Genetic Systems to Investigate Regulation of Oncogenes and Tumour Suppressor Genes in Drosophila

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
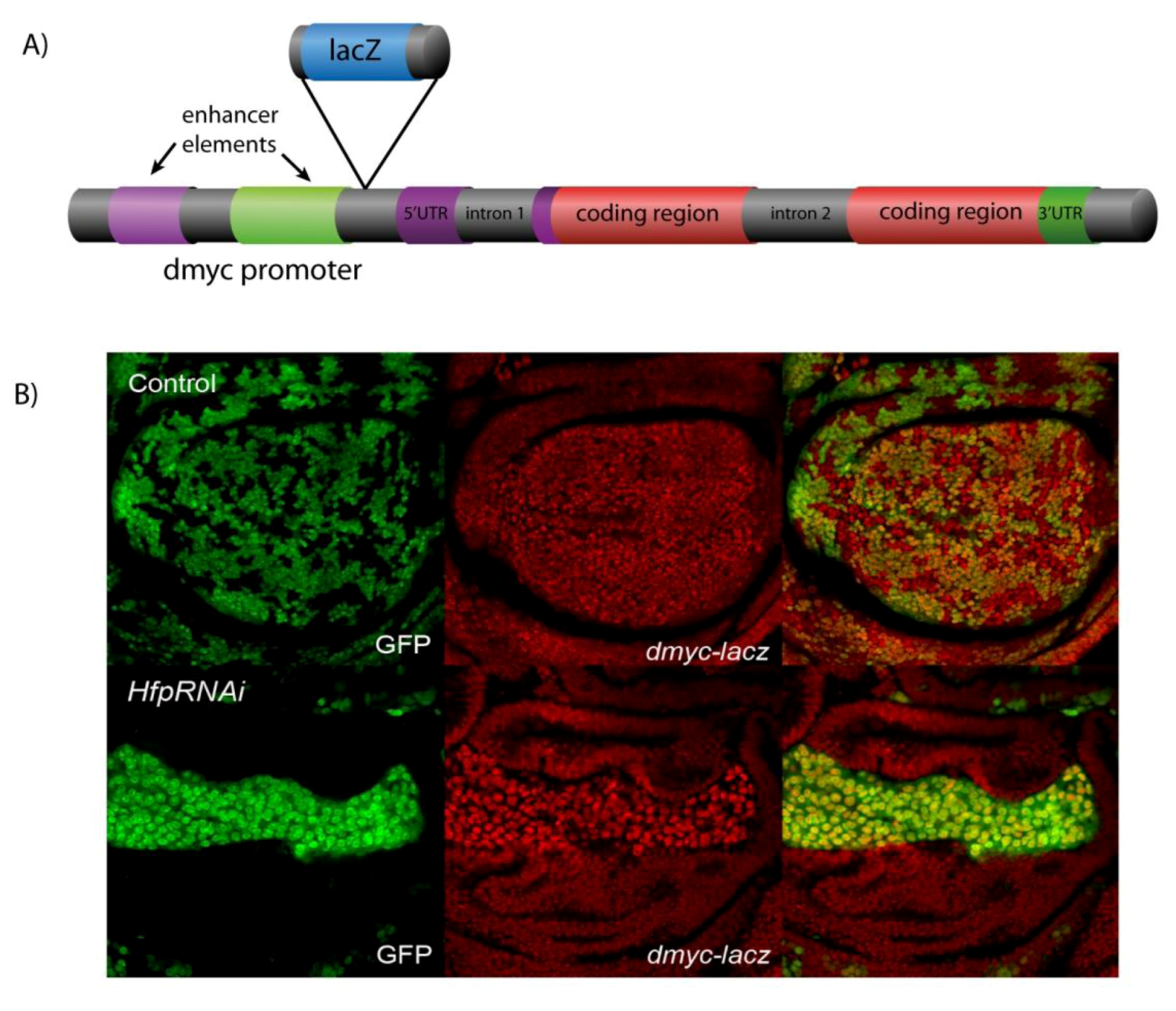
2. Cell Cycle Control is Fundamental to Development and is Disrupted in Cancer: The Myc oncogene and FIR Tumour Suppressor are Conserved between Drosophila and Mammals
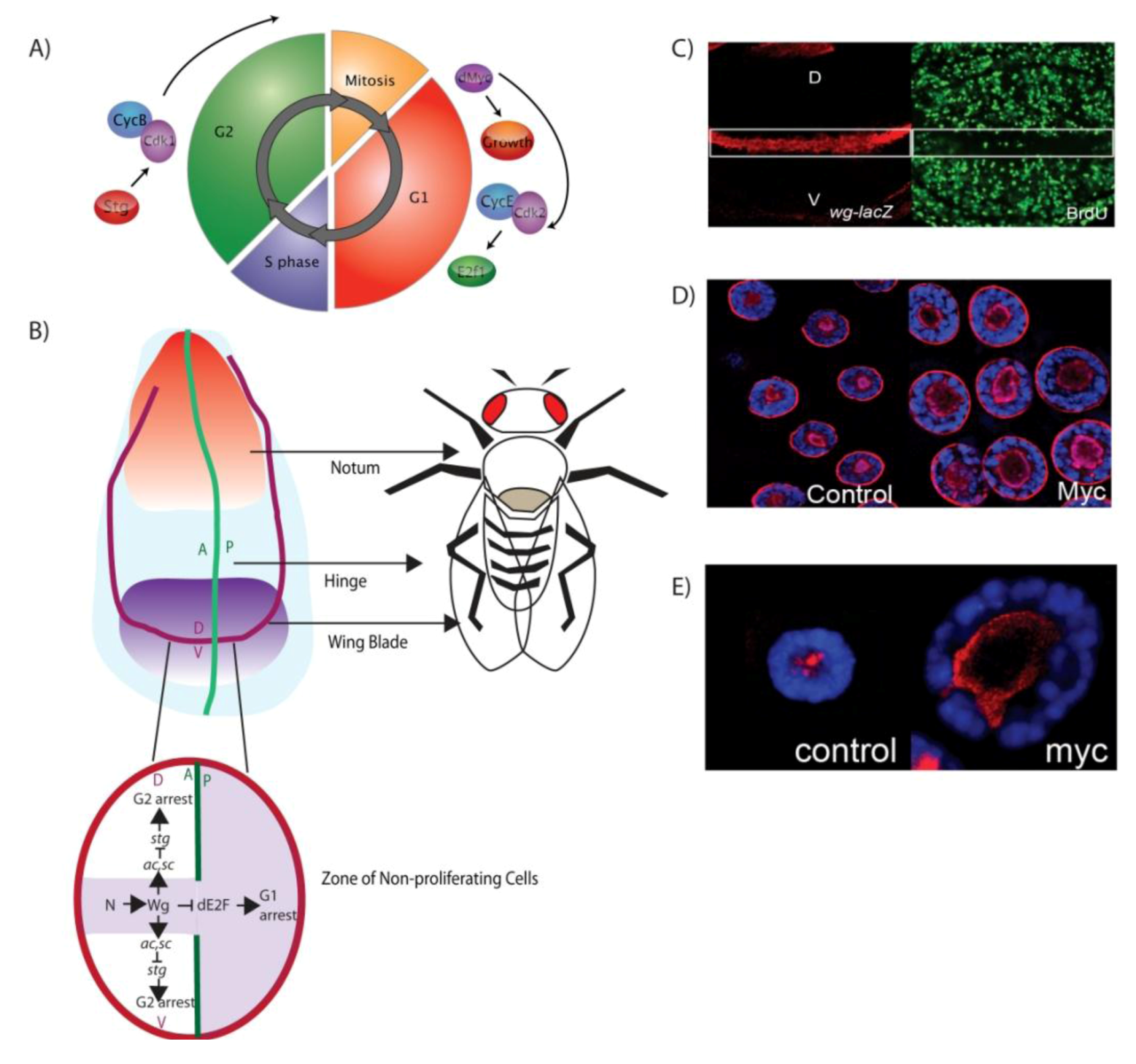
3. Drosophila Models for Understanding the Genetics of Cell Cycle Control
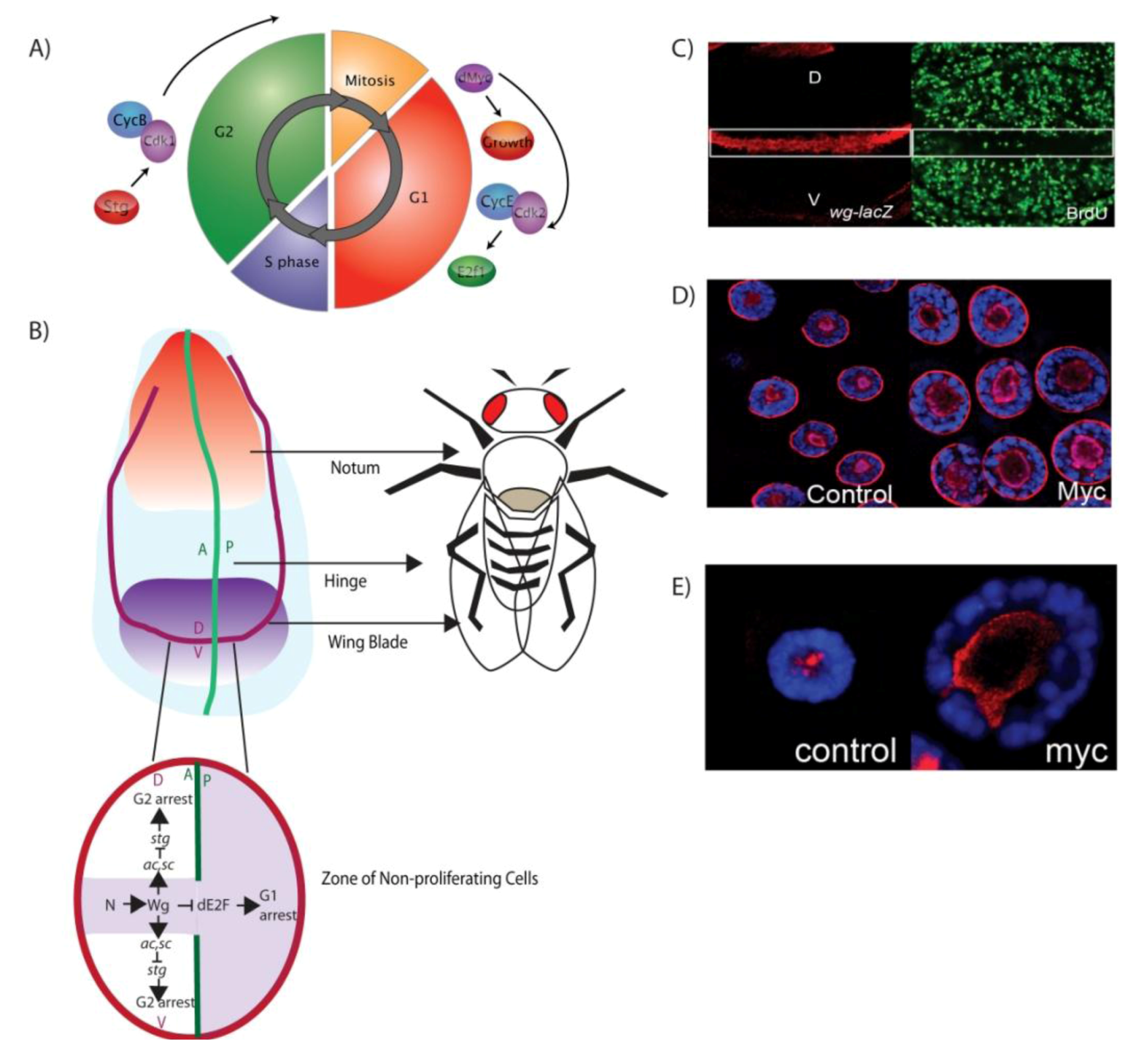
3.1. Larval Imaginal Discs: Models for Connecting Developmental Signals to Organ Growth
3.2. The Drosophila Salivary Gland: Models for Understanding Cell Growth Control
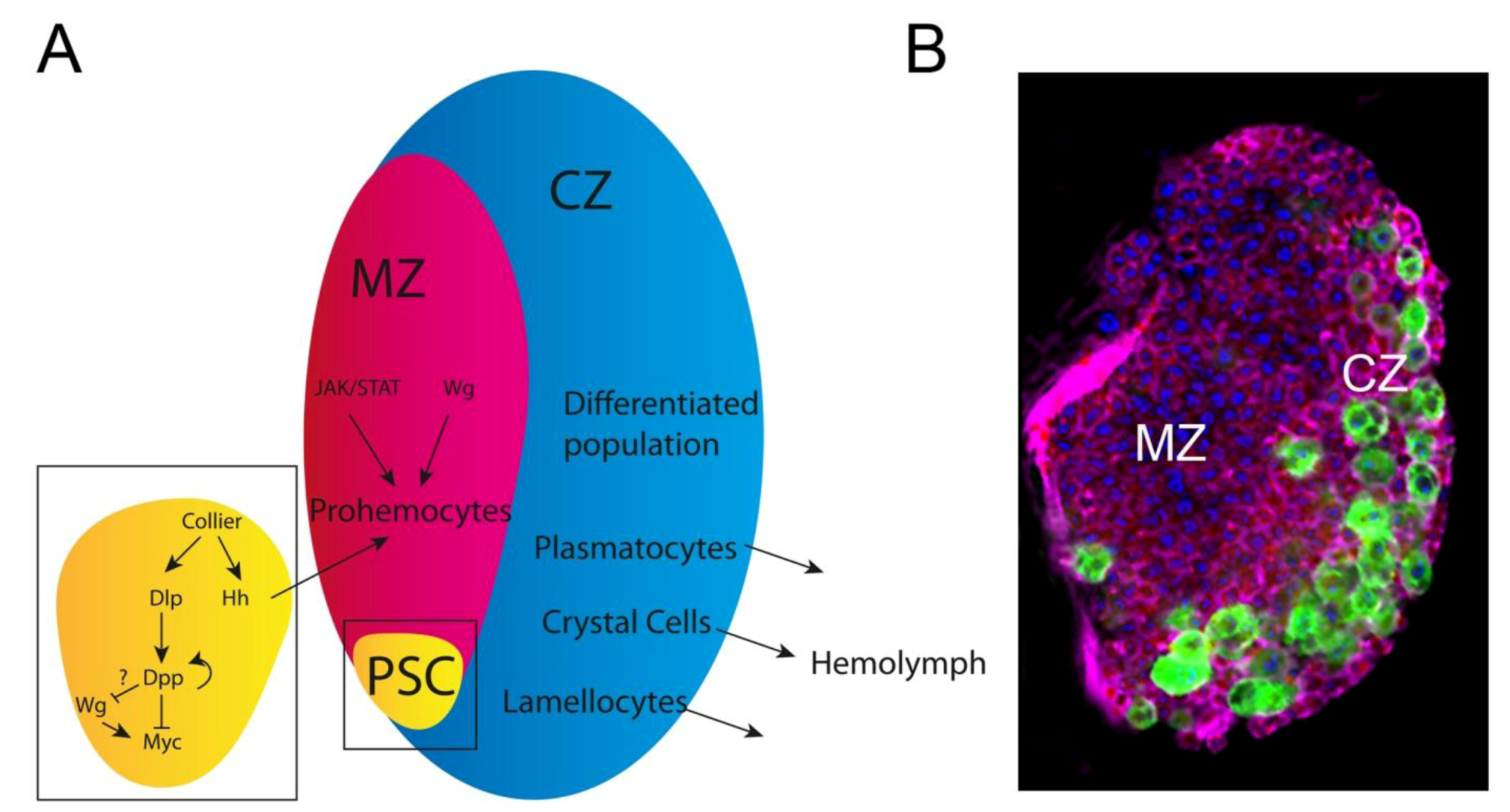
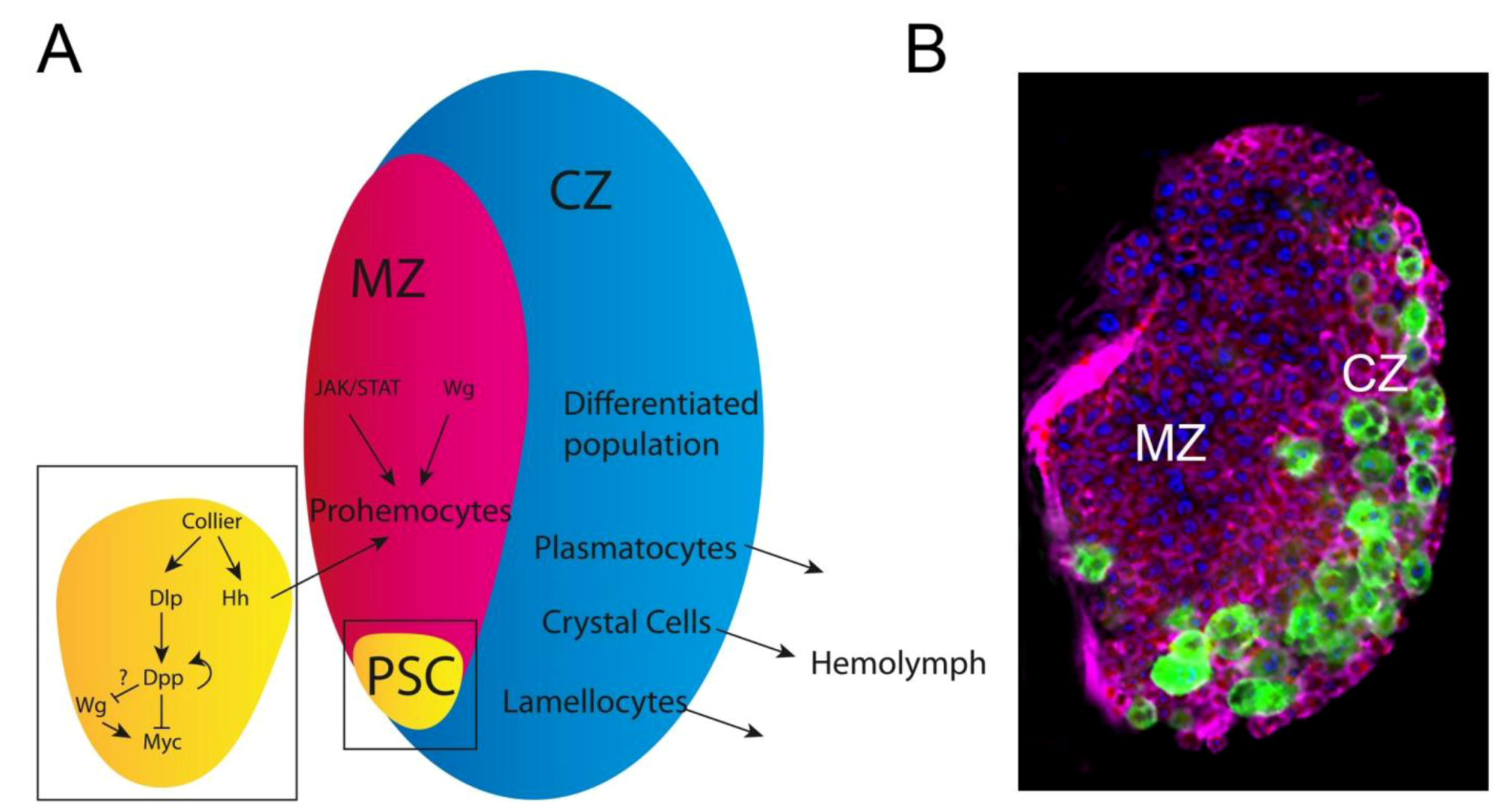
3.3. The Drosophila hemopoietic System; a Model for Mammalian Blood Development and Disease: Key Oncogenic Signaling Pathways Drive Overproliferation of Hemocytes
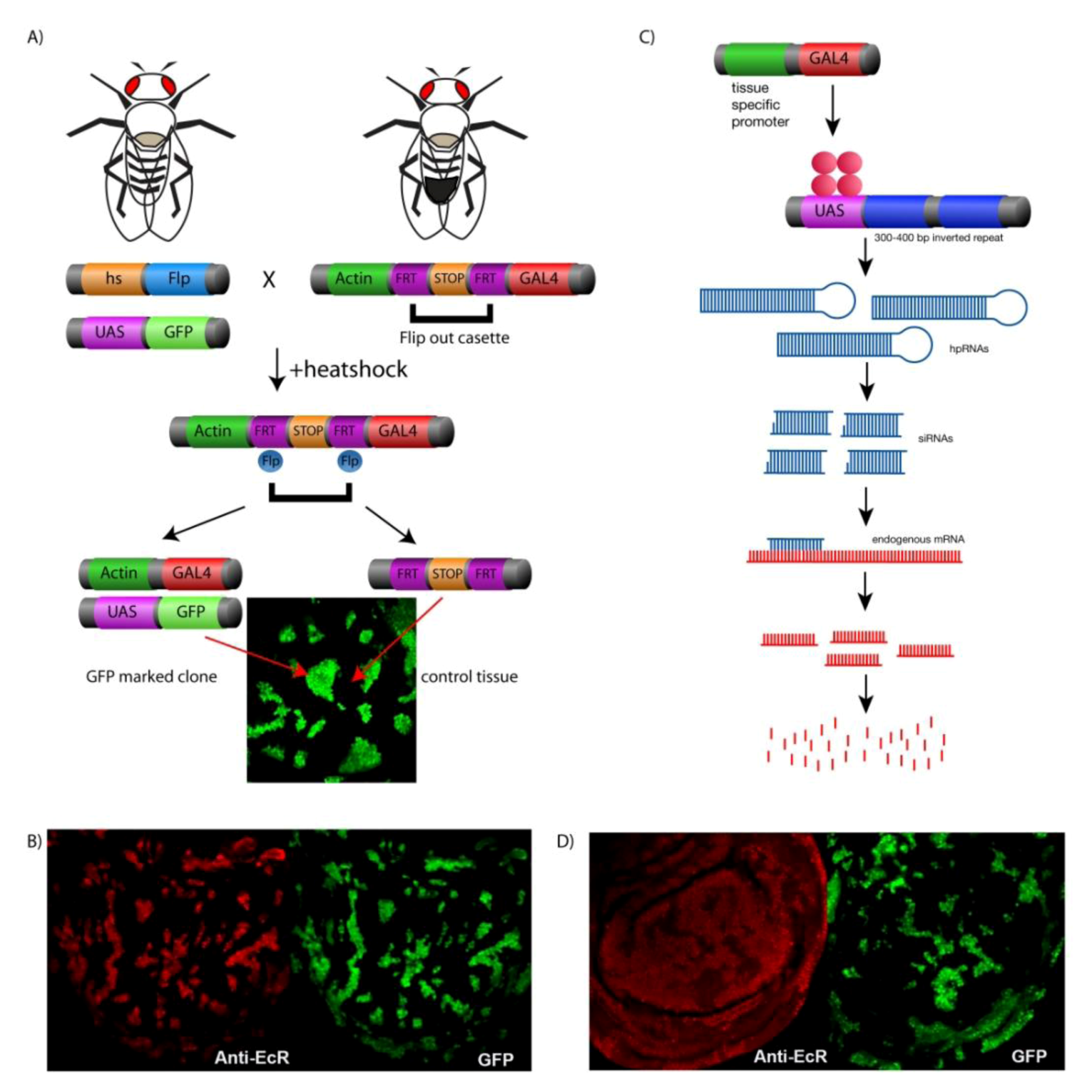
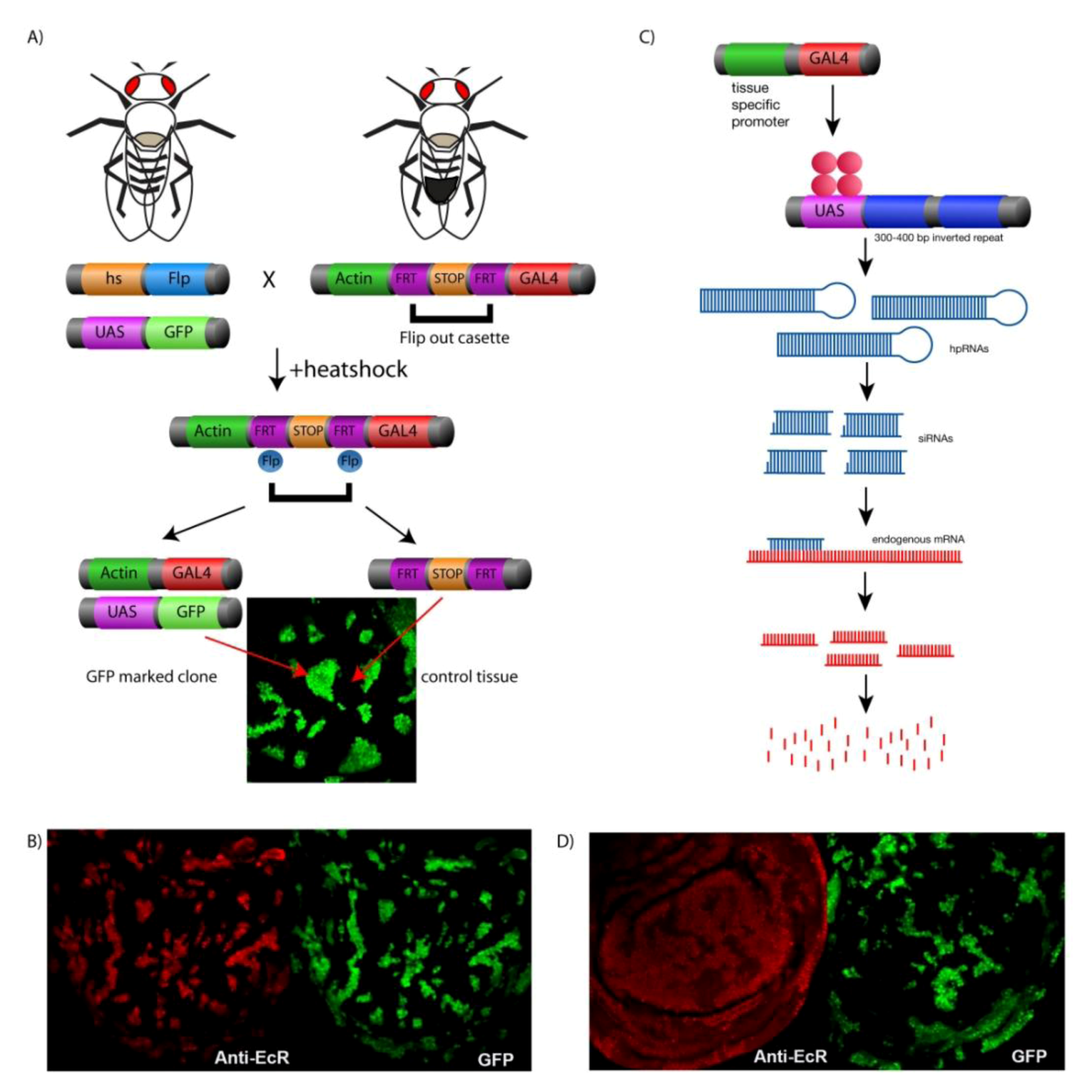
4. Genetic Tools for Manipulating Gene Expression in Drosophila
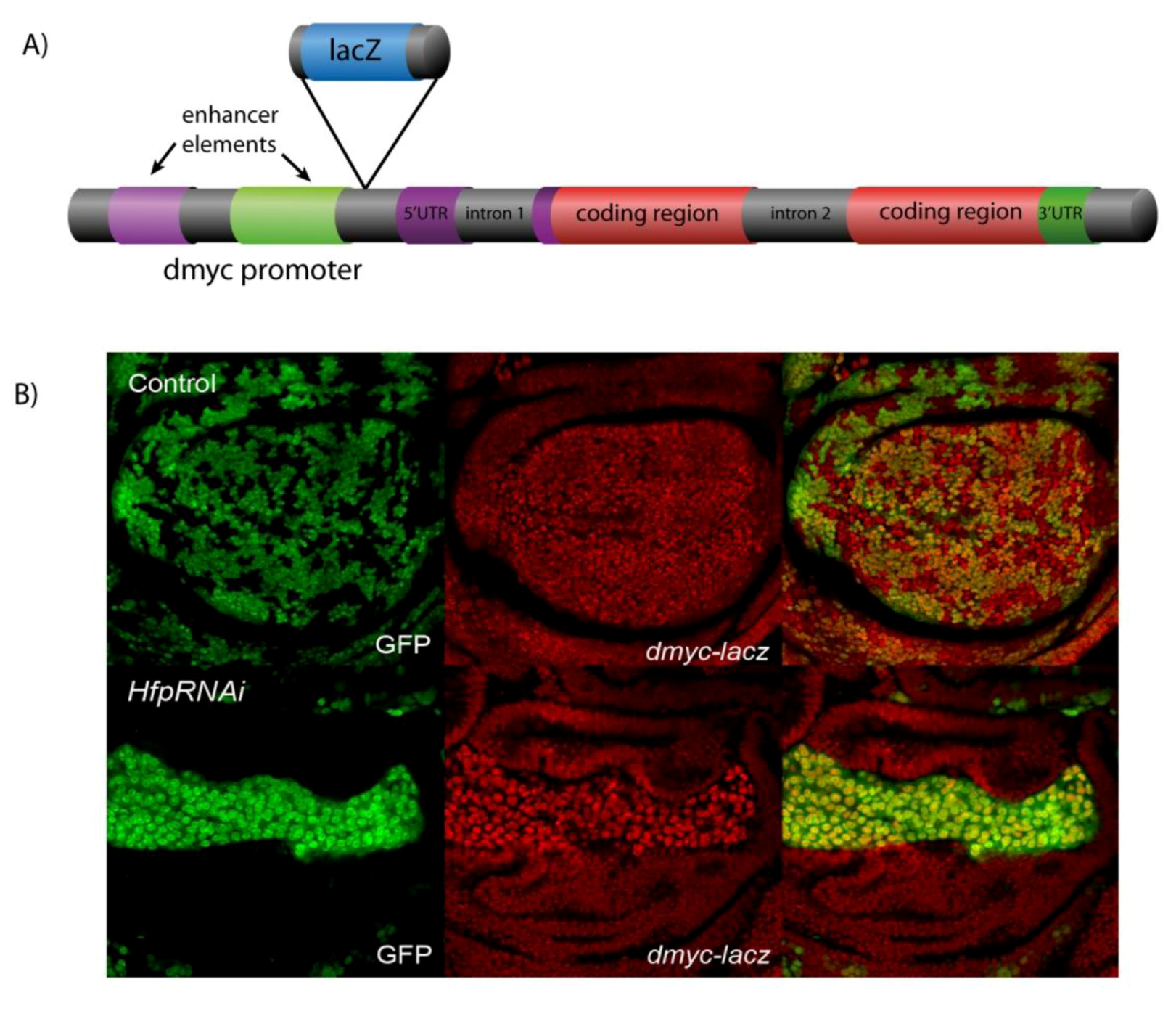
5. Conclusion
References
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila myc regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.S.; Li, L.; Orian, A.; Eisenman, R.N.; Edgar, B.A. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 2005, 7, 295–302. [Google Scholar] [CrossRef]
- Orian, A.; Grewal, S.S.; Knoepfler, P.S.; Edgar, B.A.; Parkhurst, S.M.; Eisenman, R.N. Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 299–307. [Google Scholar]
- Orian, A.; van Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.M.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Coller, H.A.; Grandori, C.; Tamayo, P.; Colbert, T.; Lander, E.S.; Eisenman, R.N.; Golub, T.R. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc. Natl. Acad. Sci. USA 2000, 97, 3260–3265. [Google Scholar]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Eilers, M.; Eisenman, R.N. Myc's broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar]
- Levens, D. Disentangling the MYC web. Proc. Natl. Acad. Sci. USA 2002, 99, 5757–5759. [Google Scholar]
- Liu, J.; Levens, D. Making myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32. [Google Scholar]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Littlewood, T.D.; Kreuzaler, P.; Evan, G.I. All things to all people. Cell 2012, 151, 11–13. [Google Scholar] [CrossRef]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef]
- Pierce, S.B.; Yost, C.; Britton, J.S.; Loo, L.W.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. dMyc is required for larval growth and endoreplication in Drosophila. Development 2004, 131, 2317–2327. [Google Scholar] [CrossRef]
- Oskarsson, T.; Trumpp, A. The Myc trilogy: lord of RNA polymerases. Nat. Cell Biol. 2005, 7, 215–217. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003, 421, 290–294. [Google Scholar]
- Levens, D. You Don't Muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef]
- Johnston, L.A.; Gallant, P. Control of growth and organ size in Drosophila. Bioessays 2002, 24, 54–64. [Google Scholar] [CrossRef]
- Maines, J.Z. Drosophila dMyc is required for ovary cell growth and endoreplication. Development 2004, 131, 775–786. [Google Scholar] [CrossRef]
- Duman-Scheel, M.; Johnston, L.A.; Du, W. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc. Natl. Acad. Sci. USA 2004, 101, 3857–3862. [Google Scholar] [CrossRef]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar]
- Zhang, H. Life without kinase: cyclin E promotes DNA replication licensing and beyond. Mol. Cell 2007, 25, 175–176. [Google Scholar]
- Araki, H. Cyclin-dependent kinase-dependent initiation of chromosomal DNA replication. Curr. Opin. Cell Biol. 2010, 22, 766–771. [Google Scholar] [CrossRef]
- Liao, D.J.; Dickson, R.B. c-Myc in breast cancer. Endocr. Relat. Cancer 2000, 7, 143–164. [Google Scholar] [CrossRef]
- Dang, C.V. Therapeutic Targeting of Myc-Reprogrammed Cancer Cell Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011. [Google Scholar]
- Eisenman, R.N.; Hann, S.R. myc-Encoded proteins of chickens and men. Curr. Top. Microbiol. Immunol. 1984, 113, 192–197. [Google Scholar]
- Chung, H.J.; Levens, D. c-myc expression: keep the noise down! Mol. Cells 2005, 20, 157–166. [Google Scholar]
- Siddall, N.A.; Lin, J.I.; Hime, G.R.; Quinn, L.M. Myc—what we have learned from flies. Current Curr. Drug Targets 2009, 10, 590–601. [Google Scholar] [CrossRef]
- Quinn, L.M.; Dickins, R.A.; Coombe, M.; Hime, G.R.; Bowtell, D.D. L.; Richardson, H. Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development 2004, 131, 1411–1423. [Google Scholar] [CrossRef]
- Mitchell, N.; Cranna, N.; Richardson, H.; Quinn, L. The Ecdysone-inducible zinc-finger transcription factor Crol regulates Wg transcription and cell cycle progression in Drosophila. Development 2008, 135, 2707–2716. [Google Scholar] [CrossRef]
- Wu, D.C.; Johnston, L.A. Control of wing size and proportions by Drosophila myc. Genetics 2010, 184, 199–211. [Google Scholar]
- Levens, D.L. Reconstructing MYC. Genes Dev. 2003, 17, 1071–1077. [Google Scholar]
- Liu, J.; Kouzine, F.; Nie, Z.; Chung, H.J.; Elisha-Feil, Z.; Weber, A.; Zhao, K.; Levens, D. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO 2006, 25, 2119–2130. [Google Scholar] [CrossRef]
- Liu, J.; Akoulitchev, S.; Weber, A.; Ge, H.; Chuikov, S.; Libutti, D.; Wang, X.W.; Conaway, J.W.; Harris, C.C.; Conaway, R.C.; et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 2001, 104, 353–363. [Google Scholar] [CrossRef]
- He, L.; Liu, J.; Collins, I.; Sanford, S.; O'Connell, B.; Benham, C.J.; Levens, D. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO 2000, 19, 1034–1044. [Google Scholar] [CrossRef]
- Matsushita, K.; Tomonaga, T.; Shimada, H.; Shioya, A.; Higashi, M.; Matsubara, H.; Harigaya, K.; Nomura, F.; Libutti, D.; Levens, D.; et al. An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res. 2006, 66, 1409–1417. [Google Scholar] [CrossRef]
- Rabenhorst, U.; Beinoraviciute-Kellner, R.; Brezniceanu, M.-L.; Joos, S.; Devens, F.; Lichter, P.; Rieker, R.J.; Trojan, J.; Chung, H.-J.; Levens, D.L.; et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology 2009, 50, 1121–1129. [Google Scholar] [CrossRef]
- Mitchell, N.C.; Johanson, T.M.; Cranna, N.J.; Er, A.L.J.; Richardson, H.E.; Hannan, R.D.; Quinn, L.M. Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development 2010, 137, 2875–2884. [Google Scholar] [CrossRef]
- Baker, N.E. Patterning signals and proliferation in Drosophila imaginal discs. Curr. Opin. Genetics Dev. 2007. [Google Scholar]
- Cranna, N.; Quinn, L. Impact of steroid hormone signals on Drosophila cell cycle during development. Cell Div. 2009, 4, 3. [Google Scholar] [CrossRef]
- Edgar, B.A.; Lehner, C.F. Developmental control of cell cycle regulators: a fly's perspective. Science 1996, 274, 1646–1652. [Google Scholar] [CrossRef]
- Johnston, L.A.; Edgar, B.A. Wingless and Notch regulate cell-cycle arrest in the developing Drosophila wing. Nature 1998, 394, 82–84. [Google Scholar]
- Johnston, L.A.; Sanders, A.L. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat. Cell Biol. 2003, 5, 827–833. [Google Scholar] [CrossRef]
- Herranz, H.; Pérez, L.; Martín, F.A.; Milán, M.A. Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. EMBO 2008, 27, 1633–1645. [Google Scholar] [CrossRef]
- Baker, N.E. Transcription of the segment-polarity gene wingless in the imaginal discs of Drosophila, and the phenotype of a pupal-lethal wg mutation. Development 1988, 102, 489–497. [Google Scholar]
- Strigini, M.; Cohen, S.M. Wingless gradient formation in the Drosophila wing. Curr. Biol. 2000, 10, 293–300. [Google Scholar] [CrossRef]
- Liao, D.J.; Thakur, A.; Wu, J.; Biliran, H.; Sarkar, F.H. Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit. Rev. Ontogenesis 2007, 13, 93–158. [Google Scholar] [CrossRef]
- Wang, C.; Lisanti, M.P.; Liao, D.J. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 2011, 10, 57–67. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar]
- Reis, T.; Edgar, B.A. Negative regulation of dE2F1 by cyclin-dependent kinases controls cell cycle timing. Cell 2004, 117, 253–264. [Google Scholar] [CrossRef]
- Edgar, B.A.; Orr-Weaver, T.L. Endoreplication cell cycles: more for less. Cell 2001, 105, 297–306. [Google Scholar] [CrossRef]
- Boisvert, F.-M.; van Koningsbruggen, S.; Navascués, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Leder, P.; Battey, J.; Lenoir, G.; Moulding, C.; Murphy, W.; Potter, H.; Stewart, T.; Taub, R. Translocations among antibody genes in human cancer. Science 1983, 222, 765–771. [Google Scholar]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Langdon, W.Y.; Harris, A.W.; Cory, S.; Adams, J.M. The c-myconcogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell 1986, 47, 11–18. [Google Scholar] [CrossRef]
- Evans, C.J.; Hartenstein, V.; Banerjee, U. Thicker than blood: conserved mechanisms in Drosophila and vertebrate hematopoiesis. Dev. Cell 2003, 5, 673–690. [Google Scholar]
- Asha, H.; Nagy, I.; Kovacs, G.; Stetson, D.; Ando, I.; Dearolf, C.R. Analysis of Ras-induced overproliferation in Drosophila hemocytes. Genetics 2003, 163, 203–215. [Google Scholar]
- Crozatier, M.; Vincent, A. Drosophila: A model for studying genetic and molecular aspects of haematopoiesis and associated leukaemias. Dis. Models Mech. 2011, 4, 439–445. [Google Scholar] [CrossRef]
- Crozatier, M.; Meister, M. Drosophila haematopoiesis. Cell. Microbiol. 2007, 9, 1117–1126. [Google Scholar]
- Minakhina, S.; Steward, R. Hematopoietic stem cells in Drosophila. Development 2009, 137, 27–31. [Google Scholar] [CrossRef]
- Pennetier, D.; Oyallon, J.; Morin-Poulard, I.; Dejean, S.; Vincent, A.; Crozatier, M. Size control of the Drosophila hematopoietic niche by bone morphogenetic protein signaling reveals parallels with mammals. PNAS 2012, 109, 3389–3394. [Google Scholar]
- Lanot, R.; Zachary, D.; Holder, F.; Meister, M. Postembryonic hematopoiesis in Drosophila. Dev. Biol. 2001, 230, 243–257. [Google Scholar] [CrossRef]
- Crozatier, M.; Ubeda, J.-M.; Vincent, A.; Meister, M. Cellular immune response to parasitization in Drosophila requires the EBF orthologue collier. PLoSBiol. 2004, 2, E196. [Google Scholar]
- Jung, S.-H.; Evans, C.J.; Uemura, C.; Banerjee, U. The Drosophila lymph gland as a developmental model of hematopoiesis. Development 2005, 132, 2521–2533. [Google Scholar] [CrossRef]
- Krzemien, J.; Dubois, L.; Makki, R.; Meister, M.; Vincent, A.; Crozatier, M. Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature 2007, 446, 325–328. [Google Scholar]
- Krzemien, J.; Oyallon, J.; Crozatier, M.; Vincent, A. Hematopoietic progenitors and hemocyte lineages in the Drosophila lymph gland. Dev. Biol. 2010, 346, 310–319. [Google Scholar] [CrossRef]
- Kocks, C.; Cho, J.H.; Nehme, N.; Ulvila, J.; Pearson, A.M.; Meister, M.; Strom, C.; Conto, S.L.; Hetru, C.; Stuart, L.M.; et al. Eater, a transmembrane protein mediating phagocytosis of bacterial pathogens in Drosophila. Cell 2005, 123, 335–346. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Tuveson, D.A. RAS in cellular transformation and senescence. Eur. J. Cancer 2009, 45 Suppl 1, 211–216. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. 2011, 11, 761–774. [Google Scholar]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Sears, R.C. The life cycle of C-myc: from synthesis to degradation. Cell Cycle 2004, 3, 1133–1137. [Google Scholar]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Nakayama, K.; Morita, I. A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2). Cancer Res. 2003, 63, 1623–1630. [Google Scholar]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.-F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef]
- Baena, E.; Ortiz, M.; Martinez, A.C.; de Alboran, I.M. c-Myc is essential for hematopoietic stem cell differentiation and regulates Lin(-)Sca-1(+)c-Kit(-) cell generation through p21. Exp. Hematology 2007, 35, 1333–1343. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Pignoni, F.; Zipursky, S.L. Induction of Drosophila eye development by decapentaplegic. Development 1997, 124, 271–278. [Google Scholar]
- Neufeld, T.P.; La Cruz de, A.F.; Johnston, L.A.; Edgar, B.A. Coordination of growth and cell division in the Drosophila wing. Cell 1998, 93, 1183–1193. [Google Scholar] [CrossRef]
- Golic, K.G. Site-specific recombination between homologous chromosomes in Drosophila. Science 1991, 252, 958–961. [Google Scholar]
- Britton, J.S.; Lockwood, W.K.; Li, L.; Cohen, S.M.; Edgar, B.A. Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev. Cell 2002, 2, 239–249. [Google Scholar] [CrossRef]
- Lam, G.; Thummel, C.S. Inducible expression of double-stranded RNA directs specific genetic interference in Drosophila. Curr. Biol. 2000, 10, 957–963. [Google Scholar] [CrossRef]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar]
- Spradling, A.C.; Stern, D.M.; Kiss, I.; Roote, J.; Laverty, T.; Rubin, G.M. Gene disruptions using P transposable elements: an integral component of the Drosophila genome project. Proc. Natl. Acad. Sci. USA 1995, 92, 10824–10830. [Google Scholar] [CrossRef]
- Bellen, H.J.; O'Kane, C.J.; Wilson, C.; Grossniklaus, U.; Pearson, R.K.; Gehring, W.J. P-element-mediated enhancer detection: a versatile method to study development in Drosophila. Genes Dev. 1989, 3, 1288–1300. [Google Scholar] [CrossRef]
- Zhang, P.; Spradling, A.C. Insertional mutagenesis of Drosophila heterochromatin with single P elements. Proc. Natl. Acad. Sci. USA 1994, 91, 3539–3543. [Google Scholar] [CrossRef]
- Peter, A.; Schottler, P.; Werner, M.; Beinert, N.; Dowe, G.; Burkert, P.; Mourkioti, F.; Dentzer, L.; He, Y.; Deak, P.; et al. Mapping and identification of essential gene functions on the X chromosome of Drosophila. EMBO Rep. 2002, 3, 34–38. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, J.E.A.; Cranna, N.J.; Chahal, A.S.; Quinn, L.M. Genetic Systems to Investigate Regulation of Oncogenes and Tumour Suppressor Genes in Drosophila. Cells 2012, 1, 1182-1196. https://doi.org/10.3390/cells1041182
Lee JEA, Cranna NJ, Chahal AS, Quinn LM. Genetic Systems to Investigate Regulation of Oncogenes and Tumour Suppressor Genes in Drosophila. Cells. 2012; 1(4):1182-1196. https://doi.org/10.3390/cells1041182
Chicago/Turabian StyleLee, Jue Er Amanda, Nicola J. Cranna, Arjun S. Chahal, and Leonie M. Quinn. 2012. "Genetic Systems to Investigate Regulation of Oncogenes and Tumour Suppressor Genes in Drosophila" Cells 1, no. 4: 1182-1196. https://doi.org/10.3390/cells1041182