Signals and Cells Involved in Regulating Liver Regeneration

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Overview
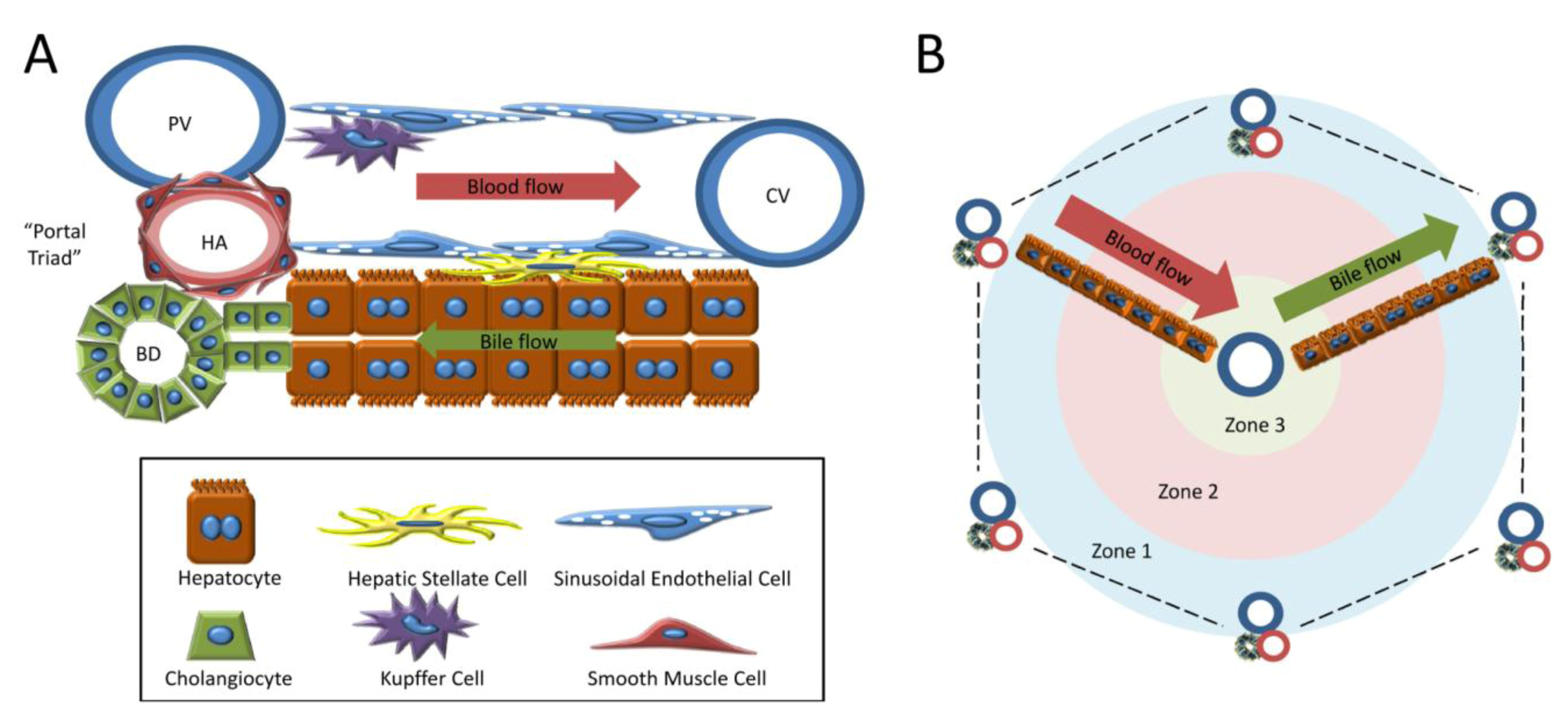
1.2. Cell Types of the Liver
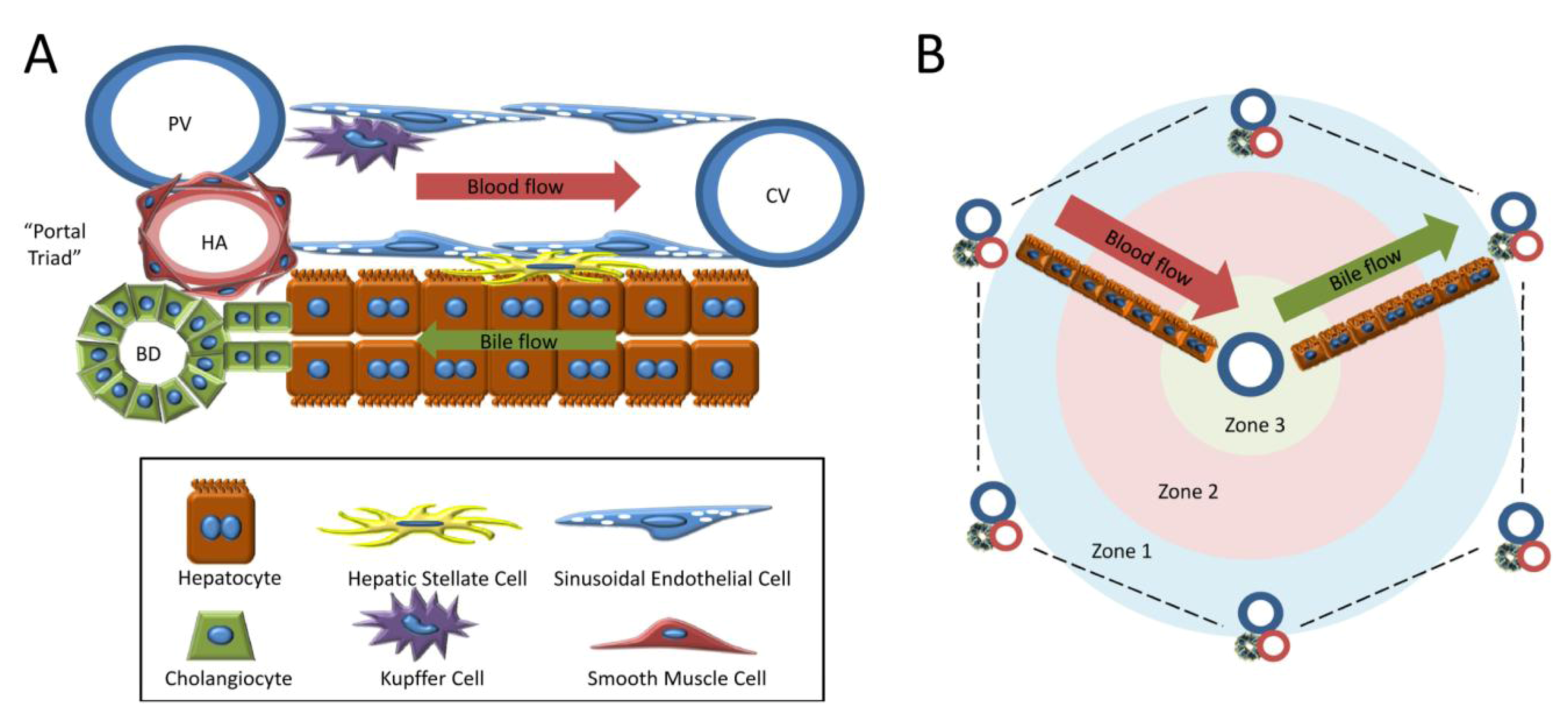
1.3. Models of Liver Regeneration and Growth
2. Molecular Signals During Liver Regeneration after PHx
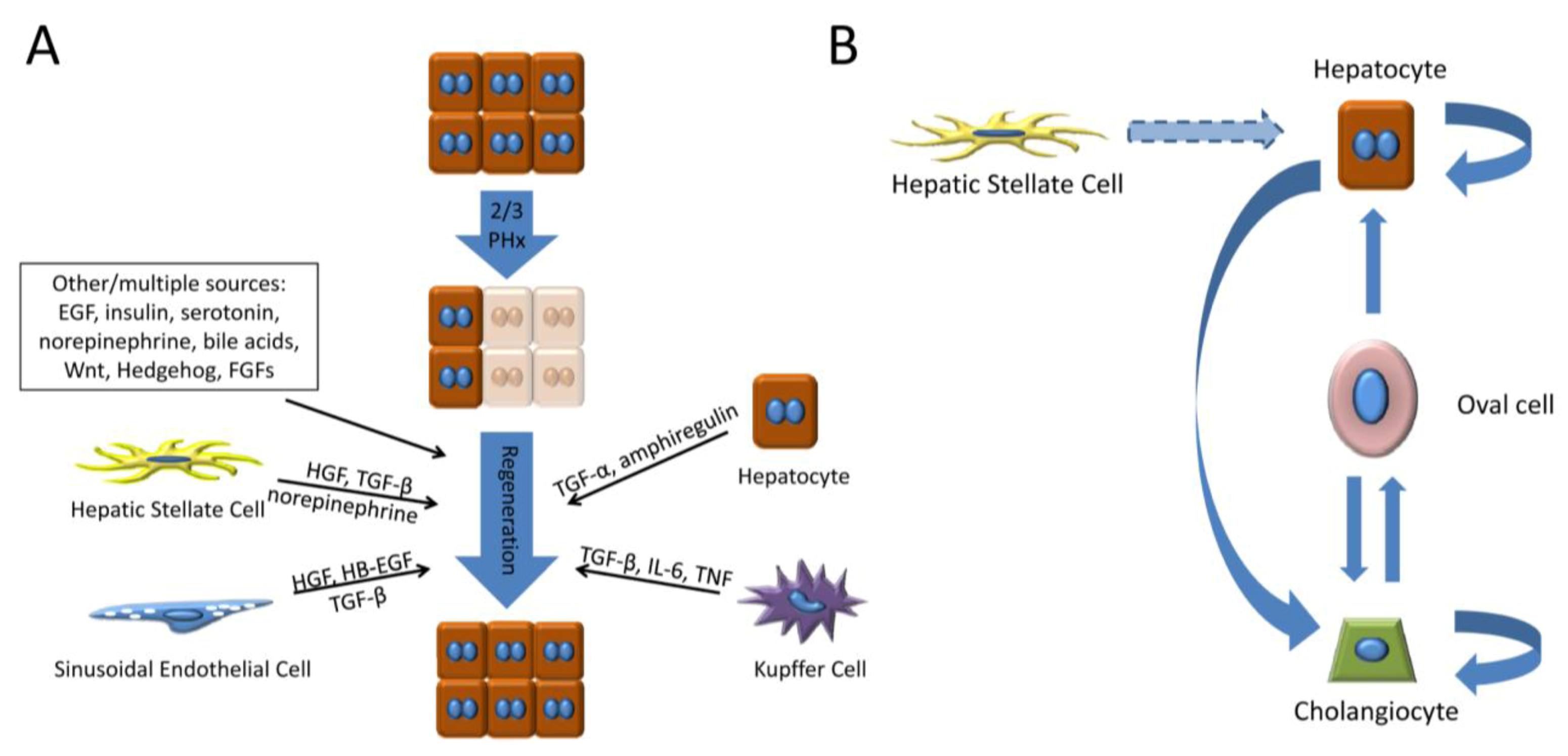
2.1. Hepatocyte Growth Factor (HGF)
2.2. Epidermal Growth Factor Receptor Ligands
2.3. Tumor Necrosis Factor (TNF)
2.4. Interleukin-6 (IL-6)
2.5. Transforming Growth Factor-β (TGF-β)
2.6. Neurotransmitters
2.7. Bile Acids
2.8. Insulin and Other Metabolic Regulators
2.9. Wnt Family Proteins/β-catenin
2.10. Hedgehog
2.11. Fibroblast Growth Factors (FGFs)
3. Contribution of cell types to successful liver regeneration
3.1. Resident Liver Cells
3.2. Progenitor Cells
3.2.1. Bone Marrow Derived Cells

3.2.2. Oval/Progenitor Cells and Transdifferentiation
4. Termination of Liver Regeneration
4.1. Integrin-Linked Kinase (ILK)
4.2. Glypican 3
4.3. TGF-β and Activins
4.4. Yes-Associated Protein (YAP)
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Wolber, E.M.; Jelkmann, W. Thrombopoietin: The novel hepatic hormone. News Physiol. Sci. 2002, 17, 6–10. [Google Scholar]
- Michalopoulos, G.K. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am. J. Pathol. 2010, 176, 2–13. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef]
- Lim, Y.S.; Kim, W.R. The global impact of hepatic fibrosis and end-stage liver disease. Clin. Liver Dis. 2008, 12, 733–746, vii.. [Google Scholar] [CrossRef]
- Parker, G.A.; Picut, C.A. Immune functioning in non lymphoid organs: The liver. Toxicol. Pathol. 2012, 40, 237–247. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Higgins, G.M.; Anderson, R.M. Experimental pathology of the liver – Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. 1931, 12, 186–202. [Google Scholar]
- Mitchell, C.; Willenbring, H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 2008, 3, 1167–1170. [Google Scholar] [CrossRef]
- de Toranzo, E.G.; Gomez, M.I.; Castro, J.A. Carbon tetrachloride activation, lipid peroxidation and liver necrosis in different strains of mice. Res. Commun. Chem. Pathol. Pharmacol. 1978, 19, 347–352. [Google Scholar]
- DeCicco, L.A.; Rikans, L.E.; Tutor, C.G.; Hornbrook, K.R. Serum and liver concentrations of tumor necrosis factor alpha and interleukin-1beta following administration of carbon tetrachloride to male rats. Toxicol. Lett. 1998, 98, 115–121. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Williams, C.D.; Ramachandran, A. Current issues with acetaminophen hepatotoxicity—a clinically relevant model to test the efficacy of natural products. Life Sci. 2011, 88, 737–745. [Google Scholar] [CrossRef]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 369–405. [Google Scholar]
- David Josephy, P. The molecular toxicology of acetaminophen. Drug Metab. Rev. 2005, 37, 581–594. [Google Scholar] [CrossRef]
- Lee, J.H.; Ilic, Z.; Sell, S. Cell kinetics of repair after allyl alcohol-induced liver necrosis in mice. Int. J. Exp. Pathol. 1996, 77, 63–72. [Google Scholar]
- Pound, A.W.; McGuire, L.J. Repeated partial hepatectomy as a promoting stimulus for carcinogenic response of liver to nitrosamines in rats. Br. J. Cancer. 1978, 37, 585–594. [Google Scholar] [CrossRef]
- Bhave, V.S.; Donthamsetty, S.; Latendresse, J.R.; Cunningham, M.L.; Mehendale, H.M. Secretory phospholipase A(2)-mediated progression of hepatotoxicity initiated by acetaminophen is exacerbated in the absence of hepatic COX-2. Toxicol. Appl. Pharmacol. 2011, 251, 173–180. [Google Scholar] [CrossRef]
- Bhave, V.S.; Donthamsetty, S.; Latendresse, J.R.; Muskhelishvili, L.; Mehendale, H.M. Secretory phospholipase A2 mediates progression of acute liver injury in the absence of sufficient cyclooxygenase-2. Toxicol. Appl. Pharmacol. 2008, 228, 225–238. [Google Scholar] [CrossRef]
- Limaye, P.B.; Bhave, V.S.; Palkar, P.S.; Apte, U.M.; Sawant, S.P.; Yu, S.; Latendresse, J.R.; Reddy, J.K.; Mehendale, H.M. Upregulation of calpastatin in regenerating and developing rat liver: role in resistance against hepatotoxicity. Hepatology 2006, 44, 379–388. [Google Scholar] [CrossRef]
- Mehendale, H.M. Once initiated, how does toxic tissue injury expand? Trends Pharmacol. Sci. 2012, 33, 200–206. [Google Scholar] [CrossRef]
- Mehendale, H.M. Tissue repair: an important determinant of final outcome of toxicant-induced injury. Toxicol. Pathol. 2005, 33, 41–51. [Google Scholar] [CrossRef]
- Columbano, A.; Ledda-Columbano, G.M. Mitogenesis by ligands of nuclear receptors: an attractive model for the study of the molecular mechanisms implicated in liver growth. Cell Death Differ. 2003, 10 Suppl. 1, S19–S21. [Google Scholar] [CrossRef]
- Columbano, A.; Simbula, M.; Pibiri, M.; Perra, A.; Deidda, M.; Locker, J.; Pisanu, A.; Uccheddu, A.; Ledda-Columbano, G.M. Triiodothyronine stimulates hepatocyte proliferation in two models of impaired liver regeneration. Cell Prolif. 2008, 41, 521–531. [Google Scholar] [CrossRef]
- Malik, R.; Habib, M.; Tootle, R.; Hodgson, H. Exogenous thyroid hormone induces liver enlargement, whilst maintaining regenerative potential—a study relevant to donor preconditioning. Am. J. Transplant. 2005, 5, 1801–1807. [Google Scholar] [CrossRef]
- Francavilla, A.; Carr, B.I.; Azzarone, A.; Polimeno, L.; Wang, Z.; Van Thiel, D.H.; Subbotin, V.; Prelich, J.G.; Starzl, T.E. Hepatocyte proliferation and gene expression induced by triiodothyronine in vivo and in vitro. Hepatology 1994, 20, 1237–1241. [Google Scholar]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar]
- Qatanani, M.; Moore, D.D. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr. Drug Metab. 2005, 6, 329–339. [Google Scholar] [CrossRef]
- Ledda-Columbano, G.M.; Coni, P.; Simbula, G.; Zedda, I.; Columbano, A. Compensatory regeneration, mitogen-induced liver growth, and multistage chemical carcinogenesis. Environ. Health Perspect. 1993, 101 Suppl. 5, 163–168. [Google Scholar]
- Bursch, W.; Taper, H.S.; Lauer, B.; Schulte-Hermann, R. Quantitative histological and histochemical studies on the occurrence and stages of controlled cell death (apoptosis) during regression of rat liver hyperplasia. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1985, 50, 153–166. [Google Scholar]
- Columbano, A.; Ledda-Columbano, G.M.; Coni, P.P.; Faa, G.; Liguori, C.; Santa Cruz, G.; Pani, P. Occurrence of cell death (apoptosis) during the involution of liver hyperplasia. Lab. Invest. 1985, 52, 670–675. [Google Scholar]
- Columbano, A.; Shinozuka, H. Liver regeneration versus direct hyperplasia. FASEB J. 1996, 10, 1118–1128. [Google Scholar]
- Apte, U.; Gkretsi, V.; Bowen, W.C.; Mars, W.M.; Luo, J.H.; Donthamsetty, S.; Orr, A.; Monga, S.P.; Wu, C.; Michalopoulos, G.K. Enhanced liver regeneration following changes induced by hepatocyte-specific genetic ablation of integrin-linked kinase. Hepatology 2009, 50, 844–851. [Google Scholar]
- Liu, B.; Bell, A.W.; Paranjpe, S.; Bowen, W.C.; Khillan, J.S.; Luo, J.H.; Mars, W.M.; Michalopoulos, G.K. Suppression of liver regeneration and hepatocyte proliferation in hepatocyte-targeted glypican 3 transgenic mice. Hepatology 2010, 52, 1060–1067. [Google Scholar] [CrossRef]
- Donthamsetty, S.; Bowen, W.; Mars, W.; Bhave, V.; Luo, J.H.; Wu, C.; Hurd, J.; Orr, A.; Bell, A.; Michalopoulos, G. Liver-specific ablation of integrin-linked kinase in mice results in enhanced and prolonged cell proliferation and hepatomegaly after phenobarbital administration. Toxicol. Sci. 2010, 113, 358–366. [Google Scholar] [CrossRef]
- Donthamsetty, S.; Bhave, V.S.; Kliment, C.S.; Bowen, W.C.; Mars, W.M.; Bell, A.W.; Stewart, R.E.; Orr, A.; Wu, C.; Michalopoulos, G.K. Excessive hepatomegaly of mice with hepatocyte-targeted elimination of integrin linked kinase following treatment with 1,4-bis [2-(3,5-dichaloropyridyloxy)] benzene. Hepatology 2011, 53, 587–595. [Google Scholar]
- Lin, C.W.; Mars, W.M.; Paranjpe, S.; Donthamsetty, S.; Bhave, V.S.; Kang, L.I.; Orr, A.; Bowen, W.C.; Bell, A.W.; Michalopoulos, G.K. Hepatocyte proliferation and hepatomegaly induced by phenobarbital and 1,4-bis [2-(3,5-dichloropyridyloxy)] benzene is suppressed in hepatocyte-targeted glypican 3 transgenic mice. Hepatology 2011, 54, 620–630. [Google Scholar] [CrossRef]
- Kodama, S.; Negishi, M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor car. Drug. Metab. Rev. 2006, 38, 75–87. [Google Scholar]
- Marubashi, S.; Sakon, M.; Nagano, H.; Gotoh, K.; Hashimoto, K.; Kubota, M.; Kobayashi, S.; Yamamoto, S.; Miyamoto, A.; Dono, K.; Nakamori, S.; Umeshita, K.; Monden, M. Effect of portal hemodynamics on liver regeneration studied in a novel portohepatic shunt rat model. Surgery 2004, 136, 1028–1037. [Google Scholar] [CrossRef]
- Francavilla, A.; Starzl, T.E.; Porter, K.; Foglieni, C.S.; Michalopoulos, G.K.; Carrieri, G.; Trejo, J.; Azzarone, A.; Barone, M.; Zeng, Q.H. Screening for candidate hepatic growth factors by selective portal infusion after canine Eck's fistula. Hepatology 1991, 14, 665–670. [Google Scholar] [Green Version]
- Kim, T.H.; Mars, W.M.; Stolz, D.B.; Petersen, B.E.; Michalopoulos, G.K. Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997, 26, 896–904. [Google Scholar] [CrossRef]
- Kim, T.H.; Mars, W.M.; Stolz, D.B.; Michalopoulos, G.K. Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology 2000, 31, 75–82. [Google Scholar] [CrossRef]
- Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am. J. Pathol. 1993, 143, 949–958. [Google Scholar]
- Mars, W.M.; Kim, T.H.; Stolz, D.B.; Liu, M.L.; Michalopoulos, G.K. Presence of urokinase in serum-free primary rat hepatocyte cultures and its role in activating hepatocyte growth factor. Cancer Res. 1996, 56, 2837–2843. [Google Scholar]
- Mars, W.M.; Liu, M.L.; Kitson, R.P.; Goldfarb, R.H.; Gabauer, M.K.; Michalopoulos, G.K. Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Hepatology 1995, 21, 1695–1701. [Google Scholar]
- Kohler, C.; Bell, A.W.; Bowen, W.C.; Monga, S.P.; Fleig, W.; Michalopoulos, G.K. Expression of Notch-1 and its ligand Jagged-1 in rat liver during liver regeneration. Hepatology 2004, 39, 1056–1065. [Google Scholar] [CrossRef]
- Monga, S.P.; Pediaditakis, P.; Mule, K.; Stolz, D.B.; Michalopoulos, G.K. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001, 33, 1098–1109. [Google Scholar] [CrossRef]
- Monga, S.P. Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int. J. Biochem. Cell Biol. 2011, 43, 1021–1029. [Google Scholar]
- Haber, B.A.; Mohn, K.L.; Diamond, R.H.; Taub, R. Induction patterns of 70 genes during nine days after hepatectomy define the temporal course of liver regeneration. J. Clin. Invest. 1993, 91, 1319–1326. [Google Scholar] [CrossRef]
- Mohn, K.L.; Laz, T.M.; Melby, A.E.; Taub, R. Immediate-early gene expression differs between regenerating liver, insulin-stimulated H-35 cells, and mitogen-stimulated Balb/c 3T3 cells. Liver-specific induction patterns of gene 33, phosphoenolpyruvate carboxykinase, and the jun, fos, and egr families. J. Biol. Chem. 1990, 265, 21914–21921. [Google Scholar]
- Strey, C.W.; Winters, M.S.; Markiewski, M.M.; Lambris, J.D. Partial hepatectomy induced liver proteome changes in mice. Proteomics 2005, 5, 318–325. [Google Scholar]
- Taub, R. Liver regeneration: from myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef]
- Zarnegar, R.; Michalopoulos, G. Purification and biological characterization of human hepatopoietin A, a polypeptide growth factor for hepatocytes. Cancer Res. 1989, 49, 3314–3320. [Google Scholar]
- Nakamura, T.; Teramoto, H.; Ichihara, A. Purification and characterization of a growth factor from rat platelets for mature parenchymal hepatocytes in primary cultures. Proc. Natl. Acad. Sci. USA 1986, 83, 6489–6493. [Google Scholar] [CrossRef]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989, 342, 440–443. [Google Scholar]
- Zarnegar, R.; Muga, S.; Enghild, J.; Michalopoulos, G. NH2-terminal amino acid sequence of rabbit hepatopoietin A, a heparin-binding polypeptide growth factor for hepatocytes. Biochem Biophys. Res. Commun. 1989, 163, 1370–1376. [Google Scholar] [CrossRef]
- Wang, X.; DeFrances, M.C.; Dai, Y.; Pediaditakis, P.; Johnson, C.; Bell, A.; Michalopoulos, G.K.; Zarnegar, R. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol. Cell. 2002, 9, 411–421. [Google Scholar] [CrossRef]
- Fafalios, A.; Ma, J.; Tan, X.; Stoops, J.; Luo, J.; Defrances, M.C.; Zarnegar, R. A hepatocyte growth factor receptor (Met)-insulin receptor hybrid governs hepatic glucose metabolism. Nat. Med. 2011, 17, 1577–1584. [Google Scholar]
- Nakamura, T. Hepatocyte growth factor as mitogen, motogen and morphogen, and its roles in organ regeneration. Princess Takamatsu Symp. 1994, 24, 195–213. [Google Scholar]
- Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Nejak-Bowen, K.; Luo, J.H.; Michalopoulos, G.K. Cell cycle effects resulting from inhibition of hepatocyte growth factor and its receptor c-Met in regenerating rat livers by RNA interference. Hepatology. 2007, 45, 1471–1477. [Google Scholar] [CrossRef]
- Wang, B.; Gao, C.; Ponder, K.P. C/EBPbeta contributes to hepatocyte growth factor-induced replication of rodent hepatocytes. J. Hepatol. 2005, 43, 294–302. [Google Scholar] [CrossRef]
- Schuppan, D.; Schmid, M.; Somasundaram, R.; Ackermann, R.; Ruehl, M.; Nakamura, T.; Riecken, E.O. Collagens in the liver extracellular matrix bind hepatocyte growth factor. Gastroenterology 1998, 114, 139–152. [Google Scholar]
- Liu, M.L.; Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Uptake and distribution of hepatocyte growth factor in normal and regenerating adult rat liver. Am. J. Pathol. 1994, 144, 129–140. [Google Scholar]
- Stolz, D.B.; Mars, W.M.; Petersen, B.E.; Kim, T.H.; Michalopoulos, G.K. Growth factor signal transduction immediately after two-thirds partial hepatectomy in the rat. Cancer Res. 1999, 59, 3954–3960. [Google Scholar]
- Pediaditakis, P.; Lopez-Talavera, J.C.; Petersen, B.; Monga, S.P.; Michalopoulos, G.K. The processing and utilization of hepatocyte growth factor/scatter factor following partial hepatectomy in the rat. Hepatology 2001, 34, 688–693. [Google Scholar] [CrossRef]
- Lindroos, P.M.; Zarnegar, R.; Michalopoulos, G.K. Hepatocyte growth factor (hepatopoietin A) rapidly increases in plasma before DNA synthesis and liver regeneration stimulated by partial hepatectomy and carbon tetrachloride administration. Hepatology 1991, 13, 743–750. [Google Scholar] [CrossRef]
- Schirmacher, P.; Geerts, A.; Pietrangelo, A.; Dienes, H.P.; Rogler, C.E. Hepatocyte growth factor/hepatopoietin A is expressed in fat-storing cells from rat liver but not myofibroblast-like cells derived from fat-storing cells. Hepatology 1992, 15, 5–11. [Google Scholar] [CrossRef]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.P.; Hillan, K.J.; Ferrara, N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science 2003, 299, 890–893. [Google Scholar] [CrossRef]
- Maher, J.J. Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J. Clin. Invest. 1993, 91, 2244–2252. [Google Scholar] [CrossRef]
- Zarnegar, R.; DeFrances, M.C.; Kost, D.P.; Lindroos, P.; Michalopoulos, G.K. Expression of hepatocyte growth factor mRNA in regenerating rat liver after partial hepatectomy. Biochem. Biophys. Res. Commun. 1991, 177, 559–565. [Google Scholar]
- Kono, S.; Nagaike, M.; Matsumoto, K.; Nakamura, T. Marked induction of hepatocyte growth factor mRNA in intact kidney and spleen in response to injury of distant organs. Biochem. Biophys. Res. Commun. 1992, 186, 991–998. [Google Scholar] [CrossRef]
- Yanagita, K.; Nagaike, M.; Ishibashi, H.; Niho, Y.; Matsumoto, K.; Nakamura, T. Lung may have an endocrine function producing hepatocyte growth factor in response to injury of distal organs. Biochem. Biophys. Res. Commun. 1992, 182, 802–809. [Google Scholar] [CrossRef]
- Broten, J.; Michalopoulos, G.; Petersen, B.; Cruise, J. Adrenergic stimulation of hepatocyte growth factor expression. Biochem. Biophys. Res. Commun. 1999, 262, 76–79. [Google Scholar] [CrossRef]
- Skrtic, S.; Wallenius, V.; Ekberg, S.; Brenzel, A.; Gressner, A.M.; Jansson, J.O. Insulin-like growth factors stimulate expression of hepatocyte growth factor but not transforming growth factor beta1 in cultured hepatic stellate cells. Endocrinology 1997, 138, 4683–4689. [Google Scholar]
- McGowan, J.A.; Strain, A.J.; Bucher, N.L. DNA synthesis in primary cultures of adult rat hepatocytes in a defined medium: effects of epidermal growth factor, insulin, glucagon, and cyclic-AMP. J. Cell. Physiol. 1981, 108, 353–363. [Google Scholar] [CrossRef]
- Bucher, N.L.; Patel, U.; Cohen, S. Hormonal factors concerned with liver regeneration. Ciba Found. Symp. 1977, 95–107. [Google Scholar]
- St Hilaire, R.J.; Hradek, G.T.; Jones, A.L. Hepatic sequestration and biliary secretion of epidermal growth factor: evidence for a high-capacity uptake system. Proc. Natl. Acad. Sci. USA 1983, 80, 3797–3801. [Google Scholar]
- St Hilaire, R.J.; Jones, A.L. Epidermal growth factor: its biologic and metabolic effects with emphasis on the hepatocyte. Hepatology 1982, 2, 601–613. [Google Scholar]
- Olsen, P.S.; Poulsen, S.S.; Kirkegaard, P. Adrenergic effects on secretion of epidermal growth factor from Brunner's glands. Gut 1985, 26, 920–927. [Google Scholar] [CrossRef]
- Jones, D.E., Jr.; Tran-Patterson, R.; Cui, D.M.; Davin, D.; Estell, K.P.; Miller, D.M. Epidermal growth factor secreted from the salivary gland is necessary for liver regeneration. Am. J. Physiol. 1995, 268, G872–G878. [Google Scholar]
- Reddy, C.C.; Wells, A.; Lauffenburger, D.A. Receptor-mediated effects on ligand availability influence relative mitogenic potencies of epidermal growth factor and transforming growth factor alpha. J. Cell. Physiol. 1996, 166, 512–522. [Google Scholar] [CrossRef]
- Webber, E.M.; FitzGerald, M.J.; Brown, P.I.; Bartlett, M.H.; Fausto, N. Transforming growth factor-alpha expression during liver regeneration after partial hepatectomy and toxic injury, and potential interactions between transforming growth factor-alpha and hepatocyte growth factor. Hepatology 1993, 18, 1422–1431. [Google Scholar]
- Luetteke, N.C.; Lee, D.C. Transforming growth factor alpha: expression, regulation and biological action of its integral membrane precursor. Semin. Cancer Biol. 1990, 1, 265–275. [Google Scholar]
- Lee, D.C.; Sunnarborg, S.W.; Hinkle, C.L.; Myers, T.J.; Stevenson, M.Y.; Russell, W.E.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; Chang, A.; Jackson, L.F. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann. N. Y. Acad. Sci. 2003, 995, 22–38. [Google Scholar] [CrossRef]
- Mead, J.E.; Fausto, N. Transforming growth factor alpha may be a physiological regulator of liver regeneration by means of an autocrine mechanism. Proc. Natl. Acad. Sci. USA 1989, 86, 1558–1562. [Google Scholar] [CrossRef]
- Russell, W.E.; Kaufmann, W.K.; Sitaric, S.; Luetteke, N.C.; Lee, D.C. Liver regeneration and hepatocarcinogenesis in transforming growth factor-alpha-targeted mice. Mol. Carcinog. 1996, 15, 183–189. [Google Scholar]
- Berasain, C.; Garcia-Trevijano, E.R.; Castillo, J.; Erroba, E.; Lee, D.C.; Prieto, J.; Avila, M.A. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology 2005, 128, 424–432. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Khan, Z. Liver regeneration, growth factors, and amphiregulin. Gastroenterology 2005, 128, 503–506. [Google Scholar] [CrossRef]
- Kiso, S.; Kawata, S.; Tamura, S.; Higashiyama, S.; Ito, N.; Tsushima, H.; Taniguchi, N.; Matsuzawa, Y. Role of heparin-binding epidermal growth factor-like growth factor as a hepatotrophic factor in rat liver regeneration after partial hepatectomy. Hepatology 1995, 22, 1584–1590. [Google Scholar]
- Ito, N.; Kawata, S.; Tamura, S.; Kiso, S.; Tsushima, H.; Damm, D.; Abraham, J.A.; Higashiyama, S.; Taniguchi, N.; Matsuzawa, Y. Heparin-binding EGF-like growth factor is a potent mitogen for rat hepatocytes. Biochem. Biophys. Res. Commun. 1994, 198, 25–31. [Google Scholar] [CrossRef]
- Mitchell, C.; Nivison, M.; Jackson, L.F.; Fox, R.; Lee, D.C.; Campbell, J.S.; Fausto, N. Heparin-binding epidermal growth factor-like growth factor links hepatocyte priming with cell cycle progression during liver regeneration. J. Biol. Chem. 2005, 280, 2562–2568. [Google Scholar]
- Kiso, S.; Kawata, S.; Tamura, S.; Inui, Y.; Yoshida, Y.; Sawai, Y.; Umeki, S.; Ito, N.; Yamada, A.; Miyagawa, J.; Higashiyama, S.; Iwawaki, T.; Saito, M.; Taniguchi, N.; Matsuzawa, Y.; Kohno, K. Liver regeneration in heparin-binding EGF-like growth factor transgenic mice after partial hepatectomy. Gastroenterology 2003, 124, 701–707. [Google Scholar]
- Paranjpe, S.; Bowen, W.C.; Tseng, G.C.; Luo, J.H.; Orr, A.; Michalopoulos, G.K. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am. J. Pathol. 2010, 176, 2669–2681. [Google Scholar] [CrossRef]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef]
- Bradham, C.A.; Plumpe, J.; Manns, M.P.; Brenner, D.A.; Trautwein, C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am. J. Physiol. 1998, 275, G387–G392. [Google Scholar]
- Hatano, E.; Bennett, B.L.; Manning, A.M.; Qian, T.; Lemasters, J.J.; Brenner, D.A. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology 2001, 120, 1251–1262. [Google Scholar]
- Hatano, E.; Brenner, D.A. Akt protects mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis through NK-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1357–G1368. [Google Scholar]
- Leist, M.; Gantner, F.; Bohlinger, I.; Tiegs, G.; Germann, P.G.; Wendel, A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am. J. Pathol. 1995, 146, 1220–1234. [Google Scholar]
- Leist, M.; Gantner, F.; Jilg, S.; Wendel, A. Activation of the 55 kDa TNF receptor is necessary and sufficient for TNF-induced liver failure, hepatocyte apoptosis, and nitrite release. J. Immunol. 1995, 154, 1307–1316. [Google Scholar]
- Leist, M.; Gantner, F.; Kunstle, G.; Bohlinger, I.; Tiegs, G.; Bluethmann, H.; Wendel, A. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol. Med. 1996, 2, 109–124. [Google Scholar]
- Akerman, P.; Cote, P.; Yang, S.Q.; McClain, C.; Nelson, S.; Bagby, G.J.; Diehl, A.M. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J. Physiol. 1992, 263, G579–G585. [Google Scholar]
- Yamada, Y.; Webber, E.M.; Kirillova, I.; Peschon, J.J.; Fausto, N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: Requirement for type 1 but not type 2 receptor. Hepatology 1998, 28, 959–970. [Google Scholar] [CrossRef]
- Iimuro, Y.; Nishiura, T.; Hellerbrand, C.; Behrns, K.E.; Schoonhoven, R.; Grisham, J.W.; Brenner, D.A. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J. Clin. Invest. 1998, 101, 802–811. [Google Scholar] [CrossRef]
- Webber, E.M.; Bruix, J.; Pierce, R.H.; Fausto, N. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology 1998, 28, 1226–1234. [Google Scholar] [CrossRef]
- Webber, E.M.; Godowski, P.J.; Fausto, N. In vivo response of hepatocytes to growth factors requires an initial priming stimulus. Hepatology 1994, 19, 489–497. [Google Scholar]
- Gauldie, J.; Northemann, W.; Fey, G.H. IL-6 functions as an exocrine hormone in inflammation. Hepatocytes undergoing acute phase responses require exogenous IL-6. J. Immunol. 1990, 144, 3804–3808. [Google Scholar]
- Castell, J.V.; Gomez-Lechon, M.J.; David, M.; Fabra, R.; Trullenque, R.; Heinrich, P.C. Acute-phase response of human hepatocytes: regulation of acute-phase protein synthesis by interleukin-6. Hepatology 1990, 12, 1179–1186. [Google Scholar] [CrossRef]
- Streetz, K.L.; Wustefeld, T.; Klein, C.; Manns, M.P.; Trautwein, C. Mediators of inflammation and acute phase response in the liver. Cell. Mol. Biol. (Noisy-le-grand) 2001, 47, 661–673. [Google Scholar]
- Peters, M.; Muller, A.M.; Rose-John, S. Interleukin-6 and soluble interleukin-6 receptor: Direct stimulation of gp130 and hematopoiesis. Blood 1998, 92, 3495–3504. [Google Scholar]
- Wang, Y.; Nesbitt, J.E.; Fuentes, N.L.; Fuller, G.M. Molecular cloning and characterization of the rat liver IL-6 signal transducing molecule, gp130. Genomics 1992, 14, 666–672. [Google Scholar] [CrossRef]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Liu, Y.; Michalopoulos, G.K.; Zarnegar, R. Structural and functional characterization of the mouse hepatocyte growth factor gene promoter. J. Biol. Chem. 1994, 269, 4152–4160. [Google Scholar]
- Sun, R.; Jaruga, B.; Kulkarni, S.; Sun, H.; Gao, B. IL-6 modulates hepatocyte proliferation via induction of HGF/p21cip1: regulation by SOCS3. Biochem. Biophys. Res. Commun. 2005, 338, 1943–1949. [Google Scholar] [CrossRef]
- Kariv, R.; Enden, A.; Zvibel, I.; Rosner, G.; Brill, S.; Shafritz, D.A.; Halpern, Z.; Oren, R. Triiodothyronine and interleukin-6 (IL-6) induce expression of HGF in an immortalized rat hepatic stellate cell line. Liver Int. 2003, 23, 187–193. [Google Scholar] [CrossRef]
- Coudriet, G.M.; He, J.; Trucco, M.; Mars, W.M.; Piganelli, J.D. Hepatocyte growth factor modulates interleukin-6 production in bone marrow derived macrophages: Implications for inflammatory mediated diseases. PLoS One 2010, 5, e15384. [Google Scholar]
- Gong, R. Multi-target anti-inflammatory action of hepatocyte growth factor. Curr. Opin. Investig. Drugs 2008, 9, 1163–1170. [Google Scholar]
- Wrana, J.L.; Attisano, L.; Carcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.F.; Massague, J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar]
- Gruppuso, P.A.; Mead, J.E.; Fausto, N. Transforming growth factor receptors in liver regeneration following partial hepatectomy in the rat. Cancer Res. 1990, 50, 1464–1469. [Google Scholar]
- Chari, R.S.; Price, D.T.; Sue, S.R.; Meyers, W.C.; Jirtle, R.L. Down-regulation of transforming growth factor beta receptor type I, II, and III during liver regeneration. Am. J. Surg. 1995, 169, 126–131, discussion 131–122.. [Google Scholar] [CrossRef]
- Bissell, D.M.; Wang, S.S.; Jarnagin, W.R.; Roll, F.J. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J. Clin. Invest. 1995, 96, 447–455. [Google Scholar] [CrossRef]
- Ikeda, H.; Nagoshi, S.; Ohno, A.; Yanase, M.; Maekawa, H.; Fujiwara, K. Activated rat stellate cells express c-met and respond to hepatocyte growth factor to enhance transforming growth factor beta1 expression and DNA synthesis. Biochem. Biophys. Res. Commun. 1998, 250, 769–775. [Google Scholar] [CrossRef]
- Nakatsukasa, H.; Evarts, R.P.; Hsia, C.C.; Thorgeirsson, S.S. Transforming growth factor-beta 1 and type I procollagen transcripts during regeneration and early fibrosis of rat liver. Lab. Invest. 1990, 63, 171–180. [Google Scholar]
- Carr, B.I.; Huang, T.H.; Itakura, K.; Noel, M.; Marceau, N. TGF beta gene transcription in normal and neoplastic liver growth. J. Cell. Biochem. 1989, 39, 477–487. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bowen, W.C.; Mule, K.; Luo, J. HGF-, EGF-, and dexamethasone-induced gene expression patterns during formation of tissue in hepatic organoid cultures. Gene Expr. 2003, 11, 55–75. [Google Scholar] [CrossRef]
- McMahon, J.B.; Richards, W.L.; del Campo, A.A.; Song, M.K.; Thorgeirsson, S.S. Differential effects of transforming growth factor-beta on proliferation of normal and malignant rat liver epithelial cells in culture. Cancer Res. 1986, 46, 4665–4671. [Google Scholar]
- Houck, K.A.; Cruise, J.L.; Michalopoulos, G. Norepinephrine modulates the growth-inhibitory effect of transforming growth factor-beta in primary rat hepatocyte cultures. J. Cell. Physiol. 1988, 135, 551–555. [Google Scholar] [CrossRef]
- Russell, W.E.; Coffey, R.J., Jr.; Ouellette, A.J.; Moses, H.L. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc. Natl. Acad. Sci. USA 1988, 85, 5126–5130. [Google Scholar]
- Jirtle, R.L.; Carr, B.I.; Scott, C.D. Modulation of insulin-like growth factor-II/mannose 6-phosphate receptors and transforming growth factor-beta 1 during liver regeneration. J. Biol. Chem. 1991, 266, 22444–22450. [Google Scholar]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar]
- Houck, K.A.; Michalopoulos, G.K. Altered responses of regenerating hepatocytes to norepinephrine and transforming growth factor type beta. J. Cell. Physiol. 1989, 141, 503–509. [Google Scholar] [CrossRef]
- O'Connor-McCourt, M.D.; Wakefield, L.M. Latent transforming growth factor-beta in serum. A specific complex with alpha 2-macroglobulin. J. Biol. Chem. 1987, 262, 14090–14099. [Google Scholar]
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming growth factor beta mRNA increases during liver regeneration: a possible paracrine mechanism of growth regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543. [Google Scholar]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Leveen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef]
- Thenappan, A.; Shukla, V.; Abdul Khalek, F.J.; Li, Y.; Shetty, K.; Liu, P.; Li, L.; Johnson, R.L.; Johnson, L.; Mishra, L. Loss of transforming growth factor beta adaptor protein beta-2 spectrin leads to delayed liver regeneration in mice. Hepatology 2011, 53, 1641–1650. [Google Scholar]
- Oben, J.A.; Roskams, T.; Yang, S.; Lin, H.; Sinelli, N.; Torbenson, M.; Smedh, U.; Moran, T.H.; Li, Z.; Huang, J.; Thomas, S.A.; Diehl, A.M. Hepatic fibrogenesis requires sympathetic neurotransmitters. Gut 2004, 53, 438–445. [Google Scholar] [CrossRef]
- Cruise, J.L.; Knechtle, S.J.; Bollinger, R.R.; Kuhn, C.; Michalopoulos, G. Alpha 1-adrenergic effects and liver regeneration. Hepatology 1987, 7, 1189–1194. [Google Scholar] [CrossRef]
- Cruise, J.L.; Michalopoulos, G. Norepinephrine and epidermal growth factor: dynamics of their interaction in the stimulation of hepatocyte DNA synthesis. J. Cell. Physiol. 1985, 125, 45–50. [Google Scholar] [CrossRef]
- Cruise, J.L.; Houck, K.A.; Michalopoulos, G.K. Induction of DNA synthesis in cultured rat hepatocytes through stimulation of alpha 1 adrenoreceptor by norepinephrine. Science 1985, 227, 749–751. [Google Scholar]
- Kanamaru, C.; Yasuda, H.; Takeda, M.; Ueda, N.; Suzuki, J.; Tsuchida, T.; Mashima, H.; Ohnishi, H.; Fujita, T. Smad7 is induced by norepinephrine and protects rat hepatocytes from activin A-induced growth inhibition. J. Biol. Chem. 2001, 276, 45636–45641. [Google Scholar]
- Han, C.; Bowen, W.C.; Michalopoulos, G.K.; Wu, T. Alpha-1 adrenergic receptor transactivates signal transducer and activator of transcription-3 (Stat3) through activation of Src and epidermal growth factor receptor (EGFR) in hepatocytes. J. Cell. Physiol. 2008, 216, 486–497. [Google Scholar] [CrossRef]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar]
- Furrer, K.; Rickenbacher, A.; Tian, Y.; Jochum, W.; Bittermann, A.G.; Kach, A.; Humar, B.; Graf, R.; Moritz, W.; Clavien, P.A. Serotonin reverts age-related capillarization and failure of regeneration in the liver through a VEGF-dependent pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 2945–2950. [Google Scholar]
- Matondo, R.B.; Punt, C.; Homberg, J.; Toussaint, M.J.; Kisjes, R.; Korporaal, S.J.; Akkerman, J.W.; Cuppen, E.; de Bruin, A. Deletion of the serotonin transporter in rats disturbs serotonin homeostasis without impairing liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G963–G968. [Google Scholar] [CrossRef]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006, 312, 233–236. [Google Scholar] [CrossRef]
- Uppal, H.; Saini, S.P.; Moschetta, A.; Mu, Y.; Zhou, J.; Gong, H.; Zhai, Y.; Ren, S.; Michalopoulos, G.K.; Mangelsdorf, D.J.; Xie, W. Activation of LXRs prevents bile acid toxicity and cholestasis in female mice. Hepatology 2007, 45, 422–432. [Google Scholar] [CrossRef]
- Keitel, V.; Haussinger, D. TGR5 in the biliary tree. Dig. Dis. 2011, 29, 45–47. [Google Scholar]
- Holeček, M. Nutritional modulation of liver regeneration by carbohydrates, lipids, and amino acids: a review. Nutrition 1999, 15, 784–788. [Google Scholar] [CrossRef]
- Rudnick, D.A.; Davidson, N.O. Functional Relationships between Lipid Metabolism and Liver Regeneration. Int. J. Hepatol. 2012, 549241. [Google Scholar]
- Michalopoulos, G.; Pitot, H.C. Primary culture of parenchymal liver cells on collagen membranes. Morphological and biochemical observations. Exp. Cell Res. 1975, 94, 70–78. [Google Scholar] [CrossRef]
- Lai, H.S.; Chung, Y.C.; Chen, W.J.; Chen, K.M. Rat liver regeneration after partial hepatectomy: effects of insulin, glucagon and epidermal growth factor. J. Formos. Med. Assoc. 1992, 91, 685–690. [Google Scholar]
- Starzl, T.E.; Francavilla, A.; Porter, K.A.; Benichou, J.; Jones, A.F. The effect of splanchnic viscera removal upon canine liver regeneration. Surg. Gynecol. Obstet. 1978, 147, 193–207. [Google Scholar]
- Weymann, A.; Hartman, E.; Gazit, V.; Wang, C.; Glauber, M.; Turmelle, Y.; Rudnick, D.A. P21 is required for dextrose-mediated inhibition of mouse liver regeneration. Hepatology 2009, 50, 207–215. [Google Scholar] [CrossRef]
- Caruana, J.A.; Whalen, D.A.; Anthony, W.P.; Sunby, C.R.; Ciechoski, M.P. Paradoxical effects of glucose feeding on liver regeneration and survival after partial hepatectomy. Endocrine Res. 1986, 12, 147–156. [Google Scholar] [CrossRef]
- McGowan, J.; Atryzek, V.; Fausto, N. Effects of protein-deprivation on the regeneration of rat liver after partial hepatectomy. Biochem. J. 1979, 180, 25–35. [Google Scholar]
- Holeček, M.; Šimek, J.; Palička, V.; Zadák, Z. Effect of glucose and branched chain amino acid (BCAA) infusion on onset of liver regeneration and plasma amino acid pattern in partially hepatectomized rats. J. Hepatol. 1991, 13, 14–20. [Google Scholar] [CrossRef]
- Glende, E.A.; Morgan, W.S. Alteration in liver lipid and lipid fatty acid composition after partial hepatectomy in the rat. Exp. Mol. Path. 1968, 8, 190–200. [Google Scholar] [CrossRef]
- Gazit, V.; Weymann, A.; Hartman, E.; Finck, B.N.; Hruz, P.W.; Tzekov, A.; Rudnick, D.A. Liver regeneration is impaired in lipodystrophic fatty liver dystrophy mice. Hepatology 2010, 52, 2109–2117. [Google Scholar] [CrossRef]
- Blaha, V.; Simek, J.; Zadak, Z. Liver regeneration in partially hepatectomized rats infused with carnitine and lipids. Exp. Toxicol. Pathol. 1992, 44, 158, 165–168. [Google Scholar]
- Nakatani, T.; Ozawa, K.; Asano, M. Differences in predominant energy substrate in relation to the resected hepatic mass in the phase immediately after hepatectomy. J. Lab. Clin. Med. 1981, 97, 887–898. [Google Scholar]
- Zeng, G.; Awan, F.; Otruba, W.; Muller, P.; Apte, U.; Tan, X.; Gandhi, C.; Demetris, A.J.; Monga, S.P. Wnt'er in liver: expression of Wnt and frizzled genes in mouse. Hepatology. 2007, 45, 195–204. [Google Scholar] [CrossRef]
- Apte, U.; Zeng, G.; Muller, P.; Tan, X.; Micsenyi, A.; Cieply, B.; Dai, C.; Liu, Y.; Kaestner, K.H.; Monga, S.P. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology 2006, 44, 992–1002. [Google Scholar] [CrossRef]
- Monga, S.P.; Mars, W.M.; Pediaditakis, P.; Bell, A.; Mule, K.; Bowen, W.C.; Wang, X.; Zarnegar, R.; Michalopoulos, G.K. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 2002, 62, 2064–2071. [Google Scholar]
- Sodhi, D.; Micsenyi, A.; Bowen, W.C.; Monga, D.K.; Talavera, J.C.; Monga, S.P. Morpholino oligonucleotide-triggered beta-catenin knockdown compromises normal liver regeneration. J. Hepatol. 2005, 43, 132–141. [Google Scholar] [CrossRef]
- Tan, X.; Behari, J.; Cieply, B.; Michalopoulos, G.K.; Monga, S.P. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology 2006, 131, 1561–1572. [Google Scholar] [CrossRef]
- Rangwala, F.; Guy, C.D.; Lu, J.; Suzuki, A.; Burchette, J.L.; Abdelmalek, M.F.; Chen, W.; Diehl, A.M. Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol. 2011, 224, 401–410. [Google Scholar] [CrossRef]
- Witek, R.P.; Yang, L.; Liu, R.; Jung, Y.; Omenetti, A.; Syn, W.K.; Choi, S.S.; Cheong, Y.; Fearing, C.M.; Agboola, K.M.; Chen, W.; Diehl, A.M. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 2009, 136, 320–330, e322. [Google Scholar]
- Ochoa, B.; Syn, W.K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; Choi, S.S.; Diehl, A.M. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef]
- Capurro, M.I.; Xu, P.; Shi, W.; Li, F.; Jia, A.; Filmus, J. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev. Cell. 2008, 14, 700–711. [Google Scholar] [CrossRef]
- Kan, M.; Wu, X.; Wang, F.; McKeehan, W.L. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J. Biol. Chem. 1999, 274, 15947–15952. [Google Scholar]
- Houck, K.A.; Zarnegar, R.; Muga, S.J.; Michalopoulos, G.K. Acidic fibroblast growth factor (HBGF-1) stimulates DNA synthesis in primary rat hepatocyte cultures. J. Cell. Physiol. 1990, 143, 129–132. [Google Scholar] [CrossRef]
- Strain, A.J.; McGuinness, G.; Rubin, J.S.; Aaronson, S.A. Keratinocyte growth factor and fibroblast growth factor action on DNA synthesis in rat and human hepatocytes: Modulation by heparin. Exp. Cell Res. 1994, 210, 253–259. [Google Scholar] [CrossRef]
- Steiling, H.; Wustefeld, T.; Bugnon, P.; Brauchle, M.; Fassler, R.; Teupser, D.; Thiery, J.; Gordon, J.I.; Trautwein, C.; Werner, S. Fibroblast growth factor receptor signalling is crucial for liver homeostasis and regeneration. Oncogene 2003, 22, 4380–4388. [Google Scholar] [CrossRef]
- Grisham, J.W. A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res. 1962, 22, 842–849. [Google Scholar]
- Widmann, J.J.; Fahimi, H.D. Proliferation of mononuclear phagocytes (Kupffer cells) and endothelial cells in regenerating rat liver. A light and electron microscopic cytochemical study. Am. J. Pathol. 1975, 80, 349–366. [Google Scholar]
- Tanaka, Y.; Mak, K.M.; Lieber, C.S. Immunohistochemical detection of proliferating lipocytes in regenerating rat liver. J. Pathol. 1990, 160, 129–134. [Google Scholar] [CrossRef]
- Stocker, E.; Heine, W.D. Regeneration of liver parenchyma under normal and pathological conditions. Beitr. Pathol. 1971, 144, 400–408. [Google Scholar]
- Stocker, E.; Heine, W.D. Proliferation and regeneration in liver and kidney of juvenile rats. Autoradiographic studies after continuous infusion of 3H-thymidine (author's transl). Verh. Dtsch. Ges. Pathol. 1971, 55, 483–488. [Google Scholar]
- Stocker, E.; Schultze, B.; Heine, W.D.; Liebscher, H. Growth and regeneration in parenchymatous organs of the rat. Autoradiographic investigations with 3 H-thymidin. Z. Zellforsch. Mikrosk. Anat. 1972, 125, 306–331. [Google Scholar] [CrossRef]
- Schmucker, D.L.; Sanchez, H. Liver regeneration and aging: a current perspective. Curr. Gerontol. Geriatr. Res. 2011, 2011, 526379. [Google Scholar]
- Stocker, E.; Wullstein, H.K.; Brau, G. Capacity of regeneration in liver epithelia of juvenile, repeated partially hepatectomized rats. Autoradiographic studies after continous infusion of 3H-thymidine (author's transl). Virchows Arch. B Cell. Pathol. 1973, 14, 93–103. [Google Scholar]
- Overturf, K.; al-Dhalimy, M.; Ou, C.N.; Finegold, M.; Grompe, M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am. J. Pathol. 1997, 151, 1273–1280. [Google Scholar]
- Bhave, V.S.; Paranjpe, S.; Bowen, W.C.; Donthamsetty, S.; Bell, A.W.; Khillan, J.S.; Michalopoulos, G.K. Genes inducing iPS phenotype play a role in hepatocyte survival and proliferation in vitro and liver regeneration in vivo. Hepatology 2011, 54, 1360–1370. [Google Scholar] [CrossRef]
- Kan, M.; Huang, J.S.; Mansson, P.E.; Yasumitsu, H.; Carr, B.; McKeehan, W.L. Heparin-binding growth factor type 1 (acidic fibroblast growth factor): a potential biphasic autocrine and paracrine regulator of hepatocyte regeneration. Proc. Natl. Acad. Sci. USA 1989, 86, 7432–7436. [Google Scholar]
- Borkham-Kamphorst, E.; Kovalenko, E.; van Roeyen, C.R.; Gassler, N.; Bomble, M.; Ostendorf, T.; Floege, J.; Gressner, A.M.; Weiskirchen, R. Platelet-derived growth factor isoform expression in carbon tetrachloride-induced chronic liver injury. Lab. Invest. 2008, 88, 1090–1100. [Google Scholar] [CrossRef]
- Ross, M.A.; Sander, C.M.; Kleeb, T.B.; Watkins, S.C.; Stolz, D.B. Spatiotemporal expression of angiogenesis growth factor receptors during the revascularization of regenerating rat liver. Hepatology 2001, 34, 1135–1148. [Google Scholar] [CrossRef]
- Martinez-Hernandez, A.; Amenta, P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995, 9, 1401–1410. [Google Scholar]
- Wack, K.E.; Ross, M.A.; Zegarra, V.; Sysko, L.R.; Watkins, S.C.; Stolz, D.B. Sinusoidal ultrastructure evaluated during the revascularization of regenerating rat liver. Hepatology 2001, 33, 363–378. [Google Scholar] [CrossRef]
- Fujii, H.; Hirose, T.; Oe, S.; Yasuchika, K.; Azuma, H.; Fujikawa, T.; Nagao, M.; Yamaoka, Y. Contribution of bone marrow cells to liver regeneration after partial hepatectomy in mice. J. Hepatol. 2002, 36, 653–659. [Google Scholar] [CrossRef]
- Zocco, M.A.; Piscaglia, A.C.; Giuliante, F.; Arena, V.; Novi, M.; Rinninella, E.; Tortora, A.; Rumi, C.; Nuzzo, G.; Vecchio, F.M.; Bombardieri, G.; Gasbarrini, A. CD133+ stem cell mobilization after partial hepatectomy depends on resection extent and underlying disease. Dig. Liver Dis. 2011, 43, 147–154. [Google Scholar]
- Wang, L.; Wang, X.; Xie, G.; Hill, C.K.; DeLeve, L.D. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J. Clin. Invest. 2012, 122, 1567–1573. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Chiu, J.D.; van de Ven, G.; Gaarde, W.A.; Deleve, L.D. Hepatic Vascular Endothelial Growth Factor Regulates Recruitment of Rat Liver Sinusoidal Endothelial Cell Progenitor Cells. Gastroenterology 2012, 143, 1555–1563. [Google Scholar]
- Lagasse, E.; Connors, H.; Al-Dhalimy, M.; Reitsma, M.; Dohse, M.; Osborne, L.; Wang, X.; Finegold, M.; Weissman, I.L.; Grompe, M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 2000, 6, 1229–1234. [Google Scholar] [CrossRef]
- Petersen, B.E.; Bowen, W.C.; Patrene, K.D.; Mars, W.M.; Sullivan, A.K.; Murase, N.; Boggs, S.S.; Greenberger, J.S.; Goff, J.P. Bone marrow as a potential source of hepatic oval cells. Science 1999, 284, 1168–1170. [Google Scholar] [CrossRef]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003, 422, 897–901. [Google Scholar]
- Kuwahara, R.; Kofman, A.V.; Landis, C.S.; Swenson, E.S.; Barendswaard, E.; Theise, N.D. The hepatic stem cell niche: identification by label-retaining cell assay. Hepatology 2008, 47, 1994–2002. [Google Scholar] [CrossRef]
- Dolle, L.; Best, J.; Mei, J.; Al Battah, F.; Reynaert, H.; van Grunsven, L.A.; Geerts, A. The quest for liver progenitor cells: a practical point of view. J. Hepatol. 2010, 52, 117–129. [Google Scholar]
- Evarts, R.P.; Nagy, P.; Nakatsukasa, H.; Marsden, E.; Thorgeirsson, S.S. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 1989, 49, 1541–1547. [Google Scholar]
- Trautwein, C.; Will, M.; Kubicka, S.; Rakemann, T.; Flemming, P.; Manns, M.P. 2-acetaminofluorene blocks cell cycle progression after hepatectomy by p21 induction and lack of cyclin E expression. Oncogene 1999, 18, 6443–6453. [Google Scholar] [CrossRef]
- Limaye, P.B.; Alarcon, G.; Walls, A.L.; Nalesnik, M.A.; Michalopoulos, G.K.; Demetris, A.J.; Ochoa, E.R. Expression of specific hepatocyte and cholangiocyte transcription factors in human liver disease and embryonic development. Lab. Invest. 2008, 88, 865–872. [Google Scholar] [CrossRef]
- Petersen, B.E.; Goff, J.P.; Greenberger, J.S.; Michalopoulos, G.K. Hepatic oval cells express the hematopoietic stem cell marker Thy-1 in the rat. Hepatology 1998, 27, 433–445. [Google Scholar] [CrossRef]
- Evarts, R.P.; Hu, Z.; Fujio, K.; Marsden, E.R.; Thorgeirsson, S.S. Activation of hepatic stem cell compartment in the rat: role of transforming growth factor alpha, hepatocyte growth factor, and acidic fibroblast growth factor in early proliferation. Cell Growth Differ. 1993, 4, 555–561. [Google Scholar]
- Darwiche, H.; Oh, S.H.; Steiger-Luther, N.C.; Williams, J.M.; Pintilie, D.G.; Shupe, T.D.; Petersen, B.E. Inhibition of Notch signaling affects hepatic oval cell response in rat model of 2AAF-PH. Hepat. Med. 2011, 3, 89–98. [Google Scholar]
- Thenappan, A.; Li, Y.; Kitisin, K.; Rashid, A.; Shetty, K.; Johnson, L.; Mishra, L. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 2010, 51, 1373–1382. [Google Scholar] [CrossRef]
- Erker, L.; Grompe, M. Signaling networks in hepatic oval cell activation. Stem Cell Res. 2007, 1, 90–102. [Google Scholar]
- Jakubowski, A.; Ambrose, C.; Parr, M.; Lincecum, J.M.; Wang, M.Z.; Zheng, T.S.; Browning, B.; Michaelson, J.S.; Baetscher, M.; Wang, B.; Bissell, D.M.; Burkly, L.C. TWEAK induces liver progenitor cell proliferation. J. Clin. Invest. 2005, 115, 2330–2340. [Google Scholar] [CrossRef]
- Mavier, P.; Martin, N.; Couchie, D.; Preaux, A.M.; Laperche, Y.; Zafrani, E.S. Expression of stromal cell-derived factor-1 and of its receptor CXCR4 in liver regeneration from oval cells in rat. Am. J. Pathol. 2004, 165, 1969–1977. [Google Scholar] [CrossRef]
- Theise, N.D.; Saxena, R.; Portmann, B.C.; Thung, S.N.; Yee, H.; Chiriboga, L.; Kumar, A.; Crawford, J.M. The canals of Hering and hepatic stem cells in humans. Hepatology 1999, 30, 1425–1433. [Google Scholar]
- Nagy, P.; Bisgaard, H.C.; Thorgeirsson, S.S. Expression of hepatic transcription factors during liver development and oval cell differentiation. J. Cell. Biol. 1994, 126, 223–233. [Google Scholar] [CrossRef]
- Petersen, B.E.; Zajac, V.F.; Michalopoulos, G.K. Bile ductular damage induced by methylene dianiline inhibits oval cell activation. Am. J. Pathol. 1997, 151, 905–909. [Google Scholar]
- Demetris, A.J.; Seaberg, E.C.; Wennerberg, A.; Ionellie, J.; Michalopoulos, G. Ductular reaction after submassive necrosis in humans. Special emphasis on analysis of ductular hepatocytes. Am. J. Pathol. 1996, 149, 439–448. [Google Scholar]
- Michalopoulos, G.K.; Barua, L.; Bowen, W.C. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology 2005, 41, 535–544. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Muller-Marbach, A.; Ale-Agha, N.; Keitel, V.; Klonowski-Stumpe, H.; Haussinger, D. CD133+ hepatic stellate cells are progenitor cells. Biochem. Biophys. Res. Commun. 2007, 352, 410–417. [Google Scholar] [CrossRef]
- Yang, L.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.; Vandongen, H.M.; Huang, J.; Alpini, G.D.; Diehl, A.M. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008, 26, 2104–2113. [Google Scholar] [CrossRef]
- Xu, C.; Yang, Y.; Yang, J.; Chen, X.; Wang, G. Analysis of the role of the integrin signaling pathway in hepatocytes during rat liver regeneration. Cell Mol Biol Lett. 2012, 17, 274–288. [Google Scholar] [CrossRef]
- Legate, K.R.; Montanez, E.; Kudlacek, O.; Fassler, R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat. Rev. Mol. Cell. Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef]
- Gkretsi, V.; Bowen, W.C.; Yang, Y.; Wu, C.; Michalopoulos, G.K. Integrin-linked kinase is involved in matrix-induced hepatocyte differentiation. Biochem. Biophys. Res. Commun. 2007, 353, 638–643. [Google Scholar]
- Gkretsi, V.; Apte, U.; Mars, W.M.; Bowen, W.C.; Luo, J.H.; Yang, Y.; Yu, Y.P.; Orr, A.; St-Arnaud, R.; Dedhar, S.; Kaestner, K.H.; Wu, C.; Michalopoulos, G.K. Liver-specific ablation of integrin-linked kinase in mice results in abnormal histology, enhanced cell proliferation, and hepatomegaly. Hepatology 2008, 48, 1932–1941. [Google Scholar] [CrossRef]
- Hippo, Y.; Watanabe, K.; Watanabe, A.; Midorikawa, Y.; Yamamoto, S.; Ihara, S.; Tokita, S.; Iwanari, H.; Ito, Y.; Nakano, K.; Nezu, J.; Tsunoda, H.; Yoshino, T.; Ohizumi, I.; Tsuchiya, M.; Ohnishi, S.; Makuuchi, M.; Hamakubo, T.; Kodama, T.; Aburatani, H. Identification of soluble NH2-terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer Res. 2004, 64, 2418–2423. [Google Scholar]
- Pilia, G.; Hughes-Benzie, R.M.; MacKenzie, A.; Baybayan, P.; Chen, E.Y.; Huber, R.; Neri, G.; Cao, A.; Forabosco, A.; Schlessinger, D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet. 1996, 12, 241–247. [Google Scholar] [CrossRef]
- Liu, B.; Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Luo, J.H.; Yu, Y.P.; Mars, W.M.; Michalopoulos, G.K. Investigation of the role of glypican 3 in liver regeneration and hepatocyte proliferation. Am. J. Pathol. 2009, 175, 717–724. [Google Scholar] [CrossRef]
- Ichikawa, T.; Zhang, Y.Q.; Kogure, K.; Hasegawa, Y.; Takagi, H.; Mori, M.; Kojima, I. Transforming growth factor beta and activin tonically inhibit DNA synthesis in the rat liver. Hepatology 2001, 34, 918–925. [Google Scholar]
- Buraschi, S.; Pal, N.; Tyler-Rubinstein, N.; Owens, R.T.; Neill, T.; Iozzo, R.V. Decorin antagonizes Met receptor activity and down-regulates {beta}-catenin and Myc levels. J. Biol. Chem. 2010, 285, 42075–42085. [Google Scholar]
- Zhu, J.X.; Goldoni, S.; Bix, G.; Owens, R.T.; McQuillan, D.J.; Reed, C.C.; Iozzo, R.V. Decorin evokes protracted internalization and degradation of the epidermal growth factor receptor via caveolar endocytosis. J. Biol. Chem. 2005, 280, 32468–32479. [Google Scholar]
- Avruch, J.; Zhou, D.; Fitamant, J.; Bardeesy, N. Mst1/2 signalling to Yap: gatekeeper for liver size and tumour development. Br. J. Cancer. 2011, 104, 24–32. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; Johnson, R.L. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; Wang, C.Y.; Gao, B.; Jiang, J.; Yang, Y. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar]
- Pahlavan, P.S.; Feldmann, R.E., Jr.; Zavos, C.; Kountouras, J. Prometheus' challenge: molecular, cellular and systemic aspects of liver regeneration. J. Surg. Res. 2006, 134, 238–251. [Google Scholar] [CrossRef]
- Humar, A.; Kosari, K.; Sielaff, T.D.; Glessing, B.; Gomes, M.; Dietz, C.; Rosen, G.; Lake, J.; Payne, W.D. Liver regeneration after adult living donor and deceased donor split-liver transplants. Liver Transpl. 2004, 10, 374–378. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kang, L.-I.; Mars, W.M.; Michalopoulos, G.K. Signals and Cells Involved in Regulating Liver Regeneration. Cells 2012, 1, 1261-1292. https://doi.org/10.3390/cells1041261
Kang L-I, Mars WM, Michalopoulos GK. Signals and Cells Involved in Regulating Liver Regeneration. Cells. 2012; 1(4):1261-1292. https://doi.org/10.3390/cells1041261
Chicago/Turabian StyleKang, Liang-I., Wendy M. Mars, and George K. Michalopoulos. 2012. "Signals and Cells Involved in Regulating Liver Regeneration" Cells 1, no. 4: 1261-1292. https://doi.org/10.3390/cells1041261