Updates from the Intestinal Front Line: Autophagic Weapons against Inflammation and Cancer

{kind=link}
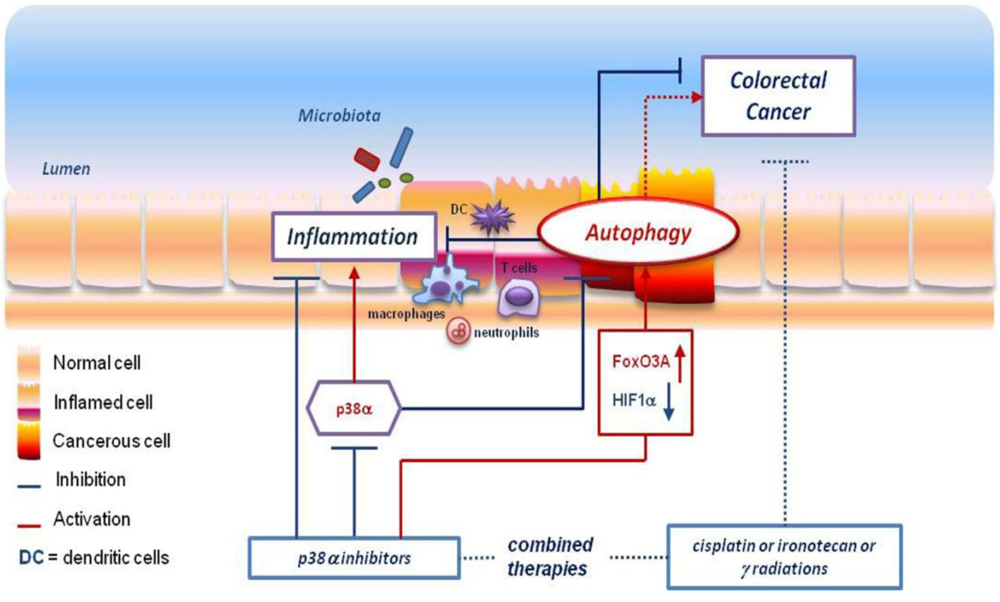
Abstract
:Abbreviations
| CRC | colorectal cancer |
| IBD | inflammatory bowel disease |
| UC | ulcerative colitis |
| CD | Crohn’s disease |
1. Notes on Colorectal Cancer
1.1. Epidemiology and Genetics
1.2. Environmental Risk Factors in CRC Development
2. Role of Autophagy in Intestinal Inflammation and Colorectal Cancer
2.1. From Inflammatory Disorders to Neoplasia
2.2. Autophagy as a Line of Defense to Environmental Insults: The Immune System Strategy

2.3. Autophagy as a Tactic to Survive: The Colorectal Cancer Strategy
2.4. Autophagy and Apoptosis: Dual Role in Colorectal Cancer
2.5 Regulators in the Autophagic Response of Colorectal Cancer
3. Modulation of Autophagy: New Strategies to Fight CRC
3.1. Current Therapies
3.2. New Approaches
4. Conclusion
Acknowledgments
Conflict of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef]
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in Europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar] [CrossRef]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar]
- Migheli, F.; Migliore, L. Epigenetics of colorectal cancer. Clin. Genet. 2012, 81, 312–318. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. New Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomaki, P.; Sistonen, P.; Aaltonen, L.A.; Nystrom-Lahti, M.; et al. Mutations of a muts homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar]
- Issa, J.P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef]
- Kondo, Y.; Issa, J.P. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 29–39. [Google Scholar] [CrossRef]
- Nosho, K.; Irahara, N.; Shima, K.; Kure, S.; Kirkner, G.J.; Schernhammer, E.S.; Hazra, A.; Hunter, D.J.; Quackenbush, J.; Spiegelman, D. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One 2008, 3, e3698. [Google Scholar]
- Howe, J.R.; Roth, S.; Ringold, J.C.; Summers, R.W.; Jarvinen, H.J.; Sistonen, P.; Tomlinson, I.P.; Houlston, R.S.; Bevan, S.; Mitros, F.A.; et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 280, 1086–1088. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Kinzler, K.W; Vogelstein, B. Life (and death) in a malignant tumour. Nature 1996, 379, 9–20. [Google Scholar] [CrossRef]
- National Cancer Institue Database. Available online: http://seer.cancer.gov/csr/1975_2009_pops09/ (accessed on 30 April 2012).
- Gruber, S.B.; Ellis, N.A.; Scott, K.K.; Almog, R.; Kolachana, P.; Bonner, J.D.; Kirchhoff, T.; Tomsho, L.P.; Nafa, K.; Pierce, H.; et al. BLM heterozygosity and the risk of colorectal cancer. Science 2002, 297, 2013. [Google Scholar]
- International Agency for Research on Caner, Fruit and Vegetables; Vainio, H.; Bianchini, F. (Eds.) International Agency for Research on Caner: Lyon, France, 2003.
- Bianchini, F.; Kaaks, R.; Vainio, H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002, 3, 565–574. [Google Scholar] [CrossRef]
- National Cancer Policy Board, Fulfilling the Potential of Cancer Prevention and Early Detection; National Academies Press: Washington, DC, USA, 2003.
- La Vecchia, C.; Gallus, S.; Talamini, R.; Decarli, A.; Negri, E.; Franceschi, S. Interaction between selected environmental factors and familial propensity for colon cancer. Eur. J. Cancer Prev. 1999, 8, 147–150. [Google Scholar] [CrossRef]
- Hall, E.H.; Crowe, S.E. Environmental and lifestyle influences on disorders of the large and small intestine: Implications for treatment. Dig. Dis. 2011, 29, 249–254. [Google Scholar] [CrossRef]
- Vannucci, L. To suppress to rescue? Changing the approach for recalling anticancer immune responses. Front. Biosci. (Schol. Ed.) 2010, 2, 1189–1197. [Google Scholar] [CrossRef]
- Blaut, M. Ecology and Physiology of the Intestinal Tract. Curr. Top. Microbiol. Immunol. 2011. [Google Scholar] [CrossRef]
- Possemiers, S.; Grootaert, C.; Vermeiren, J.; Gross, G.; Marzorati, M.; Verstraete, W.; Van de Wiele, T. The intestinal environment in health and disease—Recent insights on the potential of intestinal bacteria to influence human health. Curr. Pharm. Des. 2009, 15, 2051–2065. [Google Scholar] [CrossRef]
- Jackson, L.; Evers, B.M. Chronic inflammation and pathogenesis of GI and pancreatic cancers. Cancer Treat. Res. 2006, 130, 39–65. [Google Scholar] [CrossRef]
- Tlaskalova-Hogenova, H.; Farre-Castany, M.A.; Stepankova, R.; Kozakova, H.; Tuckova, L.; Funda, D.P.; Barot, R.; Cukrowska, B.; Sinkora, J.; Mandel, L.; et al. The gut as a lymphoepithelial organ: The role of intestinal epithelial cells in mucosal immunity. Folia Microbiol. (Praha) 1995, 40, 385–391. [Google Scholar]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004, 126, 451–459. [Google Scholar] [CrossRef]
- Waldner, M.J.; Neurath, M.F. Cytokines in colitis associated cancer: Potential drug targets? Inflamm. Allergy Drug. Targets 2008, 7, 187–194. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 2008, 118, 560–570. [Google Scholar]
- Seo, G.S. The role of NF-kappaB in colon cancer. Korean J. Gastroenterol. 2011, 57, 3–7. [Google Scholar] [CrossRef]
- Agarwal, S.; Mayer, L. Gastrointestinal manifestations in primary immune disorders. Inflamm. Bowel Dis. 2009, 16, 703–711. [Google Scholar]
- Marks, F.; Furstenberger, G.; Muller-Decker, K. Metabolic targets of cancer chemoprevention: Interruption of tumor development by inhibitors of arachidonic acid metabolism. Recent Res. Cancer 1999, 151, 45–67. [Google Scholar] [CrossRef]
- Kaur, J.; Sanyal, S.N. PI3-kinase/Wnt association mediates COX-2/PGE(2) pathway to inhibit apoptosis in early stages of colon carcinogenesis: Chemoprevention by diclofenac. Tumour Biol. 2010, 31, 623–631. [Google Scholar] [CrossRef]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar]
- Rogler, G.; Brand, K.; Vogl, D.; Page, S.; Hofmeister, R.; Andus, T.; Knuechel, R.; Baeuerle, P.A.; Scholmerich, J.; Gross, V. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 1998, 115, 357–369. [Google Scholar] [CrossRef]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Invest. New Drug. 2009, 29, 207–224. [Google Scholar] [CrossRef]
- Umesalma, S.; Sudhandiran, G. Differential inhibitory effects of the polyphenol ellagic acid on inflammatory mediators NF-kappaB, iNOS, COX-2, TNF-alpha, and IL-6 in 1,2-dimethylhydrazine-induced rat colon carcinogenesis. Basic Clin. Pharmacol. 2010, 107, 650–625. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2009, 20, 5–71. [Google Scholar]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth F.R. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Sumpter, R., Jr.; Levine, B. Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell Dev. Biol. 2010, 21, 699–711. [Google Scholar]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd ed.). Autophagy 2011, 7, 1273–1294. [Google Scholar]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Kuballa, P.; Huett, A.; Rioux, J.D.; Daly, M.J.; Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS One 2008, 3, e3391. [Google Scholar]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nunez, G.; Flavell, R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Kabi, A.; Nickerson, K.P.; Homer, C.R.; McDonald, C. Digesting the genetics of inflammatory bowel disease: Insights from studies of autophagy risk genes. Inflamm. Bowel Dis. 2011, 18, 782–792. [Google Scholar]
- Waterman, M.; Xu, W.; Stempak, J.M.; Milgrom, R.; Bernstein, C.N.; Griffiths, A.M.; Greenberg, G.R.; Steinhart, A.H.; Silverberg, M.S. Distinct and overlapping genetic loci in Crohn's disease and ulcerative colitis: Correlations with pathogenesis. Inflamm. Bowel Dis. 2011, 17, 1936–1942. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Pei, F.H.; Wang, Y.J.; Gao, S.L.; Liu, B.R.; Du, Y.J.; Liu, W.; Yu, H.Y.; Zhao, L.X.; Chi, B.R. Vitamin D receptor gene polymorphism and ulcerative colitis susceptibility in Han Chinese. J. Dig. Dis. 2011, 12, 90–98. [Google Scholar] [CrossRef]
- Naderi, N.; Farnood, A.; Habibi, M.; Derakhshan, F.; Balaii, H.; Motahari, Z.; Agah, M.R.; Firouzi, F.; Rad, M.G.; Aghazadeh, R.; et al. Association of vitamin D receptor gene polymorphisms in Iranian patients with inflammatory bowel disease. J. Gastroen. Hepatol. 2008, 23, 1816–1822. [Google Scholar] [CrossRef]
- Dresner-Pollak, R.; Ackerman, Z.; Eliakim, R.; Karban, A.; Chowers, Y.; Fidder, H.H. The BsmI vitamin D receptor gene polymorphism is associated with ulcerative colitis in Jewish Ashkenazi patients. Genet. Test. 2004, 8, 417–420. [Google Scholar] [CrossRef]
- Wang, T.T.; Dabbas, B.; Laperriere, D.; Bitton, A.J.; Soualhine, H.; Tavera-Mendoza, L.E.; Dionne, S.; Servant, M.J.; Bitton, A.; Seidman, E.G.; et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J. Biol. Chem. 2010, 285, 2227–2231. [Google Scholar]
- Groulx, J.-F.; Khalfaoui, T.; Benoit, Y.D.; Bernatchez, G.; Carrier, J.C.; Basora, N.; Beaulieu, J.-F. Autophagy is active in normal colon mucosa. Autophagy 2012, in press. [Google Scholar]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar]
- Knaevelsrud, H.; Ahlquist, T.; Merok, M.A.; Nesbakken, A.; Stenmark, H.; Lothe, R.A.; Simonsen, A. UVRAG mutations associated with microsatellite unstable colon cancer do not affect autophagy. Autophagy 2010, 6, 863–870. [Google Scholar]
- Kim, M.J.; Woo, S.J.; Yoon, C.H.; Lee, J.S.; An, S.; Choi, Y.H.; Hwang, S.G.; Yoon, G.; Lee, S.J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Ogier-Denis, E.; Houri, J.J.; Bauvy, C.; Codogno, P. Guanine nucleotide exchange on heterotrimeric Gi3 protein controls autophagic sequestration in HT-29 cells. J. Biol. Chem. 1996, 271, 28593–28600. [Google Scholar]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef]
- Diaz-Troya, S.; Perez-Perez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865. [Google Scholar]
- Habeeb, B.S.; Kitayama, J.; Nagawa, H. Adiponectin supports cell survival in glucose deprivation through enhancement of autophagic response in colorectal cancer cells. Cancer Sci. 2011, 102, 999–1006. [Google Scholar]
- Ogier-Denis, E.; Pattingre, S.; El Benna, J.; Codogno, P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J. Biol. Chem. 2000, 275, 39090–39095. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Sato, K.; Tsuchihara, K.; Fujii, S.; Sugiyama, M.; Goya, T.; Atomi, Y.; Ueno, T.; Ochiai, A.; Esumi, H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007, 67, 9677–9684. [Google Scholar]
- Watson, A.J. An overview of apoptosis and the prevention of colorectal cancer. Crit. Rev. Oncol. Hemat. 2006, 57, 107–121. [Google Scholar] [CrossRef]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Kutikhin, A.G. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum. Immunol. 2011, 72, 55–68. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef]
- Livesey, K.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J.; Li, L.; et al. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Marino, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology (Williston Park) 2011, 25, 400–410,413. [Google Scholar]
- Wang, S.; Mintz, A.; Mochizuki, K.; Dorsey, J.F.; Ackermann, J.M.; Alavi, A.; El-Deiry, W.S. Multimodality optical imaging and 18F-FDG uptake in wild-type p53-containing and p53-null human colon tumor xenografts. Cancer Biol.Ther. 2007, 6, 1649–1653. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2010, 1807, 568–576. [Google Scholar]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar]
- Rodriguez-Enriquez, S.; Vital-Gonzalez, P.A.; Flores-Rodriguez, F.L.; Marin-Hernandez, A.; Ruiz-Azuara, L.; Moreno-Sanchez, R. Control of cellular proliferation by modulation of oxidative phosphorylation in human and rodent fast-growing tumor cells. Toxicol. Appl. Pharm. 2006, 215, 208–217. [Google Scholar] [CrossRef]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar]
- Pasdois, P.; Deveaud, C.; Voisin, P.; Bouchaud, V.; Rigoulet, M.; Beauvoit, B. Contribution of the phosphorylable complex I in the growth phase-dependent respiration of C6 glioma cells in vitro. J. Bioenerg. Biomembr. 2003, 35, 439–450. [Google Scholar] [CrossRef]
- Marino, G.; Kroemer, G. Ammonia: A diffusible factor released by proliferating cells that induces autophagy. Sci. Signal. 2010, 3, e19. [Google Scholar]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Comes, F.; Matrone, A.; Lastella, P.; Nico, B.; Susca, F.C.; Bagnulo, R.; Ingravallo, G.; Modica, S.; Lo Sasso, G.; Moschetta, A.; et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007, 14, 693–702. [Google Scholar] [CrossRef]
- Matrone, A.; Grossi, V.; Chiacchiera, F.; Fina, E.; Cappellari, M.; Caringella, A.M.; Di Naro, E.; Loverro, G.; Simone, C. p38alpha is required for ovarian cancer cell metabolism and survival. Int. J. Gynecol. Cancer 2010, 20, 203–211. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. Signal-dependent regulation of gene expression as a target for cancer treatment: inhibiting p38alpha in colorectal tumors. Cancer Lett. 2008, 265, 16–26. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009, 5, 1030–1033. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Matrone, A.; Ferrari, E.; Ingravallo, G.; Lo Sasso, G.; Murzilli, S.; Petruzzelli, M.; Salvatore, L.; Moschetta, A.; Simone, C.; et al. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009, 16, 1203–1214. [Google Scholar] [CrossRef]
- Blouin, C.C.; Page, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood 2004, 103, 1124–1130. [Google Scholar]
- Hellwig-Burgel, T.; Stiehl, D.P.; Wagner, A.E.; Metzen, E.; Jelkmann, W. Review: Hypoxia-inducible factor-1 (HIF-1): A novel transcription factor in immune reactions. J. Interferon Cytokine Res. 2005, 25, 297–310. [Google Scholar] [CrossRef]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha-deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Lazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–950. [Google Scholar]
- Shacka, J.J.; Klocke, B.J.; Roth, K.A. Autophagy, bafilomycin and cell death: The "a-B-cs" of plecomacrolide-induced neuroprotection. Autophagy 2006, 2, 228–230. [Google Scholar]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F. Autophagy modulation for cancer therapy. Cancer Biol. Ther. 2011, 11, 169–176. [Google Scholar] [CrossRef]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Guo, X.L.; Bu, X.X.; Zhang, S.S.; Ma, N.N.; Song, J.R.; Hu, F.; Tao, S.F.; Sun, K.; Li, R.; et al. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Cancer Lett. 2010, 288, 68–74. [Google Scholar] [CrossRef]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A., 2nd; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell Mol. Med. 2010, 14, 2448–2459. [Google Scholar]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A., 2nd; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2010, 286, 6602–6613. [Google Scholar]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Psahoulia, F.H.; Moumtzi, S.; Roberts, M.L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. Quercetin mediates preferential degradation of oncogenic Ras and causes autophagy in Ha-RAS-transformed human colon cells. Carcinogenesis 2007, 28, 1021–1031. [Google Scholar]
- Yuk, J.M.; Shin, D.M.; Song, K.S.; Lim, K.; Kim, K.H.; Lee, S.H.; Kim, J.M.; Lee, J.S.; Paik, T.H.; Kim, J.S.; et al. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy 2010, 6, 46–60. [Google Scholar] [CrossRef]
- Kauntz, H.; Bousserouel, S.; Gosse, F.; Raul, F. Silibinin triggers apoptotic signaling pathways and autophagic survival response in human colon adenocarcinoma cells and their derived metastatic cells. Apoptosis 2011, 16, 1042–1053. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yogosawa, S.; Izutani, Y.; Watanabe, H.; Otsuji, E.; Sakai, T. A combination of indol-3-carbinol and genistein synergistically induces apoptosis in human colon cancer HT-29 cells by inhibiting Akt phosphorylation and progression of autophagy. Mol. Cancer 2009, 8, 100. [Google Scholar] [CrossRef]
- Villegas, I.; Sanchez-Fidalgo, S.; Alarcon de la Lastra, C. New mechanisms and therapeutic potential of curcumin for colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1040–1061. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. Signal-dependent control of autophagy-related gene expression. Methods Enzymol. 2009, 453, 305–324. [Google Scholar] [CrossRef]
- Fernandez de Mattos, S.; Villalonga, P.; Clardy, J.; Lam, E.W. FOXO3a mediates the cytotoxic effects of cisplatin in colon cancer cells. Mol. Cancer Ther. 2008, 7, 3237–3246. [Google Scholar] [CrossRef]
- Paillas, S.; Boissiere, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vie, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar]
- Bajbouj, K.; Poehlmann, A.; Kuester, D.; Drewes, T.; Haase, K.; Hartig, R.; Teller, A.; Kliche, S.; Walluscheck, D.; Ivanovska, J.; et al. Identification of phosphorylated p38 as a novel DAPK-interacting partner during TNFalpha-induced apoptosis in colorectal tumor cells. Am. J. Pathol. 2009, 175, 557–570. [Google Scholar] [CrossRef]
- Lee, M.R.; Dominguez, C. MAP kinase p38 inhibitors: Clinical results and an intimate look at their interactions with p38alpha protein. Curr. Med. Chem. 2005, 12, 2979–2994. [Google Scholar] [CrossRef]
- Yang, J.C.; Chien, C.T. A new approach for the prevention and treatment of Helicobacter pylori infection via upregulation of autophagy and downregulation of apoptosis. Autophagy 2009, 5, 413–414. [Google Scholar] [CrossRef]
- Barton, M.K. Daily aspirin reduces colorectal cancer incidence in patients with Lynch syndrome. CA-Cancer J. Clin. 2012, 62, 143–144. [Google Scholar]
- Lee, C.S.; McNamara, D.; O'Morain, C.A. Aspirin as a chemoprevention agent for Colorectal cancer. Curr. Drug Metab. 2012, in press. [Google Scholar]
- Bosetti, C.; Rosato, V.; Gallus, S.; Cuzick, J.; La Vecchia, C. Aspirin and cancer risk: A quantitative review to 2011. Ann. Oncol. 2012, 23, 403–415. [Google Scholar]
- Kmietowicz, Z. Study finds possible role for aspirin as treatment for colon cancer. BMJ 2012, 344, e2988. [Google Scholar] [CrossRef]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin Inhibits mTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Madia, F.; Grossi, V.; Peserico, A.; Simone, C. Updates from the Intestinal Front Line: Autophagic Weapons against Inflammation and Cancer. Cells 2012, 1, 535-557. https://doi.org/10.3390/cells1030535
Madia F, Grossi V, Peserico A, Simone C. Updates from the Intestinal Front Line: Autophagic Weapons against Inflammation and Cancer. Cells. 2012; 1(3):535-557. https://doi.org/10.3390/cells1030535
Chicago/Turabian StyleMadia, Federica, Valentina Grossi, Alessia Peserico, and Cristiano Simone. 2012. "Updates from the Intestinal Front Line: Autophagic Weapons against Inflammation and Cancer" Cells 1, no. 3: 535-557. https://doi.org/10.3390/cells1030535
APA StyleMadia, F., Grossi, V., Peserico, A., & Simone, C. (2012). Updates from the Intestinal Front Line: Autophagic Weapons against Inflammation and Cancer. Cells, 1(3), 535-557. https://doi.org/10.3390/cells1030535