Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Autophagy: A Triple-Faced Coin
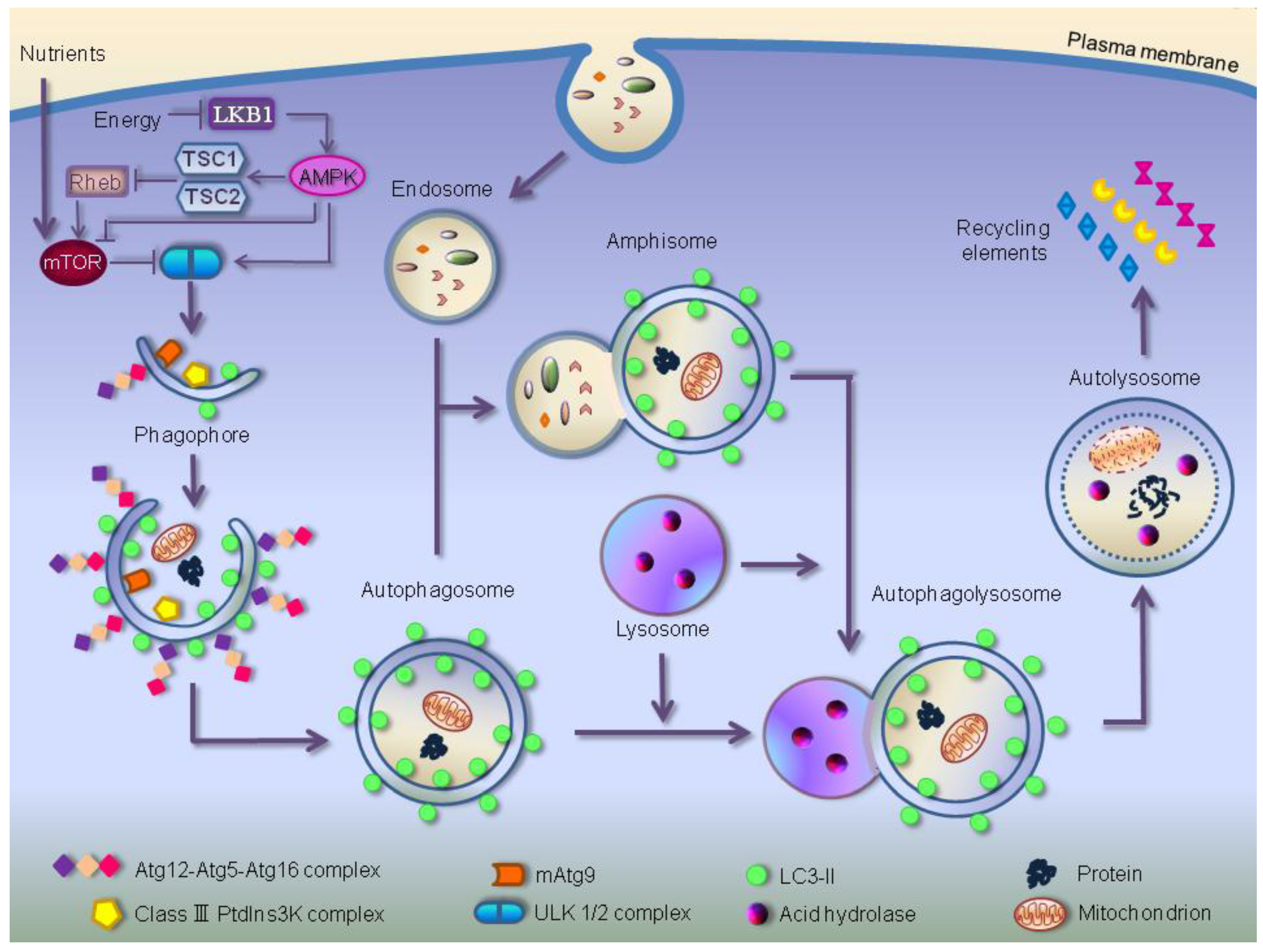
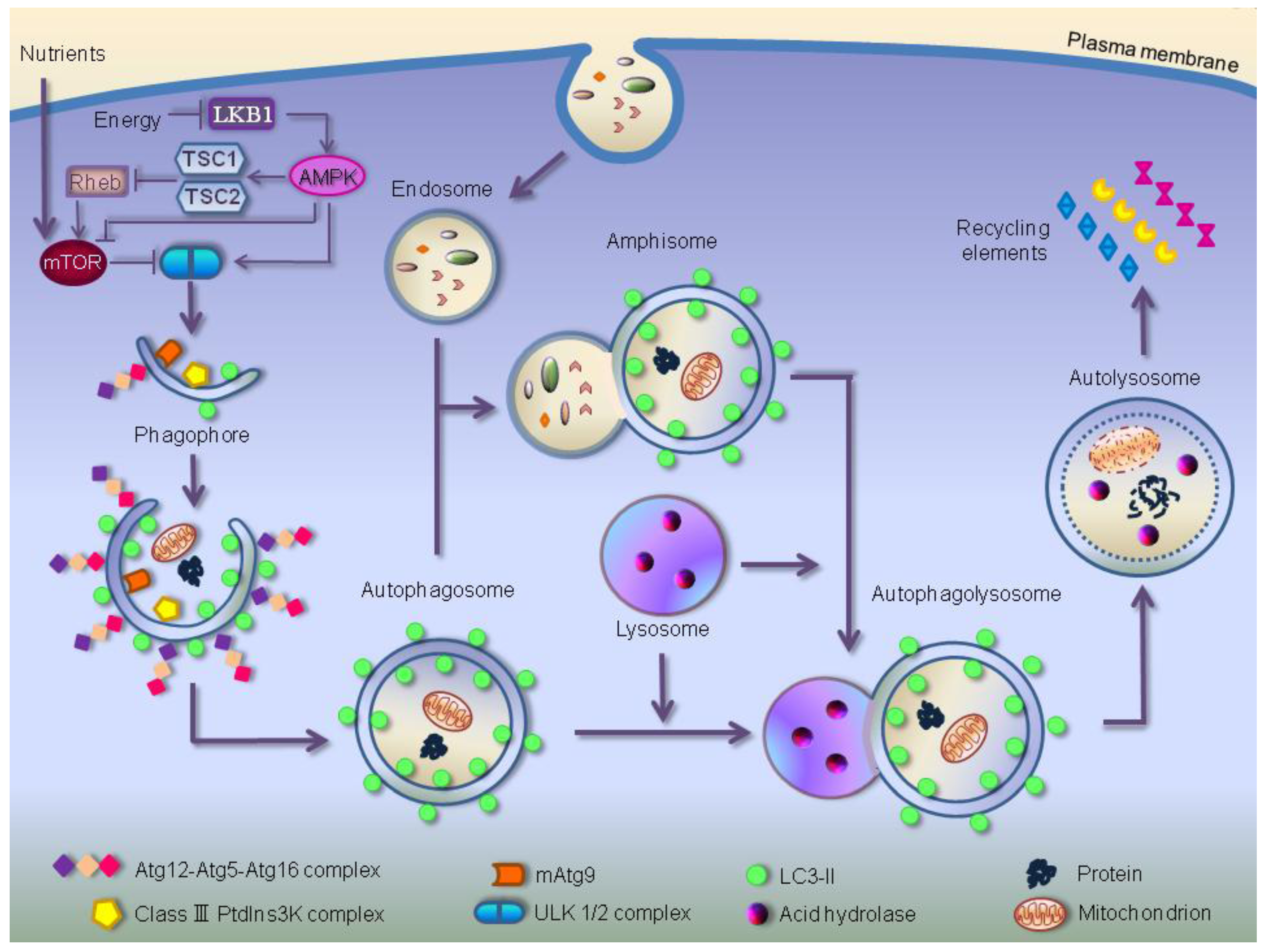
3. Autophagic Molecular Core Machinery
4. Autophagy: From Survival Program to Cell Death Mechanism
4.1. Autophagy between Life and Death
4.2. Cross-Talk between Autophagy and Apoptosis
5. Autophagy and Cancer
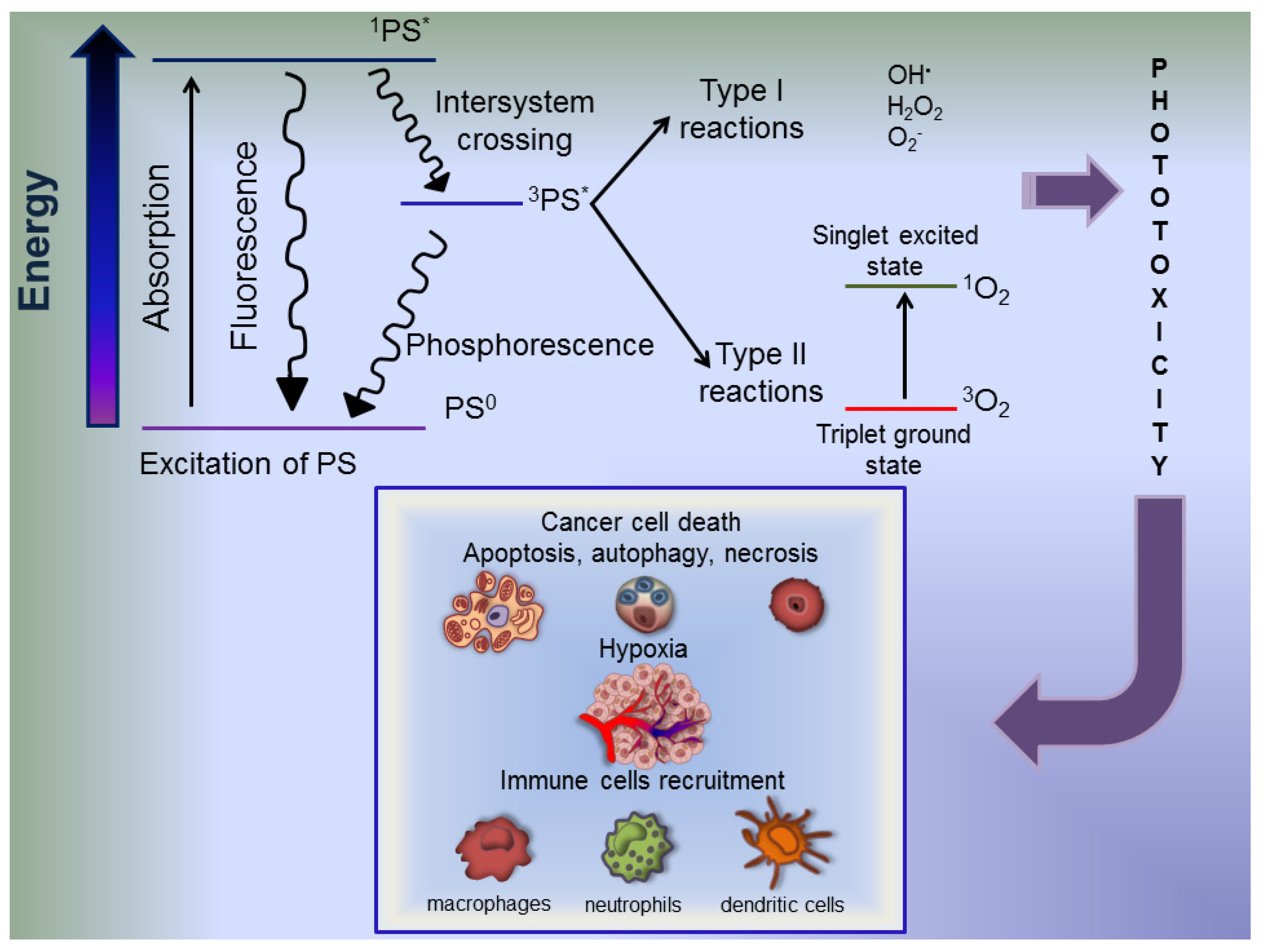
6. Photodynamic Therapy Fundamental Aspects
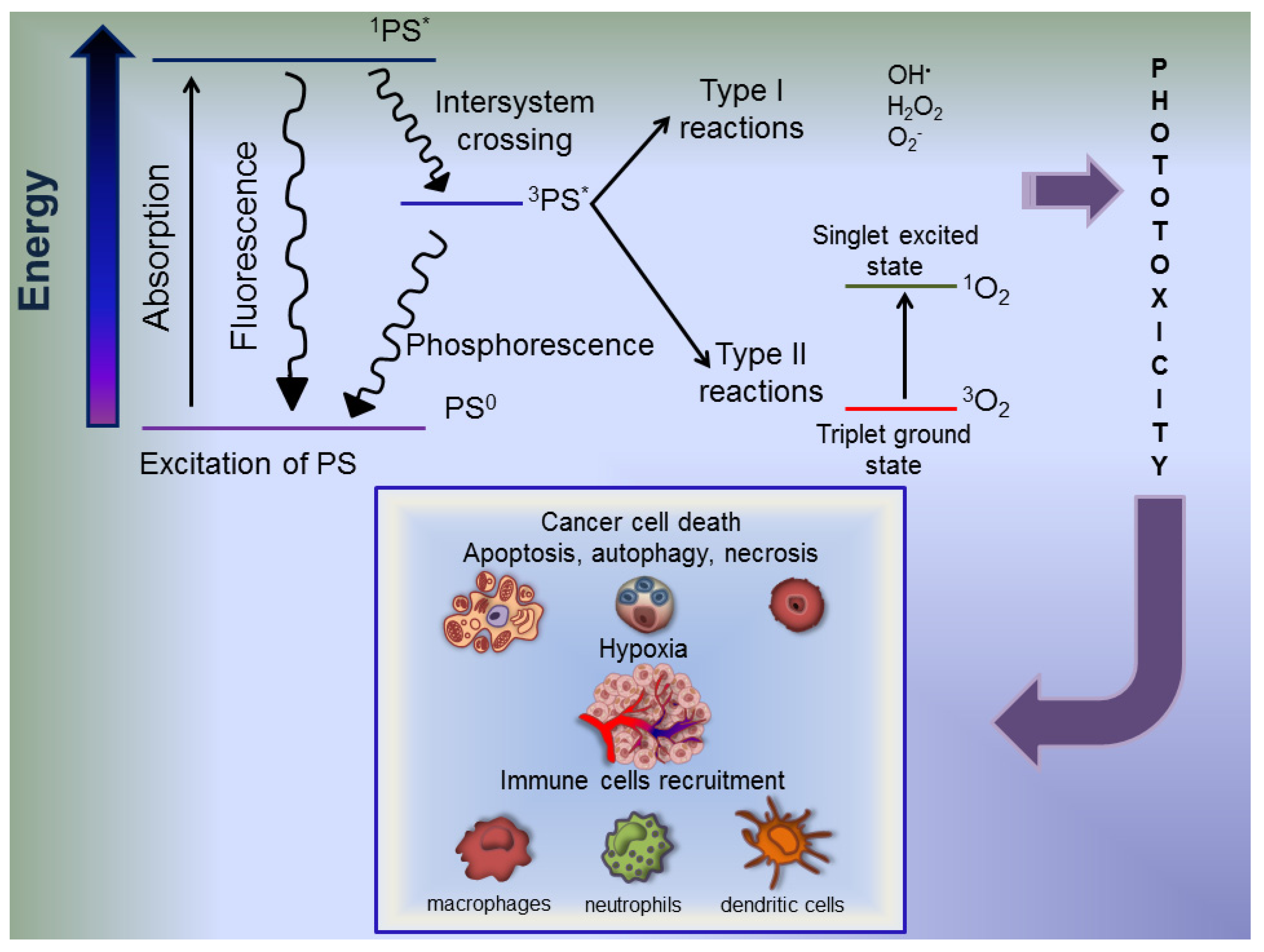
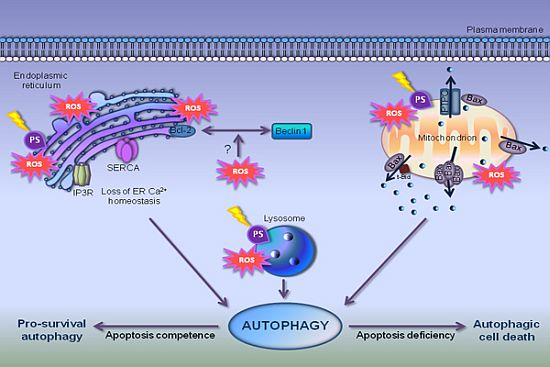
7. Autophagy in PDT-Photosensitized Cells
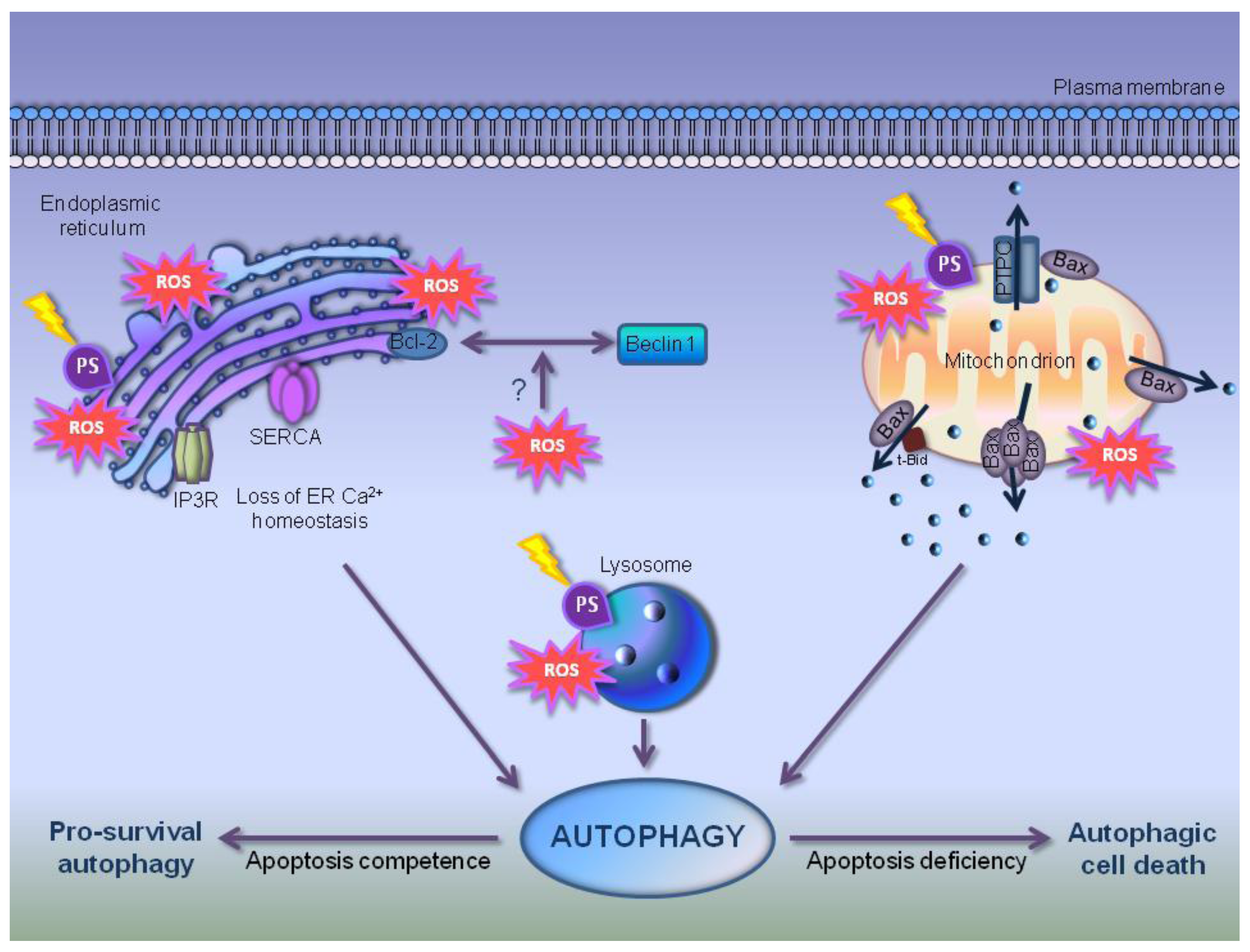
8. Does Autophagy Concur to Elicit an Immunogenic Impact in Photosensitized Cells?
9. Conclusions
Conflict of Interest
References
- Kessel, D.; Oleinick, N.L. Photodynamic therapy and cell death pathways. Methods Mol. Biol. 2010, 635, 35–46. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-Known self-eating. Cell. Mol. Life Sci. 2011, 69, 1125–1136. [Google Scholar]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULKAtg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef]
- Lindmo, K.; Stenmark, H. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 2006, 119, 605–614. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. Quantitative regulation of vesicle formation in yeast nonspecific autophagy. Autophagy 2008, 4, 955–957. [Google Scholar]
- Ohsumi, Y.; Mizushima, N. Two ubiquitin like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 2004, 15, 231–236. [Google Scholar]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–3100. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef]
- Winchester, B.G. Lysosomal membrane proteins. Eur. J. Paediatr. Neurol. 2001, 5, 11–19. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 8, 924–926. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010, 5, e15394. [Google Scholar]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy - unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohsumi, Y. Current knowledge of the pre-autophagosomal structure (PAS). FEBS Lett. 2010, 584, 1280–1286. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgó, J.; Díaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef]
- Khan, M.T.; Joseph, S.K. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 2010, 285, 16912–16920. [Google Scholar]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Harr, M.W.; McColl, K.S.; Zhong, F.; Molitoris, J.K.; Distelhorst, C.W. Glucocorticoids downregulate Fyn and inhibit IP(3)-mediated calcium signaling to promote autophagy in T lymphocytes. Autophagy 2010, 6, 912–921. [Google Scholar] [CrossRef]
- Gao, W.; Ding, W.X.; Stolz, D.B.; Yin, X.M. Induction of macroautophagy by exogenously introduced calcium. Autophagy 2008, 6, 754–761. [Google Scholar]
- Lam, D.; Kosta, A.; Luciani, M.F.; Golstein, P. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 2008, 19, 691–700. [Google Scholar]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar]
- Gastaldello, A.; Callaghan, H.; Gami, P.; Campanella, M. Ca (2+) -dependent autophagy is enhanced by the pharmacological agent PK11195. Autophagy 2010, 6, 607–613. [Google Scholar] [CrossRef]
- Grotemeier, A.; Alers, S.; Pfisterer, S.G.; Paasch, F.; Daubrawa, M.; Dieterle, A.; Viollet, B.; Wesselborg, S.; Proikas-Cezanne, T.; Stork, B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010, 22, 914–925. [Google Scholar] [CrossRef]
- Law, B.Y.; Wang, M.; Ma, D.L.; Al-Mousa, F.; Michelangeli, F.; Cheng, S.H.; Ng, M.H.; To, K.F.; Mok, A.Y.; Ko, R.Y.; et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 2010, 9, 718–730. [Google Scholar]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgó, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.B. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef]
- Cárdenas, C.; Foskett, J.K. Mitochondrial Ca(2+) signals in autophagy. Cell Calcium 2012, 52, 44–51. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. Lethal weapons: DAP-kinase, autophagy and cell death: DAP-kinase regulates autophagy. Curr. Opin. Cell Biol. 2010, 22, 199–205. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar]
- Kim, H.J.; Soyombo, A.A.; Tjon-Kon-Sang, S.; So, I.; Muallem, S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic 2009, 10, 1157–1167. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Taylor, M.P.; Jackson, W.T. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004, 2, 301–314. [Google Scholar] [CrossRef]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef]
- Colell, A.; Ricci, J.E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007, 129, 983–997. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar]
- González-Polo, R.A.; Boya, P.; Pauleau, A.L.; Jalil, A.; Larochette, N.; Souquère, S.; Eskelinen, E.L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci. 2005, 118, 3091–3102. [Google Scholar]
- Chen, Y.; Gibson, S.B. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy 2008, 4, 246–248. [Google Scholar]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar]
- Li, C.; Capan, E.; Zhao, Y.; Zhao, J.; Stolz, D.; Watkins, S.C.; Jin, S.; Lu, B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 2006, 177, 5163–5168. [Google Scholar]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar]
- Xu, Y.; Kim, S.O.; Li, Y.; Han, J. Autophagy contributes to caspase-independent macrophage cell death. J. Biol. Chem. 2006, 281, 19179–19187. [Google Scholar]
- Djavaheri Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF{kappa}B Activation Represses Tumor Necrosis Factor-{alpha}-induced Autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Freeman, M.; Hallahan, D.E.; Lu, B. Autophagy for cancer therapy through inhibition of proapoptotic proteins and mTOR signaling. J. Biol. Chem. 2006, 281, 36883–36890. [Google Scholar]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Autophagy: An emerging target for cancer therapy. Autophagy 2008, 4, 574–580. [Google Scholar]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Brech, A.; Ahlquist, T.; Lothe, R.A.; Stenmark, H. Autophagy in tumour suppression and promotion. Mol. Oncol. 2009, 3, 366–375. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar]
- Bao, X.H.; Naomoto, Y.; Hao, H.F.; Watanabe, N.; Sakurama, K.; Noma, K.; Motoki, T.; Tomono, Y.; Fukazawa, T.; Shirakawa, Y.; et al. Autophagy: Can it become a potential therapeutic target? Int. J. Mol. Med. 2010, 25, 493–503. [Google Scholar]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1176, 86–107. [Google Scholar]
- Mroz, P.; Hashmi, J.T.; Huang, Y.Y.; Lange, N.; Hamblin, M.R. Stimulation of anti-tumor immunity by photodynamic therapy. Expert Rev. Clin. Immunol. 2011, 7, 75–91. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two-photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar]
- Allison, R.R.; Sibata, C.H. Oncologic photodynamic therapy photosensitizers: A clinical review. Photodiag. Photodyn Ther. 2010, 7, 61–75. [Google Scholar] [CrossRef]
- Juzeniene, A.; Nielsen, K.P.; Moan, J. Biophysical aspects of photodynamic therapy. J. Environ. Pathol. Toxicol. Oncol. 2006, 25, 7–28. [Google Scholar] [CrossRef]
- Brancaleon, L.; Moseley, H. Laser and non-laser light sources for photodynamic therapy. Lasers Med. Sci. 2002, 17, 173–186. [Google Scholar] [CrossRef]
- Plaetzer, K.; Krammer, B.; Berlanda, J.; Berr, F.; Kiesslich, T. Photophysics and photochemistry of photodynamic therapy: Fundamental aspects. Lasers Med. Sci. 2009, 24, 259–268. [Google Scholar] [CrossRef]
- Greer, A. Christopher Foote’s discovery of the role of singlet oxygen [1O2 (1Delta g)] in photosensitized oxidation reactions. Acc. Chem. Res. 2006, 39, 797–804. [Google Scholar] [CrossRef]
- Reiners, J.J., Jr.; Agostinis, P.; Berg, K.; Oleinick, N.L.; Kessel, D. Assessing autophagy in the context of photodynamic therapy. Autophagy 2010, 6, 7–18. [Google Scholar]
- François, A.; Marchal, S.; Guillemin, F.; Bezdetnaya, L. mTHPC-based photodynamic therapy induction of autophagy and apoptosis in cultured cells in relation to mitochondria and endoplasmic reticulum stress. Int. J. Oncol. 2011, 39, 1537–1543. [Google Scholar]
- Buytaert, E.; Callewaert, G.; Hendrickx, N.; Scorrano, L.; Hartmann, D.; Missiaen, L.; Vandenheede, J.R.; Heirman, I.; Grooten, J.; Agostinis, P. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006, 20, 756–758. [Google Scholar]
- Kessel, D.; Reiners, J.J., Jr. Apoptosis and autophagy after mitochondrial or endoplasmic reticulum photodamage. Photochem. Photobiol. 2007, 83, 1024–1028. [Google Scholar] [CrossRef]
- Kessel, D.; Arroyo, A.S. Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem. Photobiol. Sci. 2007, 6, 1290–1295. [Google Scholar] [CrossRef]
- Dini, L.; Inguscio, V.; Tenuzzo, B.; Panzarini, E. Rose bengal acetate photodynamic therapy-induced autophagy. Cancer Biol. Ther. 2010, 10, 1048–1055. [Google Scholar] [CrossRef]
- Panzarini, E.; Inguscio, V.; Dini, L. Timing the multiple cell death pathways initiated by Rose Bengal acetate photodynamic therapy. Cell Death Dis. 2011, 51, 1–11. [Google Scholar]
- Panzarini, E.; Tenuzzo, B.; Palazzo, F.; Chionna, A.; Dini, L. Apoptosis induction and mitochondria alteration in human HeLa tumor cells by photoproducts of Rose Bengal acetate. J. Photochem. Photobiol. 2006, 83, 39–47. [Google Scholar] [CrossRef]
- Panzarini, E.; Tenuzzo, B.; Dini, L. Photodynamic therapy-induced apoptosis of HeLa cells. Ann. N.Y. Acad. Sci. 2009, 1171, 617–626. [Google Scholar]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Dewaele, M.; Maes, H.; Agostinis, P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010, 6, 838–854. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free RadicalBiol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Kessel, D.; Castelli, M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem. Photobiol. 2001, 74, 318–322. [Google Scholar] [CrossRef]
- Xue, L.Y.; Chiu, S.M.; Oleinick, N.L. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2001, 20, 3420–3427. [Google Scholar] [CrossRef]
- Xue, L.Y.; Chiu, S.M.; Fiebig, A.; Andrews, D.W.; Oleinick, N.L. Photodamage to multiple Bcl-xL isoforms by photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 2003, 22, 9197–9204. [Google Scholar]
- Weyergang, A.; Berg, K.; Kaalhus, O.; Peng, Q.; Selbo, P.K. Photodynamic therapy targets the mTOR signaling network in vitro and in vivo. Mol. Pharm. 2009, 6, 255–264. [Google Scholar] [CrossRef]
- Xue, L.Y.; Chiu, S.M.; Azizuddin, K.; Joseph, S.; Oleinick, N.L. Protection by Bcl-2 against apoptotic but not autophagic cell death after photodynamic therapy. Autophagy 2008, 4, 125–127. [Google Scholar]
- Usuda, J.; Azizuddin, K.; Chiu, S.M.; Oleinick, N.L. Association between the photodynamic loss of Bcl-2 and the sensitivity to apoptosis caused by phthalocyanine photodynamic therapy. Photochem. Photobiol. 2003, 78, 1–8. [Google Scholar]
- He, J.; Agarwal, M.L.; Larkin, H.E.; Friedman, L.R.; Xue, L.Y.; Oleinick, N.L. The induction of partial resistance to photodynamic therapy by the protooncogene BCL-2. Photochem. Photobiol. 1996, 64, 845–852. [Google Scholar] [CrossRef]
- Chiu, S.M.; Xue, L.Y.; Usuda, J.; Azizuddin, K.; Oleinick, N.L. Bax is essential for mitochondrion-mediated apoptosis but not for cell death caused by photodynamic therapy. Br. J. Cancer 2003, 89, 1590–1597. [Google Scholar] [CrossRef]
- Xue, L.Y.; Chiu, S.M.; Azizuddin, K.; Joseph, S.; Oleinick, N.L. The death of human cancer cells following photodynamic therapy: Apoptosis competence is necessary for Bcl-2 protection but not for induction of autophagy. Photochem. Photobiol. 2007, 83, 1016–1023. [Google Scholar] [CrossRef]
- Xue, L.Y.; Chiu, S.M.; Oleinick, N.L. Photodynamic therapy-induced death of MCF-7 human breast cancer cells: A role for caspase-3 in the late steps of apoptosis but not for the critical lethal event. Exp. Cell Res. 2001, 263, 145–155. [Google Scholar] [CrossRef]
- Ji, H.T.; Chien, L.T.; Lin, Y.H.; Chien, H.F.; Chen, C.T. 5-ALA mediated photodynamic therapy induces autophagic cell death via AMP-activated protein kinase. Mol. Cancer 2010, 91, 1–11. [Google Scholar]
- Coupienne, I.; Bontems, S.; Dewaele, M.; Rubio, N.; Habraken, Y.; Fulda, S.; Agostinis, P.; Piette, J. NF-kappaB inhibition improves the sensitivity of human glioblastoma cells to 5-aminolevulinic acid-based photodynamic therapy. Biochem. Pharmacol. 2011, 81, 606–616. [Google Scholar]
- Ramamurthy, S.; Ronnett, G.V. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J. Physiol. 2006, 574, 85–93. [Google Scholar] [CrossRef]
- Blázquez, C.; Geelen, M.J.; Velasco, G.; Guzmán, M. The AMP-activated protein kinase prevents ceramide synthesis de novo and apoptosis in astrocytes. FEBS Lett. 2001, 489, 149–153. [Google Scholar] [CrossRef]
- Russell, R.R. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 2004, 114, 495–503. [Google Scholar]
- Dagon, Y.; Avraham, Y.; Berry, E.M. AMPK activation regulates apoptosis, adipogenesis, and lipolysis by eIF2alpha in adipocytes. Biochem. Biophys. Res. Commun. 2006, 340, 43–47. [Google Scholar] [CrossRef]
- Kefas, B.A.; Cai, Y.; Ling, Z.; Heimberg, H.; Hue, L.; Pipeleers, D.; van de Casteele, M. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J. Mol. Endocrinol. 2003, 30, 151–161. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Díaz-Troya, S.; Pérez-Pérez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865. [Google Scholar]
- Dewaele, M.; Martinet, W.; Rubio, N.; Verfaillie, T.; de Witte, P.A.; Piette, J.; Agostinis, P. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell Mol. Med. 2011, 15, 1402–1014. [Google Scholar] [CrossRef]
- Andrzejak, M.; Price, M.; Kessel, D.H. Apoptotic and autophagic responses to photodynamic therapy in 1c1c7 murine hepatoma cells. Autophagy 2011, 7, 979–984. [Google Scholar] [CrossRef]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar]
- Feng, S.; Ma, L.; Yang, Y.; Wu, M. Truncated RIP3 (tRIP3) acts upstream of FADD to induce apoptosis in the human hepatocellular carcinoma cell line QGY-7703. Biochem. Biophys. Res. Commun. 2006, 347, 558–565. [Google Scholar] [CrossRef]
- Coupienne, I.; Fettweis, G.; Piette, J. RIP3 expression induces a death profile change in U2OS osteosarcoma cells after 5-ALA-PDT. Lasers Surg. Med. 2011, 43, 557–564. [Google Scholar]
- Berg, K.; Moan, J. Lysosomes as photochemical targets. Int. J. Cancer 1994, 59, 814–822. [Google Scholar] [CrossRef]
- Reiners, J.J., Jr.; Caruso, J.A.; Mathieu, P.; Chelladurai, B.; Yin, X.M.; Kessel, D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002, 9, 934–944. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K.; Bommer, J.C.; Western, A. Action spectra of phthalocyanines with respect to photosensitization of cells. Photochem. Photobiol. 1992, 56, 171–175. [Google Scholar] [CrossRef]
- Caruso, J.A.; Mathieu, P.A.; Joiakim, A.; Leeson, B.; Kessel, D.; Sloane, B.F.; Reiners, J.J., Jr. Differential susceptibilities of murine hepatoma 1c1c7 and Tao cells to the lysosomal photosensitizer NPe6: Influence of aryl hydrocarbon receptor on lysosomal fragility and protease contents. Mol. Pharmacol. 2004, 65, 1016–1028. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar]
- Arum, C.J.; Gederaas, O.A.; Larsen, E.L.; Randeberg, L.L.; Hjelde, A.; Krokan, H.E.; Svaasand, L.O.; Chen, D.; Zhao, C.M. Tissue responses to hexyl 5-aminolevulinate-induced photodynamic treatment in syngeneic orthotopic rat bladder cancer model: Possible pathways of action. J. Biomed. Opt. 2011, 16, 028001. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Clifton, G.T.; Peoples, G.E. Overcoming cancer immune tolerance and escape. Clin. Cancer Res. 2009, 15, 749–751. [Google Scholar] [CrossRef]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef]
- Vakkila, J.; Lotze, M.T. Inflammation and necrosis promote tumour growth. Nat. Rev. Immunol. 2004, 4, 641–648. [Google Scholar] [CrossRef]
- Tesniere, A.; Apetoh, L.; Ghiringhelli, F.; Joza, N.; Panaretakis, T.; Kepp, O.; Schlemmer, F.; Zitvogel, L.; Kroemer, G. Immunogenic cancer cell death: A key-lock paradigm. Curr. Opin. Immunol. 2008, 20, 504–511. [Google Scholar]
- Thorburn, J.; Horita, H.; Redzic, J.; Hansen, K.; Frankel, A.E.; Thorburn, A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009, 16, 175–183. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-Associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., 3rd; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Brackett, C.M.; Gollnick, S.O. Photodynamic therapy enhancement of anti-tumor immunity. Photochem. Photobiol. Sci. 2011, 10, 649–652. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: Surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef]
- Gollnick, S.O.; Evans, S.S.; Baumann, H.; Owczarczak, B.; Maier, P.; Vaughan, L.; Wang, W.C.; Unger, E.; Henderson, B.W. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br. J. Cancer 2003, 88, 1772–1779. [Google Scholar] [CrossRef]
- Kousis, P.C.; Henderson, B.W.; Maier, P.G.; Gollnick, S.O. Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res. 2007, 67, 10501–10510. [Google Scholar]
- Preise, D.; Oren, R.; Glinert, I.; Kalchenko, V.; Jung, S.; Scherz, A.; Salomon, Y. Systemic antitumor protection by vascular-targeted photodynamic therapy involves cellular and humoral immunity. Cancer Immunol. Immunother. 2009, 58, 71–84. [Google Scholar] [CrossRef]
- Jalili, A.; Makowski, M.; Switaj, T.; Nowis, D.; Wilczynski, G.M.; Wilczek, E.; Chorazy-Massalska, M.; Radzikowska, A.; Maslinski, W.; Biały, L.; et al. Effective photoimmunotherapy of murine colon carcinoma induced by the combination of photodynamic therapy and dendritic cells. Clin. Cancer Res. 2004, 10, 4498–4508. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Inguscio, V.; Panzarini, E.; Dini, L. Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy. Cells 2012, 1, 464-491. https://doi.org/10.3390/cells1030464
Inguscio V, Panzarini E, Dini L. Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy. Cells. 2012; 1(3):464-491. https://doi.org/10.3390/cells1030464
Chicago/Turabian StyleInguscio, Valentina, Elisa Panzarini, and Luciana Dini. 2012. "Autophagy Contributes to the Death/Survival Balance in Cancer PhotoDynamic Therapy" Cells 1, no. 3: 464-491. https://doi.org/10.3390/cells1030464