Autophagy in Skeletal Muscle Homeostasis and in Muscular Dystrophies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
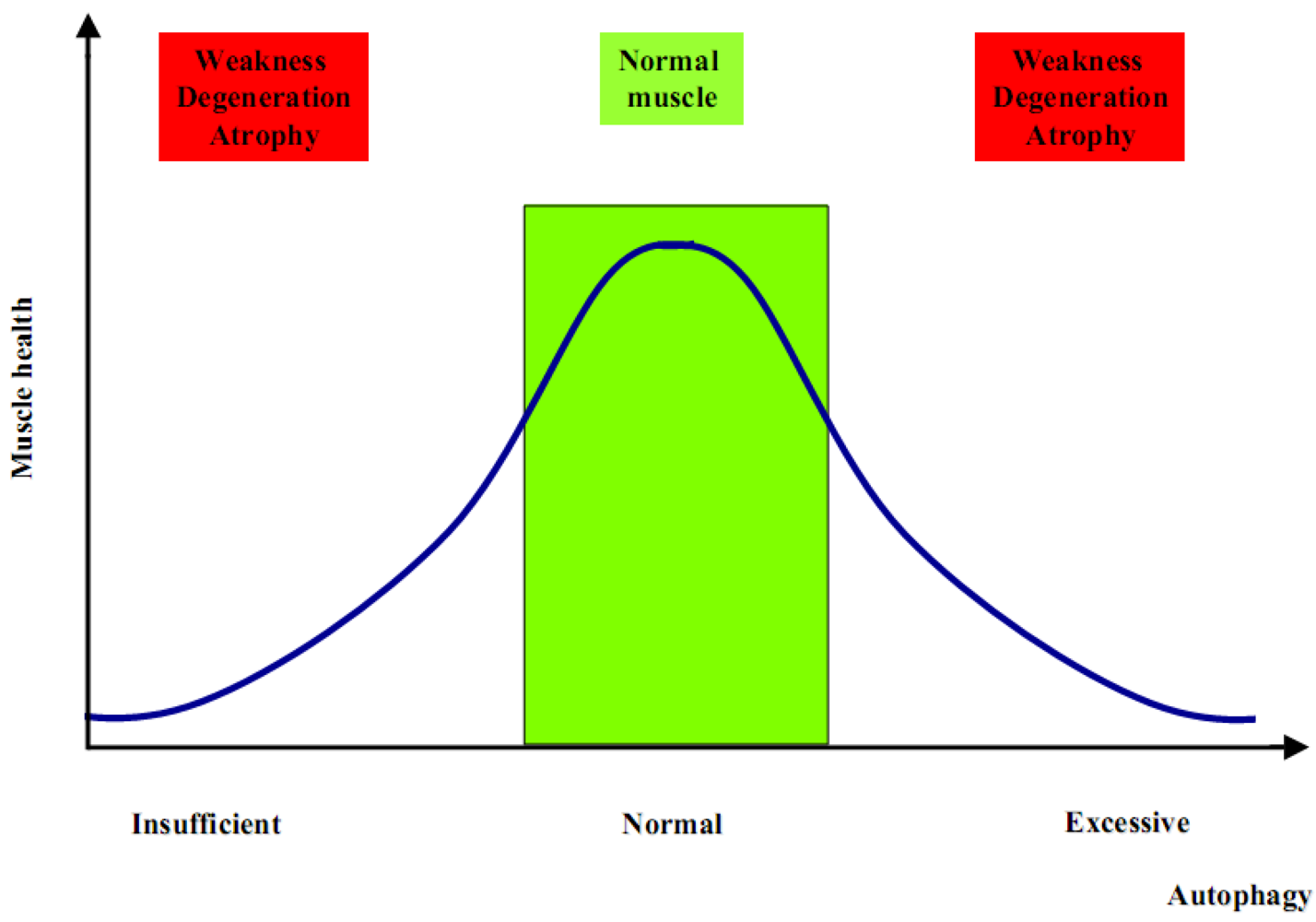
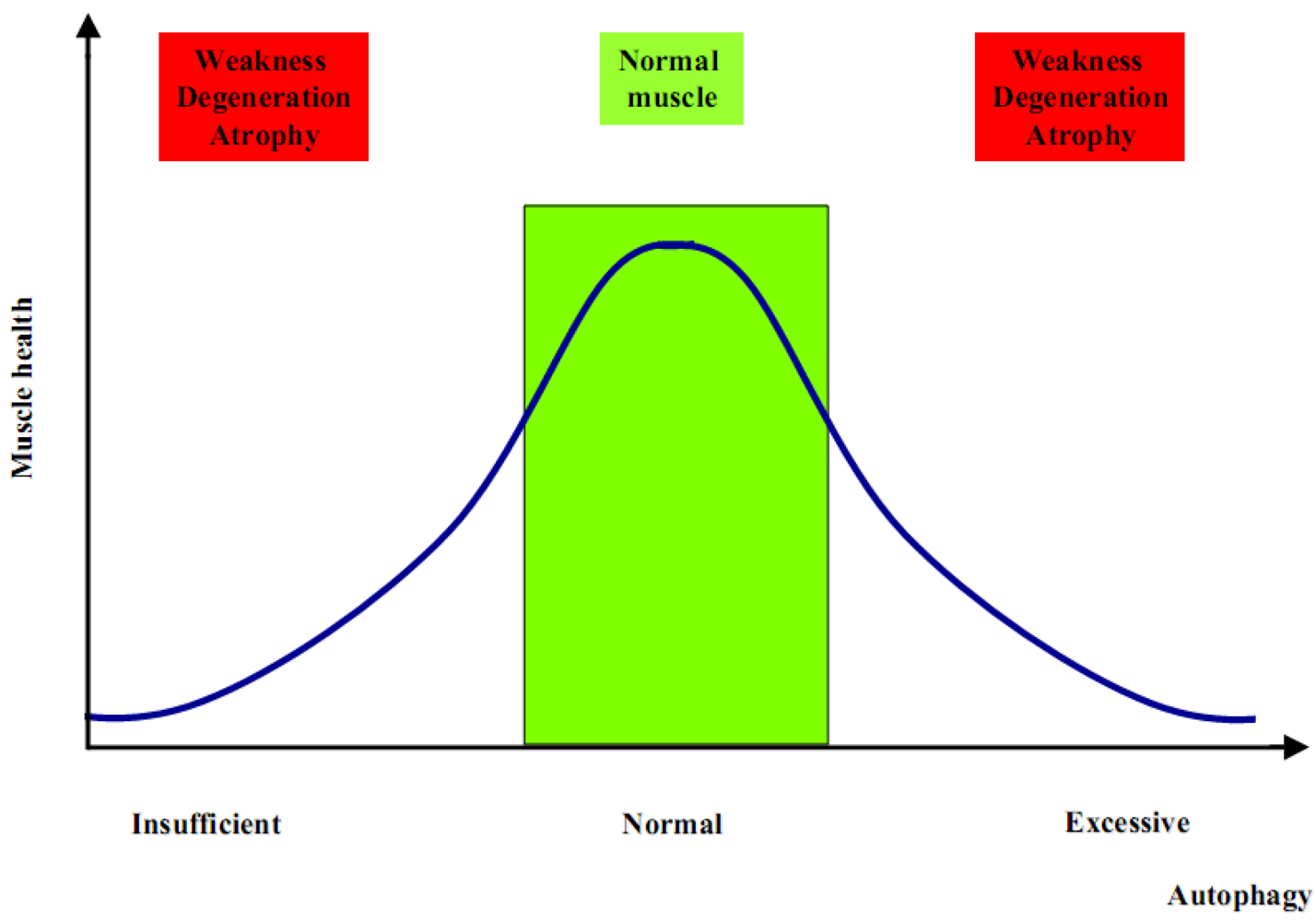
2. Autophagy in Muscle Homeostasis
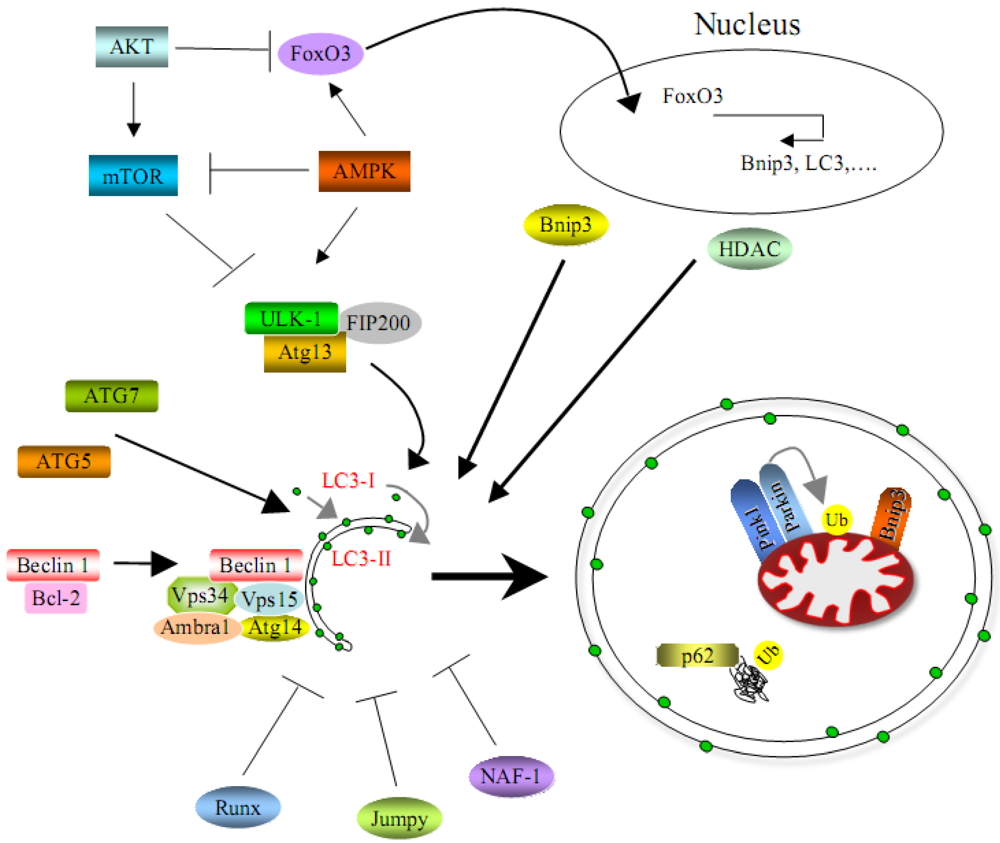
3. Molecular Mechanisms of Autophagy in Skeletal Muscles
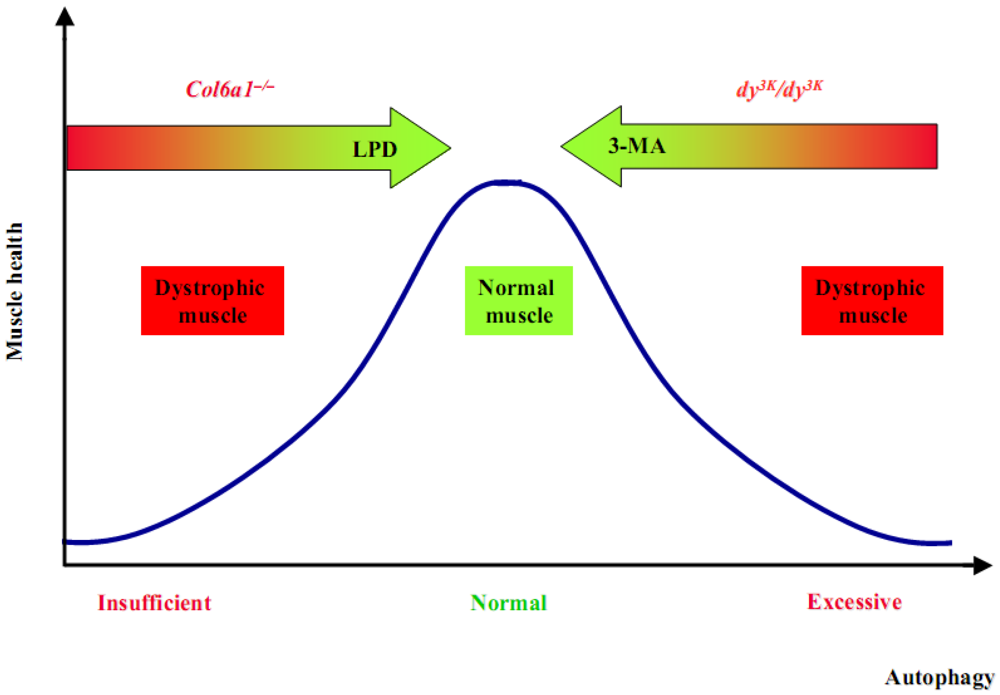
4. Autophagy in Congenital Muscle Dystrophies
5. Lysosome and Autophagy Crosstalk
6. Conclusions
Acknowledgments
References
- Sandri, M. Autophagy in skeletal muscle. FEBS Lett. 2010, 584, 1411–1416. [Google Scholar] [CrossRef]
- Sandri, M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am. J. Physiol. Cell Physiol. 2010, 298, 1291–1297. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, 11–16. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2009, 45, 138–148. [Google Scholar]
- Vergne, I.; Roberts, E.; Elmaoued, R.A.; Tosch, V.; Delgado, M.A.; Proikas-Cezanne, T.; Laporte, J.; Deretic, V. Control of autophagy initiation by phosphoinositide 3-phosphatase jumpy. EMBO J. 2009, 28, 2244–2258. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008, 23, 160–170. [Google Scholar] [CrossRef]
- Schiaffino, S.; Hanzlikova, V. Studies on the effect of denervation in developing muscle. II. The lysosomal system. J. Ultrastruct. Res. 1972, 39, 1–14. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Wang, X.; Blagden, C.; Fan, J.; Nowak, S.J.; Taniuchi, I.; Littman, D.R.; Burden, S.J. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev. 2005, 19, 1715–1722. [Google Scholar] [CrossRef]
- Nogalska, A.; Terracciano, C.; D’Agostino, C.; King Engel, W.; Askanas, V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009, 118, 407–413. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef]
- Masiero, E.; Sandri, M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 2010, 6, 307–309. [Google Scholar] [CrossRef]
- WU, J.J.; Quijano, C.; Chen, E.; Liu, H.; Cao, L.; Fergusson, M.M.; Rovira, I.I.; Gutkind, S.; Daniels, M.P.; Komatsu, M.; Finkel, T. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 2009, 1, 425–437. [Google Scholar]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; Yuan, J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar]
- de Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef]
- Demontis, F.; Perrimon, N. FOXO/4E–BP Signaling in Drosophila Muscles Regulates Organism-wide Proteostasis during Aging. Cell 2010, 143, 813–825. [Google Scholar] [CrossRef]
- Grumati, P.; Coletto, L.; Schiavinato, A.; Castagnaro, S.; Bertaggia, E.; Sandri, M.; Bonaldo, P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 2011, 7, 1415–1423. [Google Scholar]
- Jamart, C.; Benoit, N.; Raymackers, J.M.; Kim, H.J.; Kim, C.K.; Francaux, M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur. J. Appl. Physiol. 2011. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar]
- Jamart, C.; Francaux, M.; Millet, G.Y.; Deldicque, L.; Frere, D.; Féasson, L. Modulation of autophagy and ubiquitin-proteasome pathwasy during ultra-endurance running. J. Appl. Physiol. 2012. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar]
- Klionsky, D.J.; Saltiel, A.R. Autophagy works out. Cell Metab. 2012, 15, 273–274. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; Yancopoulos, G.D. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar]
- Lee, C.H.; Inoki, K.; Guan, K.L. mTOR Pathway as a Target in Tissue Hypertrophy. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 443–467. [Google Scholar]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Chan, E.Y.; Tooze, S.A. Evolution of Atg1 function and regulation. Autophagy 2009, 5, 758–765. [Google Scholar]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar]
- Chan, E.Y.; Longatti, A.; McKnight, N.C.; Tooze, S.A. Kinase-inactivated ULK proteins inhibit au- tophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell Biol. 2009, 29, 157–171. [Google Scholar] [CrossRef]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediatemTOR signaling to the autophagy machinery. Mol. Biol. Cell 2003, 20, 1992–2003. [Google Scholar]
- Ganley, I.G.; Lam, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.I.; Natsume, T.; Takehana, K.; Yamada, N.; Guan, J.L.; Oshiro, N.; Mizushima, N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Romanino, K.; Mazelin, L.; Albert, L.; Conjard–Duplany, A.; Lin, S.; Bentzinger, C.F.; Handschin, C.; Puigserver, P.; Zorzato, F.; Schaeffer, L.; et al. Myopathy caused by mammalian target of rapamycin complex 1 (mTORC1) inactivation is not reversed by restoring mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 20808–20813. [Google Scholar]
- Mordier, S.; Deval, C.; Bechet, D.; Tassa, A.; Ferrara, M. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin- independent signaling pathway. J. Biol. Chem. 2000, 275, 29900–29906. [Google Scholar]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase-Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 2003, 376, 577–586. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase- dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010, 5, e15394. [Google Scholar]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar]
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef]
- Malicdan, M.C.; Noguchi, S.; Nonaka, I.; Saftig, P.; Nishino, I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul. Disord. 2008, 18, 521–529. [Google Scholar]
- Carmignac, V.; Svensson, M.; Korner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902. [Google Scholar]
- Bruckner, P. Suprastructures of extracellular matrices: paradigms of functions controlled by aggregates rather than molecules. Cell Tissue Res. 2010, 339, 7–18. [Google Scholar] [CrossRef]
- Van Agtmael, T.; Bruckner-Tuderman, L. Basement membranes and human disease. Cell Tissue Res. 2010, 339, 167–188. [Google Scholar]
- Collins, J.; Bonnemann, C.G. Congenital muscular dystrophies: Toward molecular therapeutic interventions. Curr. Neurol. Neurosci. Rep. 2010, 10, 83–91. [Google Scholar] [CrossRef]
- Pepe, G.; Bertini, E.; Bonaldo, P.; Bushby, K.; Giusti, B.; de Visser, M.; Guicheney, P.; Lattanzi, G.; Merlini, L.; Muntoni, F.; et al. Bethlem myopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100th ENMC international workshop, 23–24 November, Naarden, The Netherlands. Neuromuscul. Disord. 2002, 12, 984–993. [Google Scholar] [CrossRef]
- Briñas, L.; Richard, P.; Quijano–Roy, S.; Gartioux, C.; Ledeuil, C.; Lacène, E.; Makri, S.; Ferreiro, A.; Maugenre, S.; Topaloglu, H.; et al. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann. Neurol. 2010, 68, 511–520. [Google Scholar]
- Mercuri, E.; Yuva, Y.; Brown, S.C.; Brockington, M.; Kinali, M.; Jungbluth, H.; Feng, L.; Sewry, C.A.; Muntoni, F. Collagen VI involvement in Ullrich syndrome: A clinical, genetic, and immunohistochemical study. Neurology 2002, 58, 1354–1359. [Google Scholar]
- Demir, E.; Ferreiro, A.; Sabatelli, P.; Allamand, V.; Makri, S.; Echenne, B.; Maraldi, M.; Merlini, L.; Topaloglu, H.; Guicheney, P. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics 2004, 35, 103–112. [Google Scholar] [CrossRef]
- Hayashi, F.L.; Chou, F.L.; Engvall, E.; Ogawa, M.; Matsuda, C.; Hirabayashi, S.; Yokochi, L.; Ziober, B.L.; Kramer, R.H.Y.K.; Kaufman, S.J.; et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet. 1998, 19, 94–97. [Google Scholar]
- Helbling-Leclerc, A.; Zhang, X.; Topaloglu, H.; Cruaud, C.; Tesson, F.; Weissenbach, J.; Tomè, F.M.S.; Schwartz, K.; Fardeau, M.; Tryggvason, K.; et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 1995, 11, 216–218. [Google Scholar]
- Hara, Y.; Balci–Hayta, B.; Yoshida-Moriguchi, T.; Kanagawa, M.; Beltrán-Valero de Bernabé, D.; Gündeşli, H.; Willer, T.; Satz, J.S.; Crawford, R.W.; Burden, S.J.; et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med. 2011, 364, 939–946. [Google Scholar]
- Lampe, A.K.; Bushby, K.M. Collagen VI related muscle disorders. J. Med. Genet. 2005, 42, 673–685. [Google Scholar] [CrossRef]
- Bernardi, P.; Bonaldo, P. Dysfunction of mitochondria and sarcoplasmic reticulum in the pathogenesis of collagen VI muscular dystrophies. Ann. NY Acad. Sci. 2008, 1147, 303–311. [Google Scholar]
- Grumati, P.; Coletto, L.; Sandri, M.; Bonaldo, P. Autophagy induction rescues muscular dystrophy. Autophagy 2011, 7, 426–428. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar]
- Okamoto, K.; Kondo–Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar]
- Narendra, D.P.; Youle, R.J. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar]
- Tolkovsky, A.M. Autophagy thwarts muscle disease. Nat. Med. 2010, 16, 1188–1190. [Google Scholar] [CrossRef]
- Carmignac, V.; Svensson, M.; Korner, Z.; Elowsson, L.; Matsumura, C.; Gawlik, K.I.; Allamand, V.; Durbeej, M. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011, 20, 4891–4902. [Google Scholar] [CrossRef]
- Carmignac, V.; Quere, R.; Durbeej, M. Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum. Mol. Genet. 2011, 20, 541–552. [Google Scholar] [CrossRef]
- Malicdan, M.C.V.; Nishino, I. Autophagy in lysosomal myopathies. Brain Pathol. 2012, 22, 82–88. [Google Scholar]
- Ramachandran, N.; Munteanu, I.; Wang, P.; Aubourg, P.; Rilstone, J.J.; Israelian, N.; Naranian, T.; Paroutis, P.; Guo, R.; Ren, Z.P.; et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell 2009, 137, 235–246. [Google Scholar]
- Fukuda, T.; Ewan, L.; Bauer, M.; Mattaliano, R.J.; Zaal, K.; Ralston, E.; Plotz, P.H.; Raben, N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann. Neurol. 2006, 59, 700–708. [Google Scholar]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–476. [Google Scholar]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome to nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Umekawa, M.; Klionsky, D.J. The cytoplasm to vacuole targeting pathway: A historical perspective. Int. J. Cell Biol. 2012, 2012, 142634. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grumati, P.; Bonaldo, P. Autophagy in Skeletal Muscle Homeostasis and in Muscular Dystrophies. Cells 2012, 1, 325-345. https://doi.org/10.3390/cells1030325
Grumati P, Bonaldo P. Autophagy in Skeletal Muscle Homeostasis and in Muscular Dystrophies. Cells. 2012; 1(3):325-345. https://doi.org/10.3390/cells1030325
Chicago/Turabian StyleGrumati, Paolo, and Paolo Bonaldo. 2012. "Autophagy in Skeletal Muscle Homeostasis and in Muscular Dystrophies" Cells 1, no. 3: 325-345. https://doi.org/10.3390/cells1030325