Stress Response Pathways in Ameloblasts: Implications for Amelogenesis and Dental Fluorosis

{kind=link}
Abstract
:1. Introduction
2. Enamel Development
2.1. The Secretory Stage
2.2. The Maturation Stage
2.3. Enamel Defects
3. Fluoride and Ameloblasts
3.1. Fluoride does not Affect the Activity of MMP20 or KLK4
3.2. Fluoride Causes Oxidative Stress
3.3. Fluoride Induces ER Stress and eIF2 Phosphorylation
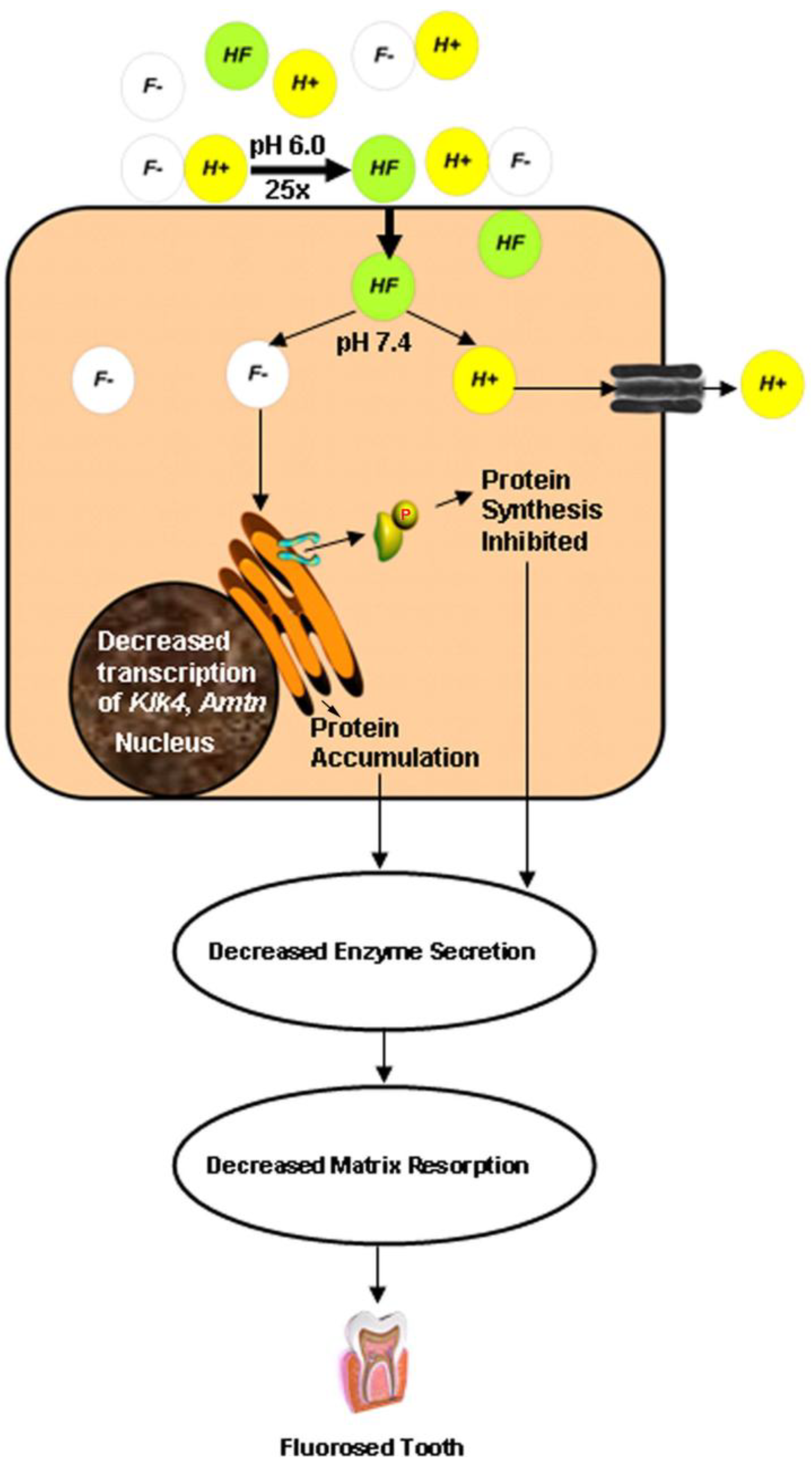
4. The Acid Hypothesis
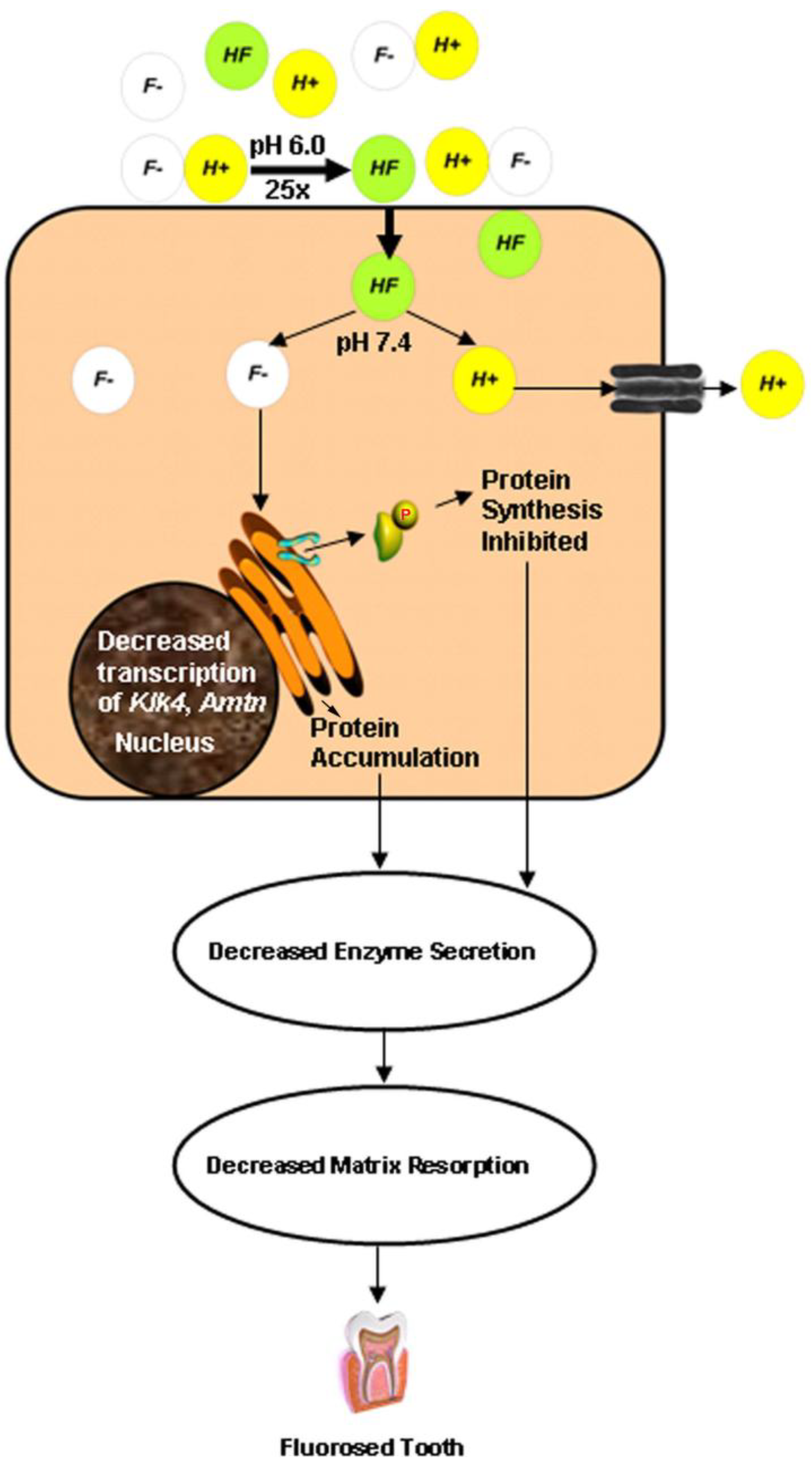
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Avery, J.K. Oral Development and Histology, 3rd ed; Thieme: New York, NY, USA, 2002; pp. 165–166. [Google Scholar]
- HHS.gov Home Page. Available online: http://www.hhs.gov/news/press/2011pres/01/20110107a.html (accessed on 2 May 2012).
- Beltrán-Aguilar, E.D.; Barker, L.; Dye, B.A. Prevalence and severity of dental fluorosis in the United States, 1999–2004. NCHS Data Brief. 2010, 53, 1–8. [Google Scholar]
- DenBesten, P.K.; Crenshaw, M.A. The effects of chronic high fluoride levels on forming enamel in the rat. Arch. Oral Biol. 1984, 29, 675–679. [Google Scholar] [CrossRef]
- Shearer, T.R.; Britton, J.L.; DeSart, D.J.; Suttie, J.W. Microhardness of molar teeth in cattle with fluorosis. Am. J. Vet. Res. 1980, 41, 1543–1545. [Google Scholar]
- Newbrun, E. Studies on the physical properties of fluorosed enamel. II. Microhardness. Arch. Oral Biol. 1960, 12, 21–27. [Google Scholar] [CrossRef]
- Sharma, R.; Tsuchiya, M.; Bartlett, J.D. Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ. Health Persp. 2008, 116, 1142–1146. [Google Scholar]
- Xu, H.; Zhou, Y.L.; Zhang, X.Y.; Lu, P.; Li, G.S. Activation of PERK signaling through fluoride-mediated endoplasmic reticulum stress in OS732 cells. Toxicology 2010, 277, 1–5. [Google Scholar] [CrossRef]
- Wei, W.; Gao, Y.; Wang, C.; Zhao, L.; Sun, D. Excessive fluoride induces endoplasmic reticulum stress and interferes enamel proteinases secretion. Environ. Toxicol. 2011. [Google Scholar]
- Hu, J.C.; Chun, Y.H.; Al Hazzazzi, T.; Simmer, J.P. Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 2007, 186, 78–85. [Google Scholar] [CrossRef]
- Smith, C.E. Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 1998, 9, 128–161. [Google Scholar] [CrossRef]
- Warshawsky, H. The fine structure of secretory ameloblasts in rat incisors. Anat. Rec. 1968, 161, 211–229. [Google Scholar] [CrossRef]
- Caterina, J.J.; Skobe, Z.; Shi, J.; Ding, Y.; Simmer, J.P.; Birkedal-Hansen, H.; Bartlett, J.D. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 2002, 277, 49598–49604. [Google Scholar]
- Fukae, M.; Tanabe, T.; Uchida, T.; Lee, S.K.; Ryu, O.H.; Murakami, C.; Wakida, K.; Simmer, J.P.; Yamada, Y.; Bartlett, J.D. Enamelysin (matrix metalloproteinase-20): Localization in the developing tooth and effects of pH and calcium on amelogenin hydrolysis. J. Dent. Res. 1998, 77, 1580–1588. [Google Scholar] [CrossRef]
- Nanci, A.; Zalzal, S.; Lavoie, P.; Kunikata, M.; Chen, W.; Krebsbach, P.H.; Yamada, Y.; Hammarström, L.; Simmer, J.P.; Fincham, A.G.; et al. Comparative immunochemical analyses of the developmental expression and distribution of ameloblastin and amelogenin in rat incisors. J. Histochem. Cytochem. 1998, 46, 911–934. [Google Scholar] [CrossRef]
- Bartlett, J.D.; Skobe, Z.; Nanci, A.; Smith, C.E. Matrix metalloproteinase 20 promotes a smooth enamel surface, a strong dentino-enamel junction, and a decussating enamel rod pattern. Eur. J. Oral Sci. 2011, 119, 199–205. [Google Scholar] [CrossRef]
- Kwak, S.Y.; Green, S.; Wiedemann-Bidlack, F.B.; Beniash, E.; Yamakoshi, Y.; Simmer, J.P.; Margolis, H.C. Regulation of calcium phosphate formation by amelogenins under physiological conditions. Eur. J. Oral Sci. 2011, 119, 103–111. [Google Scholar] [Green Version]
- Simmer, J.P.; Fukae, M.; Tanabe, T.; Yamakoshi, Y.; Uchida, T.; Xue, J.; Margolis, H.C.; Shimizu, M.; DeHart, B.C.; Hu, C.C.; et al. Purification, characterization, and cloning of enamel matrix serine proteinase 1. J. Dent. Res. 1998, 77, 377–386. [Google Scholar] [CrossRef]
- Nanci, A.; Slavkin, H.C.; Smith, C.E. Immunocytochemical and radioautographic evidence for secretion and intracellular degradation of enamel proteins by ameloblasts during the maturation stage of amelogenesis in rat incisors. Anat. Rec. 1987, 217, 107–123. [Google Scholar] [CrossRef]
- Smith, C.E.; McKee, M.D.; Nanci, A. Cyclic induction and rapid movement of sequential waves of new smooth-ended ameloblast modulation bands in rat incisors as visualized by polychrome fluorescent labeling and GBHA-staining of maturing enamel. Adv. Dent. Res. 1987, 1, 162–175. [Google Scholar]
- Josephsen, K.; Fejerskov, O. Ameloblast modulation in the maturation zone of the rat incisor enamel organ. A light and electron microscopic study. J. Anat. 1977, 124, 45–70. [Google Scholar]
- Salama, A.H.; Zaki, A.E.; Eisenmann, D.R. Tubular lysosomes in ruffle-ended ameloblasts associated with enamel maturation in rat incisor. J. Histochem. Cytochem. 1989, 37, 801–811. [Google Scholar] [CrossRef]
- Smith, C.E. Ameloblasts: Secretory and resorptive functions. J. Dent. Res. 1979, 58, 695–707. [Google Scholar] [CrossRef]
- Takano, Y.; Ozawa, H. Ultrastructural and cytochemical observations on the alternating morphologic changes of the ameloblasts at the stage of enamel maturation. Arch. Histol. Jpn. 1980, 43, 385–399. [Google Scholar]
- Sasaki, T. Endocytotic pathways at the ruffled borders of rat maturation ameloblasts. Histochemistry 1984, 80, 263–268. [Google Scholar]
- Kawamoto, T.; Shimizu, M. Pathway and speed of calcium movement from blood to mineralizing enamel. J. Histochem. Cytochem. 1997, 45, 213–230. [Google Scholar] [CrossRef]
- Smith, C.E.; Issid, M.; Margolis, H.C.; Moreno, E.C. Developmental changes in the pH of enamel fluid and its effects on matrix-resident proteinases. Adv. Dent. Res. 1996, 10, 159–169. [Google Scholar] [CrossRef]
- Wright, J.T. The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am. J. Med. Genet. A 2006, 140, 2547–2555. [Google Scholar]
- Gibson, C.W.; Yuan, Z.A.; Hall, B.; Longenecker, G.; Chen, E.; Thyagarajan, T.; Sreenath, T.; Wright, J.T.; Decker, S.; Piddington, R.; et al. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 2001, 276, 31871–31875. [Google Scholar]
- Lee, K.E.; Lee, S.K.; Jung, S.E.; Song, S.J.; Cho, S.H.; Lee, Z.H.; Kim, J.W. A novel mutation in the AMELX gene and multiple crown resorptions. Eur. J. Oral Sci. 2011, 119, 324–328. [Google Scholar] [CrossRef]
- Fukumoto, S.; Kiba, T.; Hall, B.; Iehara, N.; Nakamura, T.; Longenecker, G.; Krebsbach, P.H.; Nanci, A.; Kulkarni, A.B.; Yamada, Y. Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J. Cell Biol. 2004, 167, 973–983. [Google Scholar] [CrossRef]
- Wright, J.T.; Hart, T.C.; Hart, P.S.; Simmons, D.; Suggs, C.; Daley, B.; Simmer, J.; Hu, J.; Bartlett, J.D.; Li, Y.; et al. Human and mouse enamel phenotypes resulting from mutation or altered expression of AMEL, ENAM, MMP20 and KLK4. Cells Tissues Organs 2009, 189, 224–229. [Google Scholar] [CrossRef]
- Hu, J.C.; Hu, Y.; Smith, C.E.; McKee, M.D.; Wright, J.T.; Yamakoshi, Y.; Papagerakis, P.; Hunter, G.K.; Feng, J.Q.; Yamakoshi, F.; et al. Enamel defects and ameloblast-specific expression in Enam knock-out/lacz knock-in mice. J. Biol. Chem. 2008, 283, 10858–10871. [Google Scholar]
- Rajpar, M.H.; Harley, K.; Laing, C.; Davies, R.M.; Dixon, M.J. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum. Mol. Genet. 2001, 10, 1673–1677. [Google Scholar] [CrossRef]
- Mårdh, C.K.; Bäckman, B.; Holmgren, G.; Hu, J.C.; Simmer, J.P.; Forsman-Semb, K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2). Hum. Mol. Genet. 2002, 11, 1069–1074. [Google Scholar] [CrossRef]
- Chan, H.C.; Mai, L.; Oikonomopoulou, A.; Chan, H.L.; Richardson, A.S.; Wang, S.K.; Simmer, J.P.; Hu, J.C. Altered enamelin phosphorylation site causes amelogenesis imperfecta. J. Dent. Res. 2010, 89, 695–699. [Google Scholar] [CrossRef]
- Sharma, R.; Tye, C.E.; Arun, A.; Macdonald, D.; Chatterjee, A.; Abrazinski, T.; Everett, E.T.; Whitford, G.M.; Bartlett, J.D. Assessment of Dental Fluorosis in Mmp20+/− Mice. J. Dent. Res. 2011, 90, 788–792. [Google Scholar] [CrossRef]
- Simmer, J.P.; Hu, Y.; Lertlam, R.; Yamakoshi, Y.; Hu, J.C. Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. J. Biol. Chem. 2009, 284, 19110–19121. [Google Scholar]
- Kim, J.W.; Simmer, J.P.; Hart, T.C.; Hart, P.S.; Ramaswami, M.D.; Bartlett, J.D.; Hu, J.C. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J. Med. Genet. 2005, 42, 271–275. [Google Scholar] [CrossRef]
- Ozdemir, D.; Hart, P.S.; Ryu, O.H.; Choi, S.J.; Ozdemir-Karatas, M.; Firatli, E.; Piesco, N.; Hart, T.C. MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J. Dent. Res. 2005, 84, 1031–1035. [Google Scholar] [CrossRef]
- Papagerakis, P.; Lin, H.K.; Lee, K.Y.; Hu, Y.; Simmer, J.P.; Bartlett, J.D.; Hu, J.C. Premature stop codon in MMP20 causing amelogenesis imperfecta. J. Dent. Res. 2008, 87, 56–59. [Google Scholar] [CrossRef]
- Lee, S.K.; Seymen, F.; Kang, H.Y.; Lee, K.E.; Gencay, K.; Tuna, B.; Kim, J.W. MMP20 hemopexin domain mutation in amelogenesis imperfecta. J. Dent. Res. 2010, 89, 46–50. [Google Scholar] [CrossRef]
- Hart, P.S.; Hart, T.C.; Michalec, M.D.; Ryu, O.H.; Simmons, D.; Hong, S.; Wright, J.T. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J. Med. Genet. 2004, 41, 545–549. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, S.K.; Lee, Z.H.; Park, J.C.; Lee, K.E.; Lee, M.H.; Park, J.T.; Seo, B.M.; Hu, J.C.; Simmer, J.P. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am. J. Hum. Genet. 2008, 82, 489–494. [Google Scholar]
- O’Sullivan, J.; Bitu, C.C.; Daly, S.B.; Urquhart, J.E.; Barron, M.J.; Bhaskar, S.S.; Martelli-Júnior, H.; dos Santos Neto, P.E.; Mansilla, M.A.; Murray, J.C.; et al. Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am. J. Hum. Genet. 2011, 88, 616–620. [Google Scholar] [CrossRef]
- El-Sayed, W.; Parry, D.A.; Shore, R.C.; Ahmed, M.; Jafri, H.; Rashid, Y.; Al-Bahlani, S.; Al Harasi, S.; Kirkham, J.; Inglehearn, C.F.; et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 699–705. [Google Scholar] [CrossRef]
- Primosch, R.E. Tetracycline discoloration, enamel defects, and dental caries in patients with cystic fibrosis. Oral Surg. Oral Med. Oral Pathol. 1980, 50, 301–308. [Google Scholar] [CrossRef]
- Wright, J.T.; Kiefer, C.L.; Hall, K.I.; Grubb, B.R. Abnormal enamel development in a cystic fibrosis transgenic mouse model. J. Dent. Res. 1996, 75, 966–973. [Google Scholar] [CrossRef]
- Sui, W.; Boyd, C.; Wright, J.T. Altered pH regulation during enamel development in the cystic fibrosis mouse incisor. J. Dent. Res. 2003, 82, 388–392. [Google Scholar] [CrossRef]
- Bailleul-Forestier, I.; Molla, M.; Verloes, A.; Berdal, A. The genetic basis of inherited anomalies of the teeth. Part 1: Clinical and molecular aspects of non-syndromic dental disorders. Eur. J. Med. Genet. 2008, 51, 273–291. [Google Scholar] [CrossRef]
- Tubert-Jeannin, S.; Auclair, C.; Amsallem, E.; Tramini, P.; Gerbaud, L.; Ruffieux, C.; Schulte, A.G.; Koch, M.J.; Rège-Walther, M.; Ismail, A. Fluoride supplements (tablets, drops, lozenges or chewing gums) for preventing dental caries in children. Cochrane Database Syst. Rev. 2011, 12, CD007592. [Google Scholar]
- Aoba, T.; Fejerskov, O. Dental fluorosis: chemistry and biology. Crit. Rev. Oral. Biol. Med. 2002, 13, 155–170. [Google Scholar] [CrossRef]
- Fejerskov, O.; Thylstrup, A.; Larsen, M.J. Clinical and structural features and possible pathogenic mechanisms of dental fluorosis. Scand. J. Dent. Res. 1977, 85, 510–534. [Google Scholar]
- Wright, J.T.; Chen, S.C.; Hall, K.I.; Yamauchi, M.; Bawden, J.W. Protein characterization of fluorosed human enamel. J. Dent. Res. 1996, 75, 1936–1941. [Google Scholar] [CrossRef]
- DenBesten, P.K.; Yan, Y.; Featherstone, J.D.; Hilton, J.F.; Smith, C.E.; Li, W. Effects of fluoride on rat dental enamel matrix proteinases. Arch. Oral. Biol. 2002, 47, 763–770. [Google Scholar] [CrossRef]
- Tye, C.E.; Antone, J.V.; Bartlett, J.D. Fluoride does not inhibit enamel protease activity. J. Dent. Res. 2011, 90, 489–494. [Google Scholar] [CrossRef]
- Gerlach, R.F.; de Souza, A.P.; Cury, J.A.; Line, S.R. Fluoride effect on the activity of enamel matrix proteinases in vitro. Eur. J. Oral. Sci. 2000, 108, 48–53. [Google Scholar]
- Varol, E.; Icli, A.; Aksoy, F.; Bas, H.A.; Sutcu, R.; Ersoy, I.H.; Varol, S.; Ozaydin, M. Evaluation of total oxidative status and total antioxidant capacity in patients with endemic fluorosis. Toxicol. Ind. Health 2011, in press. [Google Scholar]
- Mittal, M.; Flora, S.J. Effects of individual and combined exposure to sodium arsenite and sodium fluoride on tissue oxidative stress, arsenic and fluoride levels in male mice. Chem. Biol. Interact. 2006, 162, 128–139. [Google Scholar] [CrossRef]
- Jin, X.Q.; Xu, H.; Shi, H.Y.; Zhang, J.M.; Zhang, H.Q. Fluoride-induced oxidative stress of osteoblasts and protective effects of baicalein against fluoride toxicity. Biol. Trace. Elem. Res. 2007, 116, 81–89. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar]
- Kulkarni, A.P.; Mittal, S.P.; Devasagayam, T.P.; Pal, J.K. Oxidative stress perturbs cell proliferation in human K562 cells by modulating protein synthesis and cell cycle. Free Radic. Res. 2009, 43, 1090–1100. [Google Scholar]
- Kulkarni, A.P.; Mittal, S.P.; Devasagayam, T.P.; Pal, J.K. Hsp90 mediates activation of the heme regulated eIF-2 alpha kinase during oxidative stress. Indian J. Biochem. Biophys. 2010, 47, 67–74. [Google Scholar]
- Mittal, M.; Flora, S.J. Vitamin E supplementation protects oxidative stress during arsenic and fluoride antagonism in male mice. Drug Chem. Toxicol. 2007, 30, 263–281. [Google Scholar] [CrossRef]
- Mansour, H.H.; Tawfik, S.S. Efficacy of lycopene against fluoride toxicity in rats. Pharm. Biol. 2011, 50, 707–711. [Google Scholar] [CrossRef]
- Feng, P.; Wei, J.R.; Zhang, Z.G. Influence of selenium and fluoride on blood antioxidant capacity of rats. Exp. Toxicol. Pathol. 2010, 64, 565–568. [Google Scholar]
- Izquierdo-Vega, J.A.; Sanchez-Gutierrez, M.; Del Razo, L.M. NADPH oxidase participates in the oxidative damage caused by fluoride in rat spermatozoa: Protective role of alpha-tocopherol. J. Appl. Toxicol. 2010, 31, 579–588. [Google Scholar]
- Feng, P.; Wei, J.; Zhang, Z. Intervention of Selenium on Chronic Fluorosis-Induced Injury of Blood Antioxidant Capacity in Rats. Biol. Trace Elem. Res. 2011, 144, 1024–1031. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Smith, C.E.; Chen, Y.B.; Hubbard, M.J.; Hacia, J.G.; Paine, M.L. Gene-expression analysis of early- and late-maturation-stage rat enamel organ. Eur. J. Oral. Sci. 2011, 119, 149–157. [Google Scholar] [CrossRef]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry 2011, 76, 407–422. [Google Scholar]
- Merchant, A.A.; Singh, A.; Matsui, W.; Biswal, S. The redox-sensitive transcription factor, Nrf2, regulates murine hematopoietic stem cell survival independent of ROS levels. Blood 2011, 118, 6572–6579. [Google Scholar] [CrossRef]
- Yanagawa, T.; Itoh, K.; Uwayama, J.; Shibata, Y.; Yamaguchi, A.; Sano, T.; Ishii, T.; Yoshida, H.; Yamamoto, M. Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells 2004, 9, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Ratner, S. The Iron Content of Teeth of Normal and Anemic Rats. J. Dent. Res. 1935, 15, 89–92. [Google Scholar] [CrossRef]
- Lindemann, G. Pigment alterations and other disturbances in rat incisor enamel in chronic fluorosis and in recovery. Acta Odontol. Scand. 1967, 25, 525–539. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar]
- Levonen, A.L.; Landar, A.; Ramachandran, A.; Ceaser, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar]
- Kubota, K.; Lee, D.H.; Tsuchiya, M.; Young, C.S.; Everett, E.T.; Martinez-Mier, E.A.; Snead, M.L.; Nguyen, L.; Urano, F.; Bartlett, J. D. Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J. Biol. Chem. 2005, 280, 23194–23202. [Google Scholar]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell. Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Riksen, E.A.; Kalvik, A.; Brookes, S.; Hynne, A.; Snead, M.L.; Lyngstadaas, S.P.; Reseland, J.E. Fluoride reduces the expression of enamel proteins and cytokines in an ameloblast-derived cell line. Arch. Oral Biol. 2010, 56, 324–330. [Google Scholar]
- Sharma, R.; Tsuchiya, M.; Skobe, Z.; Tannous, B.A.; Bartlett, J.D. The acid test of fluoride: How pH modulates toxicity. PLoS One 2010, 5, e10895. [Google Scholar]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell. Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.J.; Anderson, P.; Kaufman, R.J. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar]
- Wehner, K.A.; Schutz, S.; Sarnow, P. OGFOD1, a novel modulator of eukaryotic translation initiation factor 2alpha phosphorylation and the cellular response to stress. Mol. Cell. Biol. 2010, 30, 2006–2016. [Google Scholar] [CrossRef]
- Flora, S.J.; Mittal, M.; Pachauri, V.; Dwivedi, N. A possible mechanism for combined arsenic and fluoride induced cellular and DNA damage in mice. Metallomics 2012, 4, 78–90. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar]
- Sasaki, S.; Takagi, T.; Suzuki, M. Cyclical changes in pH in bovine developing enamel as sequential bands. Arch. Oral Biol. 1991, 36, 227–231. [Google Scholar] [CrossRef]
- Tsuji, T.; Onuma, K.; Yamamoto, A.; Iijima, M.; Shiba, K. Direct transformation from amorphous to crystalline calcium phosphate facilitated by motif-programmed artificial proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 16866–16870. [Google Scholar]
- Smith, C.E.; Chong, D.L.; Bartlett, J.D.; Margolis, H.C. Mineral acquisition rates in developing enamel on maxillary and mandibular incisors of rats and mice: Implications to extracellular acid loading as apatite crystals mature. J. Bone Miner. Res. 2005, 20, 240–249. [Google Scholar]
- Kawase, T.; Suzuki, A. Studies on the transmembrane migration of fluoride and its effects on proliferation of L-929 fibroblasts (L cells) in vitro. Arch. Oral Biol. 1989, 34, 103–107. [Google Scholar] [CrossRef]
- He, H.; Ganapathy, V.; Isales, C.M.; Whitford, G.M. pH-dependent fluoride transport in intestinal brush border membrane vesicles. Biochim. Biophys. Acta 1998, 1372, 244–254. [Google Scholar]
- Zhou, R.; Zaki, A.E.; Eisenmann, D.R. Morphometry and autoradiography of altered rat enamel protein processing due to chronic exposure to fluoride. Arch. Oral Biol. 1996, 41, 739–747. [Google Scholar] [CrossRef]
- Lyaruu, D.M.; de Jong, M.; Bronckers, A.L.; Wöltgens, J.H. Ultrastructure of in-vitro recovery of mineralization capacity of fluorotic enamel matrix in hamster tooth germs pre-exposed to fluoride in organ culture during the secretory phase of amelogenesis. Arch. Oral Biol. 1987, 32, 107–115. [Google Scholar] [CrossRef]
- Denbesten, P.K.; Crenshaw, M.A.; Wilson, M.H. Changes in the fluoride-induced modulation of maturation stage ameloblasts of rats. J. Dent. Res. 1985, 64, 1365–1370. [Google Scholar] [CrossRef]
- Smith, C.E.; Nanci, A.; Denbesten, P.K. Effects of chronic fluoride exposure on morphometric parameters defining the stages of amelogenesis and ameloblast modulation in rat incisors. Anat. Rec. 1993, 237, 243–258. [Google Scholar] [CrossRef]
- Whitford, G.M.; Reynolds, K.E. Plasma and developing enamel fluoride concentrations during chronic acid-base disturbances. J. Dent. Res. 1979, 58, 2058–2065. [Google Scholar] [CrossRef]
- Reynolds, K.E.; Whitford, G.M.; Pashley, D.H. Acute fluoride toxicity: the influence of acid-base status. Toxicol. Appl. Pharmacol. 1978, 45, 415–427. [Google Scholar] [CrossRef]
- Whitford, G.M.; Angmar-Månsson, B. Fluorosis-like effects of acidosis, but not NH4+, on rat incisor enamel. Caries Res. 1995, 29, 20–25. [Google Scholar] [CrossRef]
- Angmar-Månsson, B.; Lindh, U.; Whitford, G.M. Enamel and dentin fluoride levels and fluorosis following single fluoride doses: A nuclear microprobe study. Caries Res. 1990, 24, 258–262. [Google Scholar] [CrossRef]
- Den Besten, P.K. Effects of fluoride on protein secretion and removal during enamel development in the rat. J. Dent. Res. 1986, 65, 1272–1277. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sierant, M.L.; Bartlett, J.D. Stress Response Pathways in Ameloblasts: Implications for Amelogenesis and Dental Fluorosis. Cells 2012, 1, 631-645. https://doi.org/10.3390/cells1030631
Sierant ML, Bartlett JD. Stress Response Pathways in Ameloblasts: Implications for Amelogenesis and Dental Fluorosis. Cells. 2012; 1(3):631-645. https://doi.org/10.3390/cells1030631
Chicago/Turabian StyleSierant, Megan L., and John D. Bartlett. 2012. "Stress Response Pathways in Ameloblasts: Implications for Amelogenesis and Dental Fluorosis" Cells 1, no. 3: 631-645. https://doi.org/10.3390/cells1030631