LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy

Abstract
:1. Introduction

2. Autophagy
3. LC3-Associated Phagocytosis
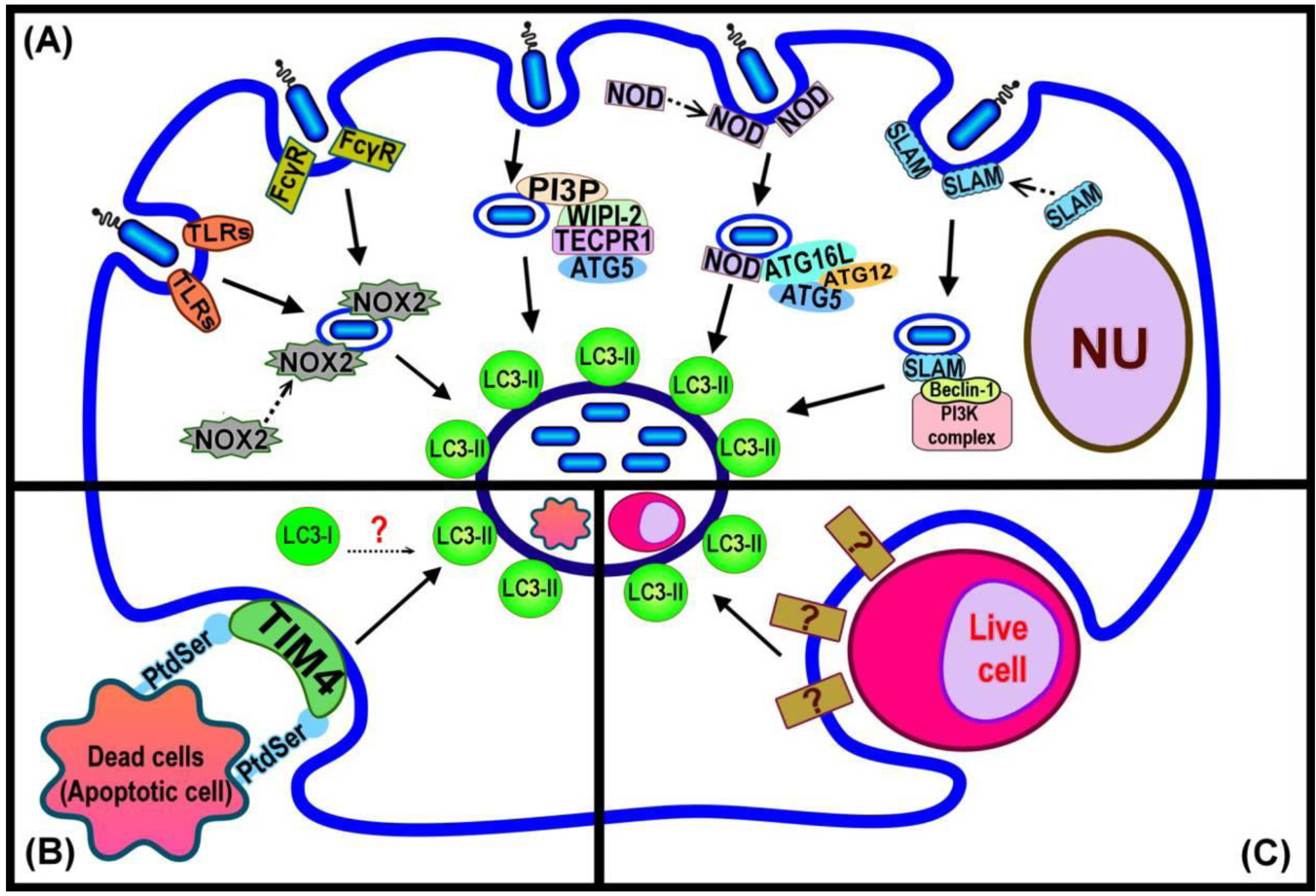
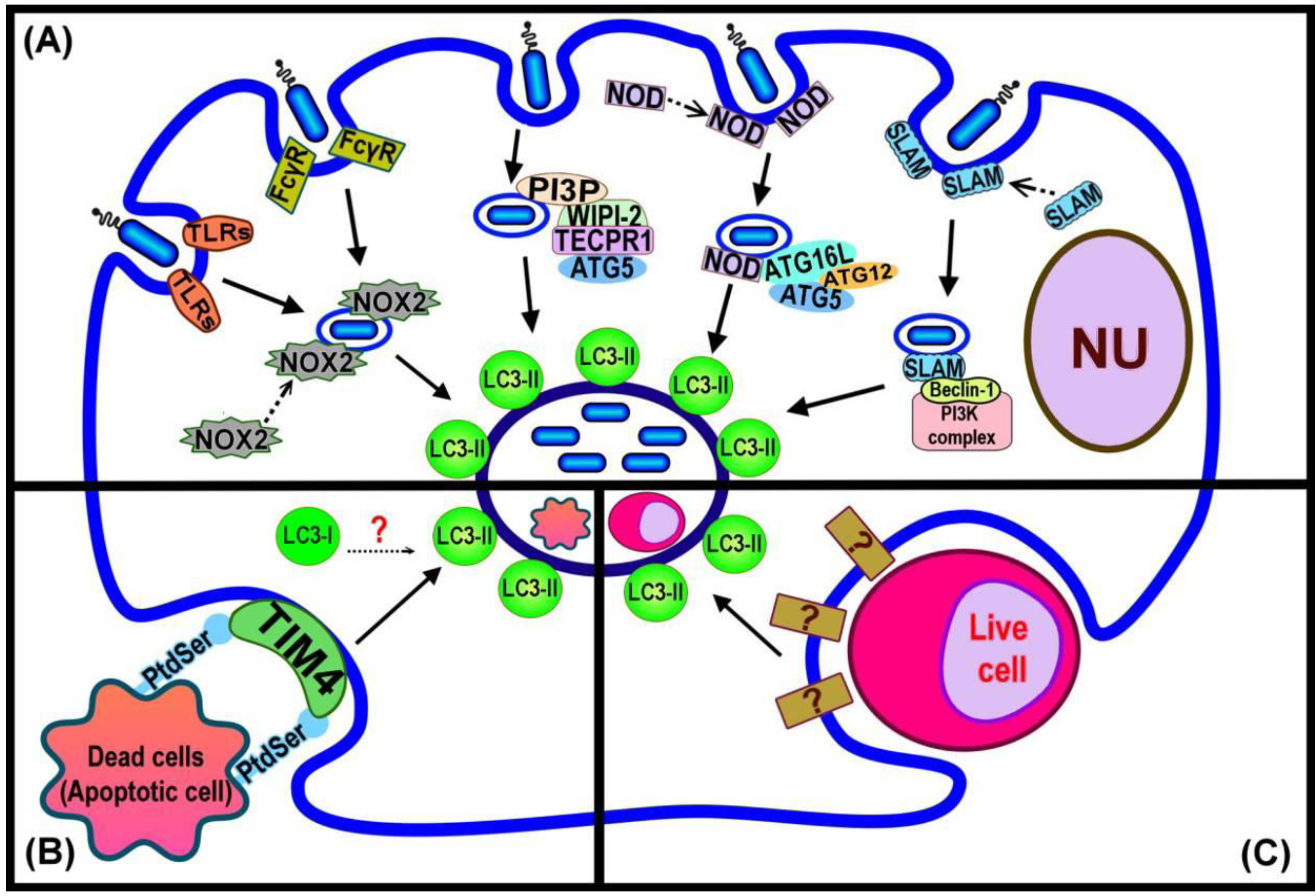
4. Initiating LAP

{kind=link}
{kind=link}
{kind=link}
| Bacterial ligands or cellular signal | Receptor | Link to autophagic-related pathway | Reference |
|---|---|---|---|
| Lipopolysaccharides (LPS) | TLRs (TLR2, TLR4) | Co-localization with LC3 on phagosomal membrane | [18] |
| Peptidoglycan | NLRs (NOD1, NOD2) | Direct binding with Atg16L | [42] |
| Reactive oxygen species (ROS) | TLR2, FcγR | Inhibition of NADPH oxidase decreased LC3 recruitment | [14] |
| Outer membrane proteins C and F of E. coli | SLAM | React with Vps34-Beclin 1-Vps15 complex; possibly direct binding with Beclin 1 | [40] |
| Tecpr1 | WIPI-2 | An interaction between PI3P, WIPI-2, Tecpr1 and Atg5 followed by recruitment of LC3 | [43] |
| Ubiquitin | Adaptor protein (P62, NDP52) | LIR and UBA motifs facilitate binding of ubiquitinated material to LC3 | [47,48,49] |
| PS of dead cell membranes | TIM4 | LC3 translocation triggered by phagocytosis of PS-containing liposomes in macrophages | [16] |
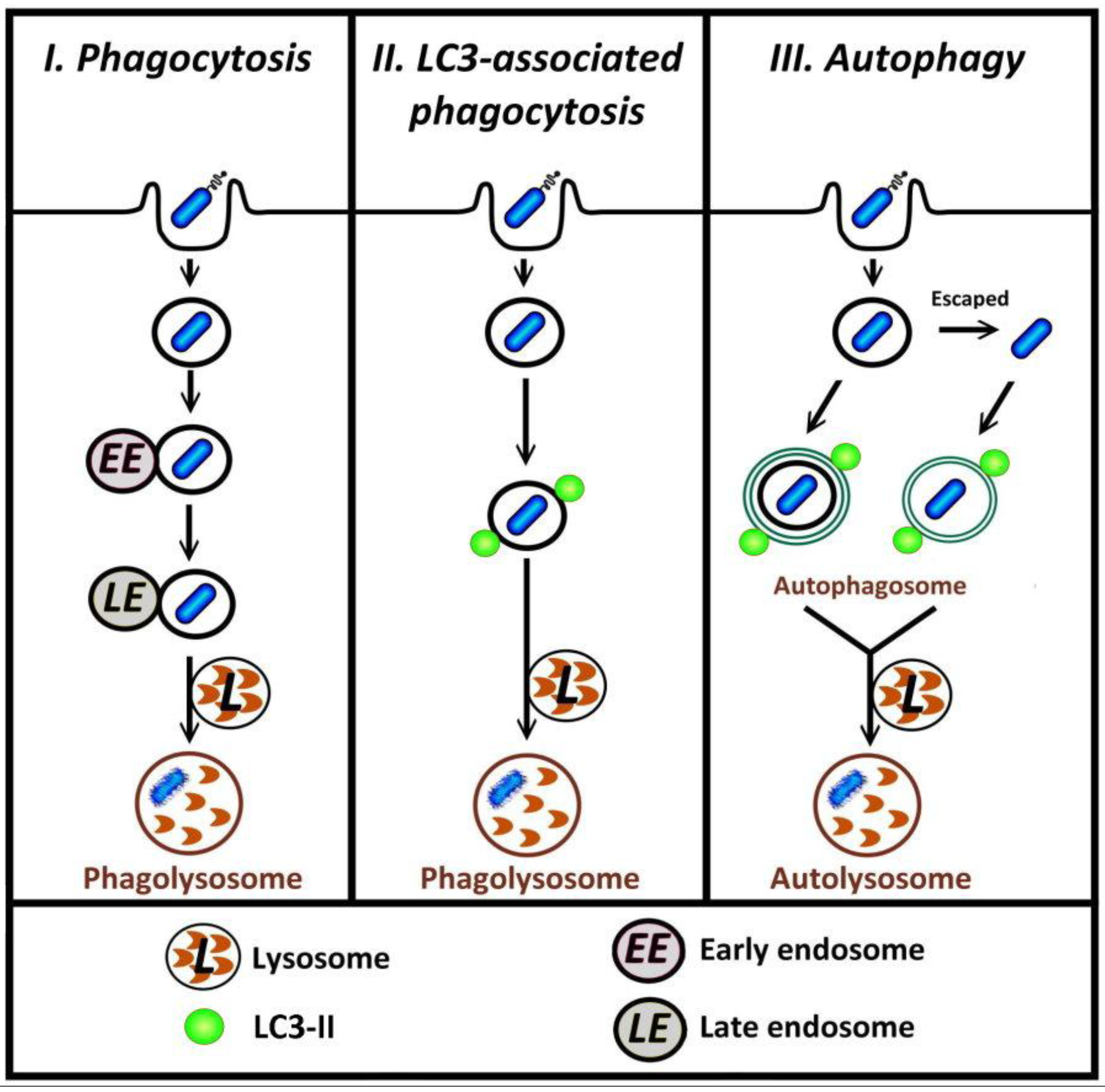
5. Distinguishing LAP from Autophagy

| Phagocytic cells | |||
|---|---|---|---|
| Phagocytosed Material | Cell Type | Morphological Features | Reference |
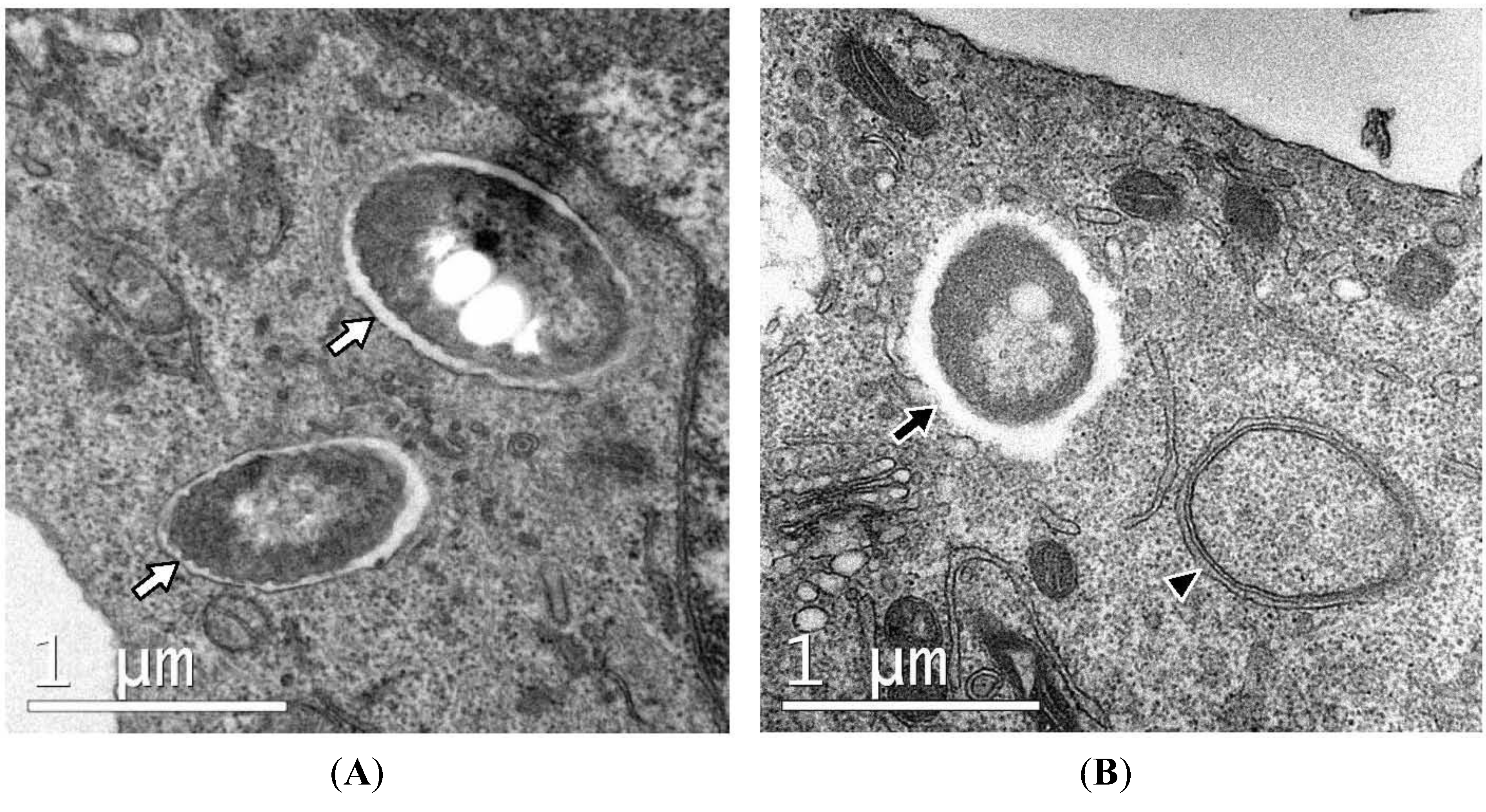
| Zymosan (dead yeast) | RAW264.7 murine macrophages expressing GFP-LC3 | Cells are engulfed within single-membrane phagosomes even after starvation or treatment with rapamycin | [18] |
| Dead cells | CD11b+ F4/80+ primary macrophages derived from bone marrow of GFP-LC3+ mice | The vesicular structures containing apoptotic or necrotic cells are observed as being single-membraned by TEM observation at the time that the majority of dead cell-containing phagosomes co-localized with LC3-II | [16] |
| Burkholderia pseudomallei | RAW264.7-GFP-LC3 | The LC3-positive structures that co-localize with bacteria are revealed as phagosomes by TEM. | [13] |
| Mycobacterium marinum | RAW264.7-GFP-LC3 | Bacteria were within single-membrane components with the presence of typical autophagosomes nearby. | [15] |
| Non-phagocytic cells | |||
| Endocytosed Materials | Cell Type | Ultrastructure | Reference |
| Salmonella typhimurium | MEF-GFP-LC3 | Correlative light microscopy-electron microscopy (CLEM) observation of GFP-LC3 signals after infection displayed a single-membrane structure surrounding bacteria. | [33] |
| Entotic cells | MCF10A-GFP-LC3 | Correlative video-light-electron microscopy demonstrated that the internalized cells were within single-membrane vacuoles. | [12] |
6. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Stuart, L.M.; Ezekowitz, R.A. Phagocytosis and comparative innate immunity: Learning on the fly. Nat. Rev. Immunol. 2008, 8, 131–141. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Grinstein, S. Pathogen destruction versus intracellular survival: The role of lipids as phagosomal fate determinants. J. Clin. Invest. 2008, 118, 2002–2011. [Google Scholar] [CrossRef]
- Kinchen, J.M.; Ravichandran, K.S. Phagosome maturation: Going through the acid test. Nat. Rev. Mol. Cell Biol. 2008, 9, 781–795. [Google Scholar]
- Mijaljica, D.; Prescott, M.; Klionsky, D.J.; Devenish, R.J. Autophagy and vacuole homeostasis: A case for self-degradation? Autophagy 2007, 3, 417–421. [Google Scholar]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar]
- Campoy, E.; Colombo, M.I. Autophagy in intracellular bacterial infection. Biochim. et Biophys. Acta Mol. Cell Res. 2009, 1793, 1465–1477. [Google Scholar] [CrossRef]
- Ogawa, M.; Mimuro, H.; Yoshikawa, Y.; Ashida, H.; Sasakawa, C. Manipulation of autophagy by bacteria for their own benefit. Microbiol. Immunol. 2011, 55, 459–471. [Google Scholar]
- Ogawa, M.; Sasakawa, C. Intracellular survival of Shigella. Cell Microbiol. 2006, 8, 177–184. [Google Scholar] [CrossRef]
- Vergne, I.; Singh, S.; Roberts, E.; Kyei, G.; Master, S.; Harris, J.; de Haro, S.; Naylor, J.; Davis, A.; Delgado, M.; et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy 2006, 2, 175–178. [Google Scholar]
- Yano, T.; Kurata, S. Intracellular recognition of pathogens and autophagy as an innate immune host defence. J. Biochem. 2011, 150, 143–149. [Google Scholar] [CrossRef]
- Lerena, M.C.; Vázquez, C.L.; Colombo, M.I. Bacterial pathogens and the autophagic response. Cell. Microbiol. 2010, 12, 10–18. [Google Scholar] [CrossRef]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef]
- Gong, L.; Cullinane, M.; Treerat, P.; Ramm, G.; Prescott, M.; Adler, B.; Boyce, J.D.; Devenish, R.J. The Burkholderia pseudomallei Type III Secretion System and BopA Are Required for Evasion of LC3-Associated Phagocytosis. PLoS One 2011, 6, e17852. [Google Scholar]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar]
- Lerena, M.C.; Colombo, M.I. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol. 2011, 13, 814–835. [Google Scholar]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar]
- Sanjuan, M.A.; Green, D.R. Eating for good health: Linking autophagy and phagocytosis in host defense. Autophagy 2008, 4, 607–611. [Google Scholar]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar]
- Al-Younes, H.M.; Al-Zeer, M.A.; Khalil, H.; Gussmann, J.; Karlas, A.; Machuy, N.; Brinkmann, V.; Braun, P.R.; Meyer, T.F. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy 2011, 7, 814–828. [Google Scholar]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar]
- Inoue, Y.; Klionsky, D.J. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 2010, 21, 664–670. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar]
- Noda, T.; Fujita, N.; Yoshimori, T. The Ubi brothers reunited. Autophagy 2008, 4, 540–541. [Google Scholar]
- He, H.; Dang, Y.; Dai, F.; Guo, Z.; Wu, J.; She, X.; Pei, Y.; Chen, Y.; Ling, W.; Wu, C.; et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J. Biol. Chem. 2003, 278, 29278–29287. [Google Scholar]
- Xin, Y.; Yu, L.; Chen, Z.; Zheng, L.; Fu, Q.; Jiang, J.; Zhang, P.; Gong, R.; Zhao, S. Cloning, expression patterns, and chromosome localization of three human and two mouse homologues of GABA(A) receptor-associated protein. Genomics 2001, 74, 408–413. [Google Scholar]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar]
- Kageyama, S.; Omori, H.; Saitoh, T.; Sone, T.; Guan, J.L.; Akira, S.; Imamoto, F.; Noda, T.; Yoshimori, T. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol. Biol. Cell 2011, 22, 2290–2300. [Google Scholar]
- Li, W.; Zou, W.; Yang, Y.; Chai, Y.; Chen, B.; Cheng, S.; Tian, D.; Wang, X.; Vale, R.D.; Ou, G. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar]
- Freche, B.; Reig, N.; van der Goot, F.G. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin. Immunopathol. 2007, 29, 249–260. [Google Scholar]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef]
- Shui, W.; Sheu, L.; Liu, J.; Smart, B.; Petzold, C.J.; Hsieh, T.Y.; Pitcher, A.; Keasling, J.D.; Bertozzi, C.R. Membrane proteomics of phagosomes suggests a connection to autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 16952–16957. [Google Scholar]
- Berger, S.B.; Romero, X.; Ma, C.; Wang, G.; Faubion, W.A.; Liao, G.; Compeer, E.; Keszei, M.; Rameh, L.; Wang, N.; et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat. Immunol. 2010, 11, 920–927. [Google Scholar]
- Ramjeet, M.; Hussey, S.; Philpott, D.J.; Travassos, L.H. ‘Nodophagy’: New crossroads in Crohn disease pathogenesis. Gut Microbes 2010, 1, 307–315. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar]
- Ogawa, M.; Yoshikawa, Y.; Kobayashi, T.; Mimuro, H.; Fukumatsu, M.; Kiga, K.; Piao, Z.; Ashida, H.; Yoshida, M.; Kakuta, S.; et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 2011, 9, 376–389. [Google Scholar]
- Obara, K.; Ohsumi, Y. PtdIns 3-Kinase Orchestrates Autophagosome Formation in Yeast. J. Lipids 2011, 2011. [Google Scholar] [CrossRef]
- Ogawa, M.; Sasakawa, C. The role of Tecpr1 in selective autophagy as a cargo receptor. Autophagy 2011, 7, 1389–1391. [Google Scholar] [CrossRef]
- Ravichandran, K.S.; Lorenz, U. Engulfment of apoptotic cells: Signals for a good meal. Nat. Rev. Immunol. 2007, 7, 964–974. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar]
- Mijaljica, D.; Nazarko, T.; Brumell, J.; Huang, W.-P.; Komatsu, M.; Prescott, M.; Simonsen, A.; Yamamoto, A.; Zhang, H.; Klionsky, D.J.; et al. Receptor protein complexes are in control of autophagy. Autophagy 2012, 8. in press. [Google Scholar]
- Gelman, A.; Elazar, Z. Autophagic factors cut to the bone. Dev. Cell 2011, 21, 808–810. [Google Scholar] [CrossRef]
- Moreau, K.; Ravikumar, B.; Renna, M.; Puri, C.; Rubinsztein, D.C. Autophagosome precursor maturation requires homotypic fusion. Cell 2011, 146, 303–317. [Google Scholar]
- Mousavi, S.A.; Malerod, L.; Berg, T.; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J. 2004, 377, 1–16. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelarova, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar]
- Li, X.; Prescott, M.; Adler, B.; Boyce, J.D.; Devenish, R.J. Beclin 1 is required for starvation-enhanced, but not rapamycin-enhanced, LC3-associated phagocytosis of Burkholderia pseudomallei in RAW 264.7 cells. 2012; Unpublished word. [Google Scholar]
- Florey, O.; Overholtzer, M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012, 22, 374–380. [Google Scholar] [CrossRef]
- Razi, M.; Tooze, S.A. Correlative light and electron microscopy. Methods Enzymol. 2009, 452, 261–275. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lai, S.-c.; Devenish, R.J. LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy. Cells 2012, 1, 396-408. https://doi.org/10.3390/cells1030396
Lai S-c, Devenish RJ. LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy. Cells. 2012; 1(3):396-408. https://doi.org/10.3390/cells1030396
Chicago/Turabian StyleLai, Shu-chin, and Rodney J. Devenish. 2012. "LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy" Cells 1, no. 3: 396-408. https://doi.org/10.3390/cells1030396