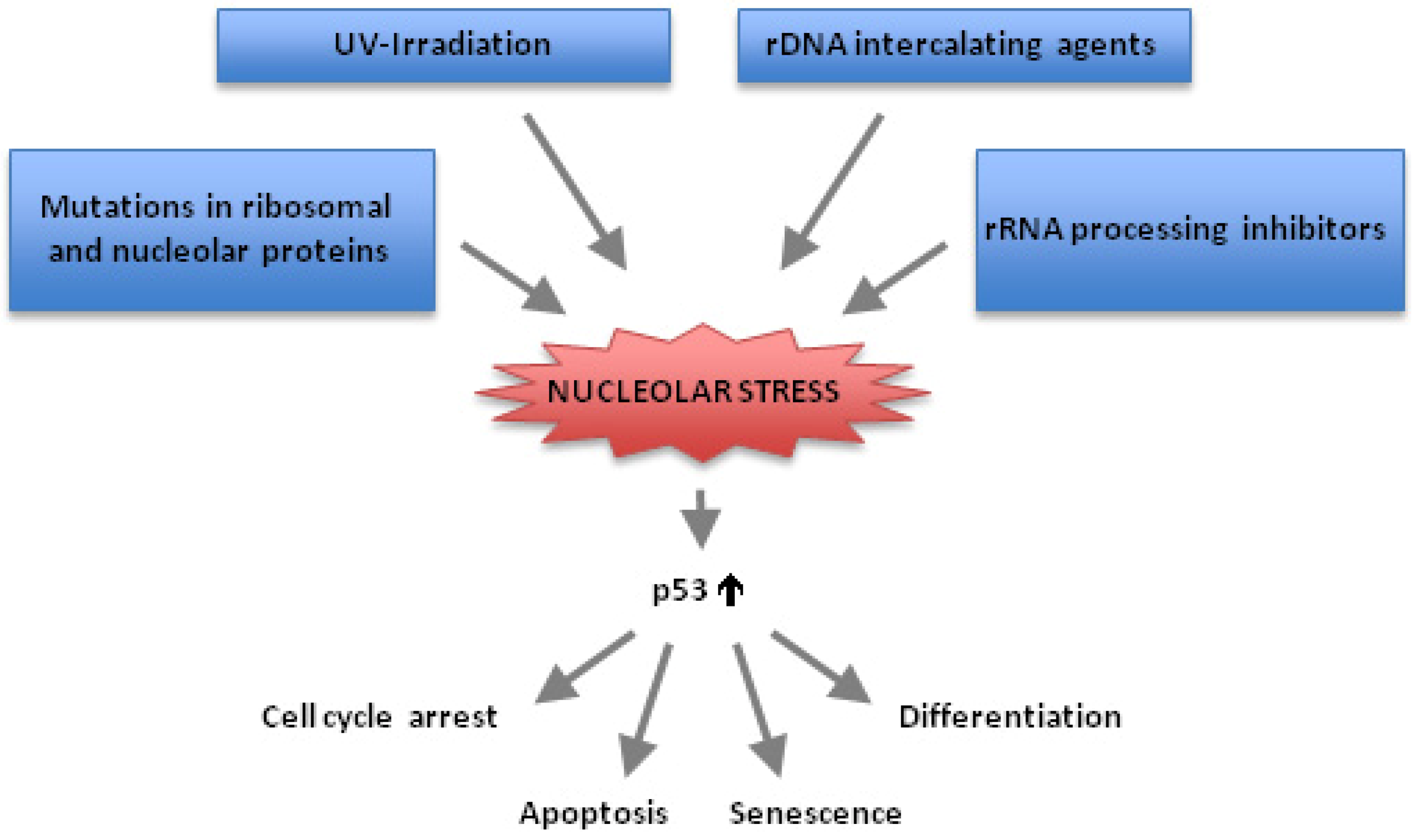
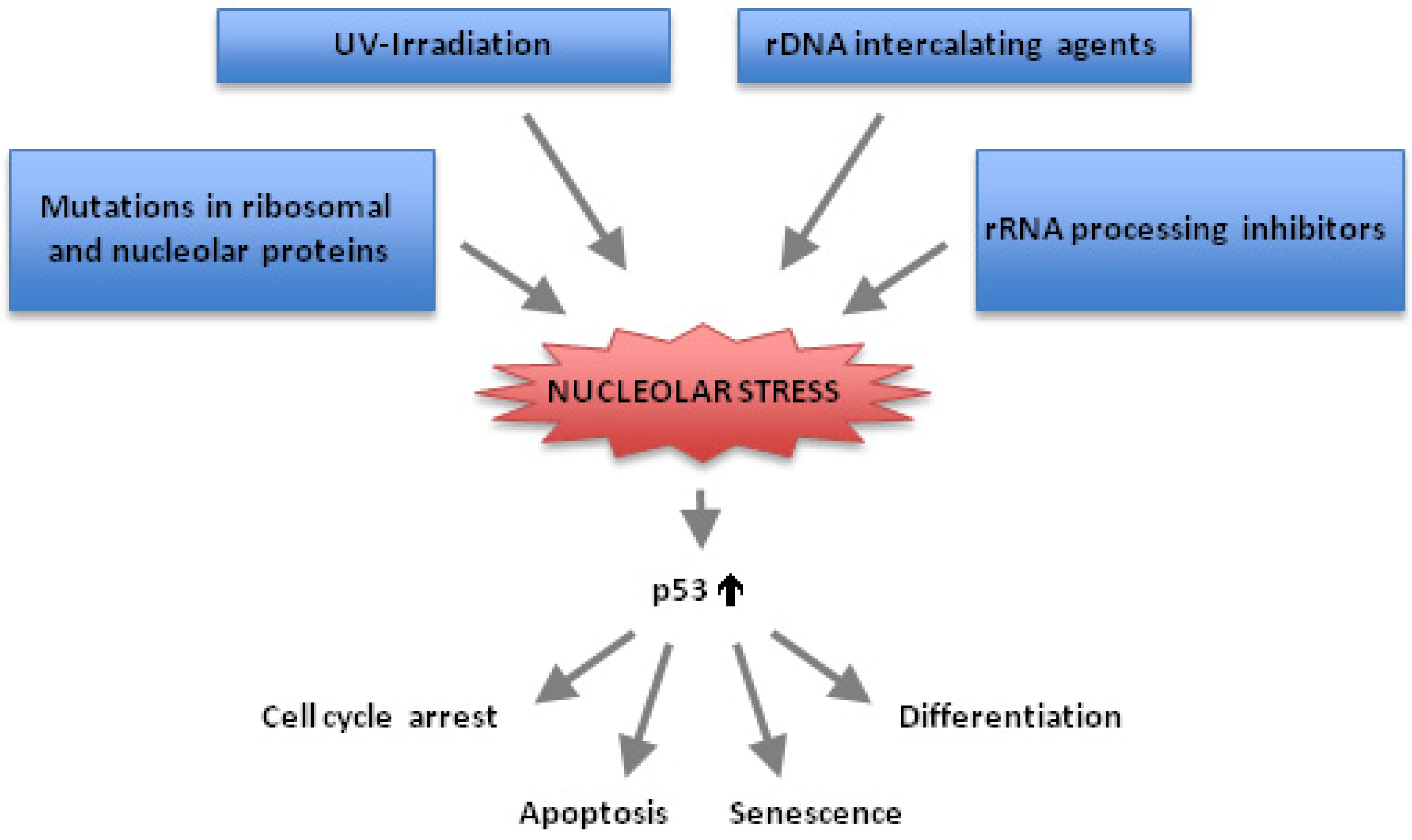
:1. Introduction
2. p53 and Nucleolar Stress
3. Chemotherapeutic Drugs Often Block Ribosome Biogenesis and Induce Nucleolar Stress
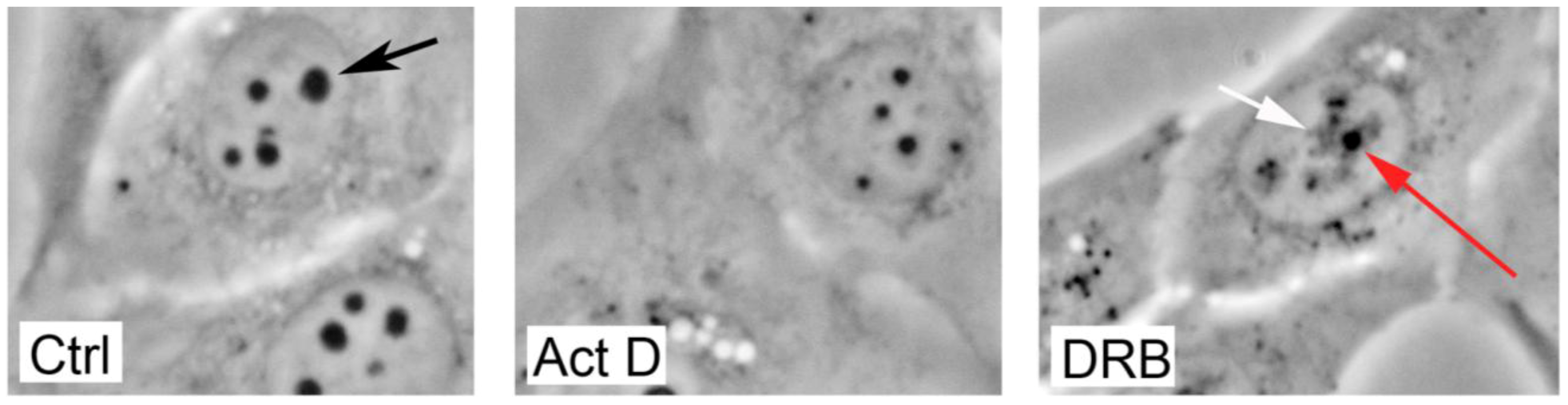
4. Changes In Nucleolar Morphology Following Cellular Stress
5. Early Evidence From Mouse Models Reveal That Ribosome Biogenesis Defects Activate p53
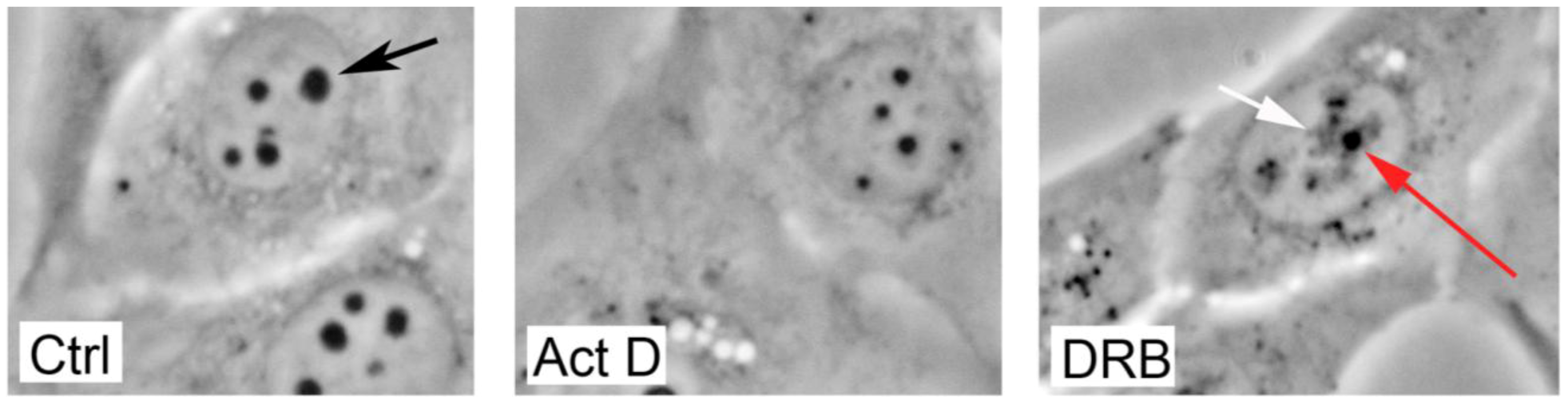
6. Human Ribosomopathies—the Rle of p53
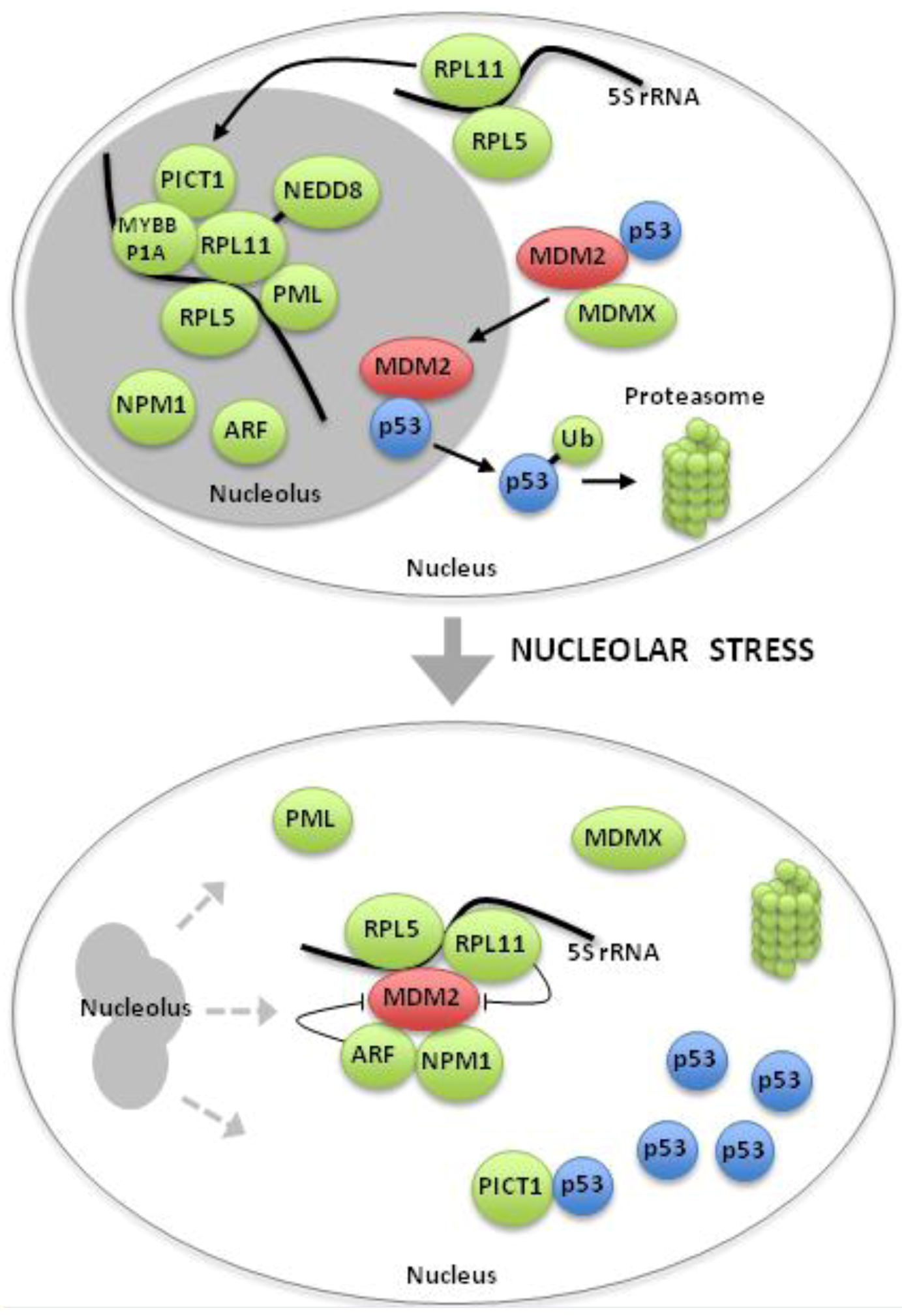
7. Mechanisms of p53 Activation by Nucleolar Stress
7.1. p53 Protein Stabilization By Default
7.2. Inhibition of the MDM2 E3 Ligase Activity by Ribosomal Proteins
8. Role of Ribosomal Protein-MDM2 Signaling in Cancer and DBA
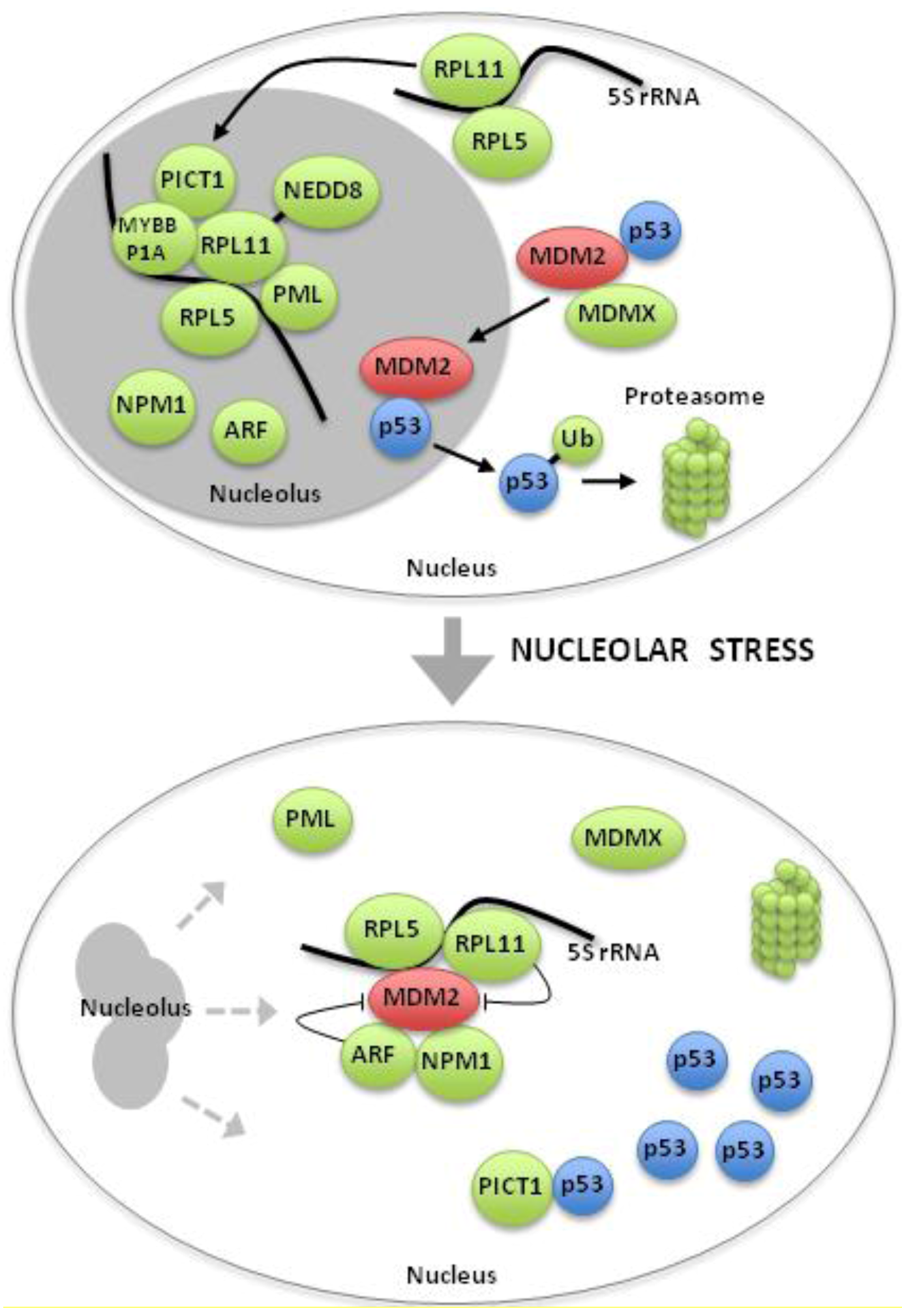
9. Novel regulators of The Ribosomal Protein-MDM2 Complex
10. p53-Independent Nucleolar Stress Pathways
11. Conclusions
Acknowledgements
Conflict of Interest
References
- Hernandez-Verdun, D. Nucleolus: From structure to dynamics. Histochem. Cell Biol. 2006, 125, 127–137. [Google Scholar] [CrossRef]
- Ahmad, Y.; Boisvert, F.M.; Gregor, P.; Cobley, A.; Lamond, A.I. Nopdb: Nucleolar proteome database-2008 update. Nucleic Acids Res. 2009, 37, D181–D184. [Google Scholar] [CrossRef]
- Andersen, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11. [Google Scholar] [CrossRef]
- Andersen, J.S.; Lam, Y.W.; Leung, A.K.; Ong, S.E.; Lyon, C.E.; Lamond, A.I.; Mann, M. Nucleolar proteome dynamics. Nature 2005, 433, 77–83. [Google Scholar] [CrossRef]
- Lam, Y.W.; Lamond, A.I.; Mann, M.; Andersen, J.S. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 2007, 17, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Brown, J. Nucleoli: Composition, function, and dynamic. Plant Physiol. 2012, 158, 44–51. [Google Scholar] [CrossRef]
- Boisvert, F.M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Cisterna, B.; Biggiogera, M. Ribosome biogenesis: From structure to dynamics. Int. Rev. Cell Mol. Biol. 2010, 284, 67–111. [Google Scholar] [CrossRef]
- Panse, V.G.; Johnson, A.W. Maturation of eukaryotic ribosomes: Acquisition of functionality. Trends Biochem. Sci. 2010, 35, 260–266. [Google Scholar] [CrossRef]
- Leung, A.K.; Andersen, J.S.; Mann, M.; Lamond, A.I. Bioinformatic analysis of the nucleolus. Biochem J. 2003, 376, 553–569. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.O. Sensing cellular stress: Another new function for the nucleolus? Sci. STKE 2004, 2004, pe10. [Google Scholar] [CrossRef]
- Olson, M.O.; Dundr, M.; Szebeni, A. The nucleolus: An old factory with unexpected capabilities. Trends Cell Biol. 2000, 10, 189–196. [Google Scholar] [CrossRef]
- Pederson, T. The plurifunctional nucleolus. Nucleic Acids Res. 1998, 26, 3871–3876. [Google Scholar] [CrossRef]
- Pederson, T.; Tsai, R.Y. In search of nonribosomal nucleolar protein function and regulation. J. Cell Biol. 2009, 184, 771–776. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Bartova, E.; Horakova, A.H.; Uhlirova, R.; Raska, I.; Galiova, G.; Orlova, D.; Kozubek, S. Structure and epigenetics of nucleoli in comparison with non-nucleolar compartments. J. Histochem. Cytochem. 2010, 58, 391–403. [Google Scholar] [CrossRef]
- McKeown, P.C.; Shaw, P.J. Chromatin: Linking structure and function in the nucleolus. Chromosoma 2009, 118, 11–23. [Google Scholar] [CrossRef]
- Nemeth, A.; Langst, G. Genome organization in and around the nucleolus. Trends Genet. 2011, 27, 149–156. [Google Scholar] [CrossRef]
- Ruggero, D.; Pandolfi, P.P. Does the ribosome translate cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer p53 guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Itahana, K.; Mao, H.; Jin, A.; Itahana, Y.; Clegg, H.V.; Lindström, M.S.; Bhat, K.P.; Godfrey, V.L.; Evan, G.I.; Zhang, Y. Targeted inactivation of Mdm2 ring finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007, 12, 355–366. [Google Scholar] [CrossRef]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Cellular stress and nucleolar function. Cell Cycle 2005, 4, 1036–1038. [Google Scholar] [CrossRef]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. Embo. J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef]
- Lindström, M.S.; Zhang, Y. Ribosomal protein S9 is a novel B23/NPM-binding protein required for normal cell proliferation. J. Biol. Chem. 2008, 283, 15568–15576. [Google Scholar] [CrossRef]
- Morgado-Palacin, L.; Llanos, S.; Serrano, M. Ribosomal stress induces L11- and p53-dependent apoptosis in mouse pluripotent stem cells. Cell Cycle 2012, 11, 503–510. [Google Scholar] [CrossRef]
- Liu, J.J.; Huang, B.H.; Zhang, J.; Carson, D.D.; Hooi, S.C. Repression of HIP/RPL29 expression induces differentiation in colon cancer cells. J. Cell Physiol. 2006, 207, 287–292. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O'Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O'Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar]
- Raska, I.; Shaw, P.J.; Cmarko, D. New insights into nucleolar architecture and activity. Int. Rev. Cytol. 2006, 255, 177–235. [Google Scholar] [CrossRef]
- Bhat, K.P.; Itahana, K.; Jin, A.; Zhang, Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. Embo. J. 2004, 23, 2402–2412. [Google Scholar] [CrossRef]
- Sun, X.X.; Dai, M.S.; Lu, H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 2007, 282, 8052–8059. [Google Scholar] [CrossRef]
- Sun, X.X.; Dai, M.S.; Lu, H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J. Biol Chem 2008, 283, 12387–12392. [Google Scholar] [CrossRef]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef]
- Abd El-Aal, H.H.; Habib, E.E.; Mishrif, M.M. Wilms' tumor: The experience of the pediatric unit of kasr el-aini center of radiation oncology and nuclear medicine (nemrock). J. Egyptian Natl. Cancer Inst. 2005, 17, 308–314. [Google Scholar]
- Jaffe, N.; Paed, D.; Traggis, D.; Salian, S.; Cassady, J.R. Improved outlook for Ewing's sarcoma with combination chemotherapy (vincristine, actinomycind and cyclophosphamide) and radiation therapy. Cancer 1976, 38, 1925–1930. [Google Scholar] [CrossRef]
- Turan, T.; Karacay, O.; Tulunay, G.; Boran, N.; Koc, S.; Bozok, S.; Kose, M.F. Results with ema/co (etoposide, methotrexate, actinomycin d, cyclophosphamide, vincristine) chemotherapy in gestational trophoblastic neoplasia. Int. J. Gynecol. Cancer 2006, 16, 1432–1438. [Google Scholar] [CrossRef]
- Ochs, R.L. Methods used to study structure and function of the nucleolus. Methods Cell Biol. 1998, 53, 303–321. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Persistent synthesis of 5S RNA when production of 28S and 18S ribosomal RNA is inhibited by low doses of Actinomycind. J. Cell Physiol. 1968, 72, 235–246. [Google Scholar] [CrossRef]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by Actinomycin d: Characteristic dose-response of different RNA species. J. Cell Physiol. 1970, 76, 127–139. [Google Scholar] [CrossRef]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef]
- Donati, G.; Brighenti, E.; Vici, M.; Mazzini, G.; Trere, D.; Montanaro, L.; Derenzini, M. Selective inhibition of rRNA transcription downregulates E2F-1: A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 2012, 124, 3017–3028. [Google Scholar]
- Haaf, T.; Ward, D.C. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp. Cell Res. 1996, 224, 163–173. [Google Scholar] [CrossRef]
- Scheer, U.; Hugle, B.; Hazan, R.; Rose, K.M. Drug-induced dispersal of transcribed rRNA genes and transcriptional products: Immunolocalization and silver staining of different nucleolar components in rat cells treated with 5,6-dichloro-beta-d-ribofuranosylbenzimidazole. J. Cell Biol. 1984, 99, 672–679. [Google Scholar] [CrossRef]
- Shav-Tal, Y.; Blechman, J.; Darzacq, X.; Montagna, C.; Dye, B.T.; Patton, J.G.; Singer, R.H.; Zipori, D. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell 2005, 16, 2395–2413. [Google Scholar] [CrossRef]
- Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; Natt, F.; Schwemberger, S.; Hall, J.; Babcock, G.F.; Bernardi, R.; Pandolfi, P.P.; Thomas, G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an RPL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 2009, 11, 501–508. [Google Scholar] [CrossRef]
- O'Donohue, M.F.; Choesmel, V.; Faubladier, M.; Fichant, G.; Gleizes, P.E. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J. Cell Biol. 2010, 190, 853–866. [Google Scholar] [CrossRef]
- Holzel, M.; Orban, M.; Hochstatter, J.; Rohrmoser, M.; Harasim, T.; Malamoussi, A.; Kremmer, E.; Langst, G.; Eick, D. Defects in 18S or 28S rRNA processing activate the p53 pathway. J. Biol. Chem. 2010, 285, 6364–6370. [Google Scholar]
- Golomb, L.; Bublik, D.R.; Wilder, S.; Nevo, R.; Kiss, V.; Grabusic, K.; Volarevic, S.; Oren, M. Importin 7 and exportin 1 link c-myc and p53 to regulation of ribosomal biogenesis. Mol. Cell 2012, 45, 222–232. [Google Scholar] [CrossRef]
- Ferreira-Cerca, S.; Poll, G.; Gleizes, P.E.; Tschochner, H.; Milkereit, P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18s rRNA and ribosome function. Mol. Cell 2005, 20, 263–275. [Google Scholar] [CrossRef]
- Ferreira-Cerca, S.; Poll, G.; Kuhn, H.; Neueder, A.; Jakob, S.; Tschochner, H.; Milkereit, P. Analysis of the in vivo assembly pathway of eukaryotic 40s ribosomal proteins. Mol. Cell 2007, 28, 446–457. [Google Scholar] [CrossRef]
- Robledo, S.; Idol, R.A.; Crimmins, D.L.; Ladenson, J.H.; Mason, P.J.; Bessler, M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 2008, 14, 1918–1929. [Google Scholar] [CrossRef]
- Volarevic, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef]
- Panic, L.; Tamarut, S.; Sticker-Jantscheff, M.; Barkic, M.; Solter, D.; Uzelac, M.; Grabusic, K.; Volarevic, S. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol. Cell Biol. 2006, 26, 8880–8891. [Google Scholar] [CrossRef]
- Sulic, S.; Panic, L.; Barkic, M.; Mercep, M.; Uzelac, M.; Volarevic, S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 2005, 19, 3070–3082. [Google Scholar] [CrossRef]
- McGowan, K.A.; Li, J.Z.; Park, C.Y.; Beaudry, V.; Tabor, H.K.; Sabnis, A.J.; Zhang, W.; Fuchs, H.; de Angelis, M.H.; Myers, R.M.; et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 2008, 40, 963–970. [Google Scholar] [CrossRef]
- Barkic, M.; Crnomarkovic, S.; Grabusic, K.; Bogetic, I.; Panic, L.; Tamarut, S.; Cokaric, M.; Jeric, I.; Vidak, S.; Volarevic, S. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol. Cell Biol. 2009, 29, 2489–2504. [Google Scholar] [CrossRef]
- Zhang, J.; Tomasini, A.J.; Mayer, A.N. Rbm19 is essential for preimplantation development in the mouse. BMC Dev. Biol. 2008, 8, 115. [Google Scholar] [CrossRef]
- Fumagalli, S.; Thomas, G. The role of p53 in ribosomopathies. Semin. Hematol. 2011, 48, 97–105. [Google Scholar] [CrossRef]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef]
- Choesmel, V.; Fribourg, S.; Aguissa-Toure, A.H.; Pinaud, N.; Legrand, P.; Gazda, H.T.; Gleizes, P.E. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum. Mol. Genet. 2008, 17, 1253–1263. [Google Scholar] [CrossRef]
- Farrar, J.E.; Nater, M.; Caywood, E.; McDevitt, M.A.; Kowalski, J.; Takemoto, C.M.; Talbot, C.C., Jr.; Meltzer, P.; Esposito, D.; Beggs, A.H.; et al. Abnormalities of the large ribosomal subunit protein, RPL35a, in Diamond-Blackfan anemia. Blood 2008, 112, 1582–1592. [Google Scholar] [CrossRef]
- Gazda, H.T.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O'Donohue, M.F.; Schneider, H.; Darras, N.; Hasman, C.; Sieff, C.A.; Newburger, P.E.; et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 2008, 83, 769–780. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Invest. 2012, 122, 2439–2443. [Google Scholar] [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef]
- Chakraborty, A.; Uechi, T.; Kenmochi, N. Guarding the 'translation apparatus': Defective ribosome biogenesis and the p53 signaling pathway. Wiley Interdiscip. Rev. RNA 2011, 2, 507–522. [Google Scholar] [CrossRef]
- Barlow, J.L.; Drynan, L.F.; Hewett, D.R.; Holmes, L.R.; Lorenzo-Abalde, S.; Lane, A.L.; Jolin, H.E.; Pannell, R.; Middleton, A.J.; Wong, S.H.; et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 2010, 16, 59–66. [Google Scholar]
- McGowan, K.A.; Pang, W.W.; Bhardwaj, R.; Perez, M.G.; Pluvinage, J.V.; Glader, B.E.; Malek, R.; Mendrysa, S.M.; Weissman, I.L.; Park, C.Y.; et al. Reduced ribosomal protein gene dosage and p53 activation in low-risk myelodysplastic syndrome. Blood 2011, 118, 3622–3633. [Google Scholar] [CrossRef]
- Jones, N.C.; Lynn, M.L.; Gaudenz, K.; Sakai, D.; Aoto, K.; Rey, J.P.; Glynn, E.F.; Ellington, L.; Du, C.; Dixon, J.; et al. Prevention of the neurocristopathy treacher collins syndrome through inhibition of p53 function. Nat. Med. 2008, 14, 125–133. [Google Scholar] [CrossRef]
- Chakraborty, A.; Uechi, T.; Higa, S.; Torihara, H.; Kenmochi, N. Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. Plos One 2009, 4, e4152. [Google Scholar]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008, 112, 5228–5237. [Google Scholar] [CrossRef]
- Fumagalli, S.; Ivanenkov, V.V.; Teng, T.; Thomas, G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012, 26, 1028–1040. [Google Scholar] [CrossRef]
- Lindström, M.S.; Nistér, M. Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. Plos One 2010. [Google Scholar] [CrossRef]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef]
- Macias, E.; Jin, A.; Deisenroth, C.; Bhat, K.; Mao, H.; Lindström, M.S.; Zhang, Y. An Arf-independent c-Myc-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 interaction. Cancer Cell 2010, 18, 231–243. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Komarova, E.A. Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol. 2010, 2, a001180. [Google Scholar] [CrossRef]
- Miliani de Marval, P.L.; Zhang, Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget 2011, 2, 234–238. [Google Scholar]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-mdm2-p53 pathway. Oncogene 2011, 29, 4253–4260. [Google Scholar] [CrossRef]
- Boyd, M.T.; Vlatkovic, N.; Rubbi, C.P. The nucleolus directly regulates p53 export and degradation. J. Cell Biol. 2011, 194, 689–703. [Google Scholar] [CrossRef]
- Warner, J.R.; McIntosh, K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef]
- Kuroda, T.; Murayama, A.; Katagiri, N.; Ohta, Y.M.; Fujita, E.; Masumoto, H.; Ema, M.; Takahashi, S.; Kimura, K.; Yanagisawa, J. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J. 2011, 30, 1054–1066. [Google Scholar] [CrossRef]
- MacInnes, A.W.; Amsterdam, A.; Whittaker, C.A.; Hopkins, N.; Lees, J.A. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Pro.c Natl. Acad. Sci. USA 2008, 105, 10408–10413. [Google Scholar] [CrossRef]
- Castro, M.E.; Leal, J.F.; Lleonart, M.E.; Ramon, Y.C.S.; Carnero, A. Loss-of-function genetic screening identifies a cluster of ribosomal proteins regulating p53 function. Carcinogenesis 2008, 29, 1343–1350. [Google Scholar] [CrossRef]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef]
- Tao, W.; Levine, A.J. P19(Arf) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef]
- Marechal, V.; Elenbaas, B.; Piette, J.; Nicolas, J.C.; Levine, A.J. The ribosomal L5 protein is associated with Mdm-2 and Mdm-2-p53 complexes. Mol. Cell Biol. 1994, 14, 7414–7420. [Google Scholar]
- Dai, M.S.; Lu, H. Inhibition of Mdm2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar]
- Dai, M.S.; Zeng, S.X.; Jin, Y.; Sun, X.X.; David, L.; Lu, H. Ribosomal protein L23 activates p53 by inhibiting Mdm2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell Biol. 2004, 24, 7654–7668. [Google Scholar] [CrossRef]
- Jin, A.; Itahana, K.; O'Keefe, K.; Zhang, Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef]
- Lindström, M.S.; Deisenroth, C.; Zhang, Y. Putting a finger on growth surveillance: Insight into Mdm2 zinc finger-ribosomal protein interactions. Cell Cycle 2007, 6, 434–437. [Google Scholar] [CrossRef]
- Lindström, M.S.; Jin, A.; Deisenroth, C.; White Wolf, G.; Zhang, Y. Cancer-associated mutations in the Mdm2 zinc finger domain disrupt ribosomal protein interaction and attenuate Mdm2-induced p53 degradation. Mol. Cell Biol. 2007, 27, 1056–1068. [Google Scholar] [CrossRef]
- Lohrum, M.A.; Ludwig, R.L.; Kubbutat, M.H.; Hanlon, M.; Vousden, K.H. Regulation of Hdm2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein Mdm2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef]
- Yadavilli, S.; Mayo, L.D.; Higgins, M.; Lain, S.; Hegde, V.; Deutsch, W.A. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and Mdm2 through its KH domain. DNA Repair 2009, 8, 1215–1224. [Google Scholar] [CrossRef]
- Zhu, Y.; Poyurovsky, M.V.; Li, Y.; Biderman, L.; Stahl, J.; Jacq, X.; Prives, C. Ribosomal protein S7 is both a regulator and a substrate of Mdm2. Mol. Cell 2009, 35, 316–326. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Liao, J.; Zhang, Q.; Lu, H. Ribosomal protein S14 unties the Mdm2-p53 loop upon ribosomal stress. Oncogene 2012. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Wang, H.; Wang, M.H.; Xu, W.; Zhang, R. Identification of ribosomal protein S25 (RPS25)-Mdm2-p53 regulatory feedback loop. Oncogene 2012. [Google Scholar] [CrossRef]
- He, H.; Sun, Y. Ribosomal protein S27L is a direct p53 target that regulates apoptosis. Oncogene 2007, 26, 2707–2716. [Google Scholar] [CrossRef]
- Li, J.; Tan, J.; Zhuang, L.; Banerjee, B.; Yang, X.; Chau, J.F.; Lee, P.L.; Hande, M.P.; Li, B.; Yu, Q. Ribosomal protein S27-like, a p53-inducible modulator of cell fate in response to genotoxic stress. Cancer Res. 2007, 67, 11317–11326. [Google Scholar]
- Xiong, X.; Zhao, Y.; He, H.; Sun, Y. Ribosomal protein S27-like and S27 interplay with p53-Mdm2 axis as a target, a substrate and a regulator. Oncogene 2011, 30, 1798–1811. [Google Scholar] [CrossRef]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef]
- Dai, M.S.; Shi, D.; Jin, Y.; Sun, X.X.; Zhang, Y.; Grossman, S.R.; Lu, H. Regulation of the Mdm2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J. Biol. Chem. 2006, 281, 24304–24313. [Google Scholar]
- Zhang, Q.; Xiao, H.; Chai, S.C.; Hoang, Q.Q.; Lu, H. Hydrophilic residues are crucial for ribosomal protein L11 (RPL11) interaction with zinc finger domain of Mdm2 and p53 protein activation. J. Biol. Chem. 2012, 286, 38264–38274. [Google Scholar]
- Sun, X.X.; Wang, Y.G.; Xirodimas, D.P.; Dai, M.S. Perturbation of 60S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J. Biol. Chem. 2010, 285, 25812–25821. [Google Scholar] [CrossRef]
- Candeias, M.M.; Malbert-Colas, L.; Powell, D.J.; Daskalogianni, C.; Maslon, M.M.; Naski, N.; Bourougaa, K.; Calvo, F.; Fahraeus, R. p53 mRNA controls p53 activity by managing Mdm2 functions. Nat. Cell Biol. 2008, 10, 1098–1105. [Google Scholar] [CrossRef]
- Naski, N.; Gajjar, M.; Bourougaa, K.; Malbert-Colas, L.; Fahraeus, R.; Candeias, M.M. The p53 mRNA-Mdm2 interaction. Cell Cycle 2009, 8, 31–34. [Google Scholar] [CrossRef]
- Sundqvist, A.; Liu, G.; Mirsaliotis, A.; Xirodimas, D.P. Regulation of nucleolar signalling to p53 through neddylation of L11. EMBO Rep. 2009, 10, 1132–1139. [Google Scholar] [CrossRef]
- Janas, M.M.; Wang, E.; Love, T.; Harris, A.S.; Stevenson, K.; Semmelmann, K.; Shaffer, J.M.; Chen, P.H.; Doench, J.G.; Yerramilli, S.V.; et al. Reduced expression of ribosomal proteins relieves microrna-mediated repression. Mol. Cell 2012, 46, 171–186. [Google Scholar] [CrossRef]
- Schlott, T.; Reimer, S.; Jahns, A.; Ohlenbusch, A.; Ruschenburg, I.; Nagel, H.; Droese, M. Point mutations and nucleotide insertions in the Mdm2 zinc finger structure of human tumours. J. Pathol. 1997, 182, 54–61. [Google Scholar] [CrossRef]
- Tamborini, E.; Della Torre, G.; Lavarino, C.; Azzarelli, A.; Carpinelli, P.; Pierotti, M.A.; Pilotti, S. Analysis of the molecular species generated by Mdm2 gene amplification in liposarcomas. Int. J. Cancer 2001, 92, 790–796. [Google Scholar] [CrossRef]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef]
- Donati, G.; Bertoni, S.; Brighenti, E.; Vici, M.; Trere, D.; Volarevic, S.; Montanaro, L.; Derenzini, M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene 2011, 30, 3274–3288. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Chen, L.; Chen, J. Mdmx regulation of p53 response to ribosomal stress. EMBO. J. 2006, 25, 5614–5625. [Google Scholar] [CrossRef]
- Bernardi, R.; Scaglioni, P.P.; Bergmann, S.; Horn, H.F.; Vousden, K.H.; Pandolfi, P.P. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat. Cell Biol. 2004, 6, 665–672. [Google Scholar] [CrossRef]
- Li, M.; Gu, W. A critical role for noncoding 5S rRNA in regulating Mdmx stability. Mol. Cell 2011, 43, 1023–1032. [Google Scholar] [CrossRef]
- Smith, J.S.; Tachibana, I.; Lee, H.K.; Qian, J.; Pohl, U.; Mohrenweiser, H.W.; Borell, T.J.; Hosek, S.M.; Soderberg, C.L.; von Deimling, A.; et al. Mapping of the chromosome 19q-arm glioma tumor suppressor gene using fluorescence in situ hybridization and novel microsatellite markers. Genes Chromosomes Cancer 2000, 29, 16–25. [Google Scholar]
- Merritt, M.A.; Parsons, P.G.; Newton, T.R.; Martyn, A.C.; Webb, P.M.; Green, A.C.; Papadimos, D.J.; Boyle, G.M. Expression profiling identifies genes involved in neoplastic transformation of serous ovarian cancer. BMC Cancer 2009, 9, 378. [Google Scholar] [CrossRef]
- Kim, Y.J.; Cho, Y.E.; Kim, Y.W.; Kim, J.Y.; Lee, S.; Park, J.H. Suppression of putative tumour suppressor gene GLTSCR2 expression in human glioblastomas. J. Pathol. 2008, 216, 218–224. [Google Scholar] [CrossRef]
- Yim, J.H.; Kim, Y.J.; Ko, J.H.; Cho, Y.E.; Kim, S.M.; Kim, J.Y.; Lee, S.; Park, J.H. The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ. 2007, 14, 1872–1879. [Google Scholar] [CrossRef]
- Sasaki, M.; Kawahara, K.; Nishio, M.; Mimori, K.; Kogo, R.; Hamada, K.; Itoh, B.; Wang, J.; Komatsu, Y.; Yang, Y.R.; et al. Regulation of the Mdm2-p53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 2011, 17, 944–951. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.Y.; Kim, Y.J.; Seok, K.O.; Kim, J.H.; Chang, Y.J.; Kang, H.Y.; Park, J.H. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 2012, 19, 1613–1622. [Google Scholar] [CrossRef]
- Dai, M.S.; Sun, X.X.; Lu, H. Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of Mdm2. Mol. Cell Biol. 2008, 28, 4365–4376. [Google Scholar] [CrossRef]
- Ma, H.; Pederson, T. Nucleostemin: A multiplex regulator of cell-cycle progression. Trends Cell Biol. 2008, 18, 575–579. [Google Scholar] [CrossRef]
- Thomson, E.; Tollervey, D. Nop53p is required for late 60S ribosome subunit maturation and nuclear export in yeast. RNA 2005, 11, 1215–1224. [Google Scholar] [CrossRef]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Yeh, E.T. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 1997, 272, 28557–28562. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-ring ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar]
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. Fbxo11 promotes the neddylation of p53 and inhibits its transcriptional activity. J. Biol. Chem. 2007, 282, 1797–1804. [Google Scholar]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated nedd8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef]
- Mahata, B.; Sundqvist, A.; Xirodimas, D.P. Recruitment of RPL11 at promoter sites of p53-regulated genes upon nucleolar stress through NEDD8 and in an Mdm2-dependent manner. Oncogene 2011, 31, 3060–3071. [Google Scholar]
- Kurki, S.; Peltonen, K.; Latonen, L.; Kiviharju, T.M.; Ojala, P.M.; Meek, D.; Laiho, M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475. [Google Scholar] [CrossRef]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14Arf without relocation of Mdm2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef]
- Iadevaia, V.; Caldarola, S.; Biondini, L.; Gismondi, A.; Karlsson, S.; Dianzani, I.; Loreni, F. PIM1 kinase is destabilized by ribosomal stress causing inhibition of cell cycle progression. Oncogene 2010, 29, 5490–5499. [Google Scholar] [CrossRef]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef]
- Ganguli, G.; Wasylyk, B. p53-independent functions of Mdm2. Mol. Cancer Res. 2003, 1, 1027–1035. [Google Scholar]
- Dai, M.S.; Arnold, H.; Sun, X.X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. Embo. J. 2007, 26, 3332–3345. [Google Scholar] [CrossRef]
- Dai, M.S.; Sun, X.X.; Lu, H. Ribosomal protein L11 associates with c-Myc at 5S rRNA and tRNA genes and regulates their expression. J. Biol. Chem. 2010, 285, 12587–12594. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Sun, X.X.; Zhang, X.; DeVine, T.; Zhang, Q.; Sears, R.C.; Dai, M.S. Ribosomal protein L11 recruits mir-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell Biol. 2011, 31, 4007–4021. [Google Scholar] [CrossRef]
- Lindström, M.S. Emerging functions of ribosomal proteins in gene-specific transcription and translation. Biochem. Biophys. Res. Commun. 2009, 379, 167–170. [Google Scholar] [CrossRef]
- Wool, I.G. Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci. 1996, 21, 164–165. [Google Scholar]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef]
- Budde, A.; Grummt, I. p53 represses ribosomal gene transcription. Oncogene 1999, 18, 1119–1124. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Holmberg Olausson, K.; Nistér, M.; Lindström, M.S. p53 -Dependent and -Independent Nucleolar Stress Responses. Cells 2012, 1, 774-798. https://doi.org/10.3390/cells1040774
Holmberg Olausson K, Nistér M, Lindström MS. p53 -Dependent and -Independent Nucleolar Stress Responses. Cells. 2012; 1(4):774-798. https://doi.org/10.3390/cells1040774
Chicago/Turabian StyleHolmberg Olausson, Karl, Monica Nistér, and Mikael S. Lindström. 2012. "p53 -Dependent and -Independent Nucleolar Stress Responses" Cells 1, no. 4: 774-798. https://doi.org/10.3390/cells1040774