Genome-Wide Mining and Identification of Protein Kinase Gene Family Impacts Salinity Stress Tolerance in Highly Dense Genetic Map Developed from Interspecific Cross between G. hirsutum L. and G. darwinii G. Watt

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Gene Mining within SSR Markers
2.3. Phylogenetic Analysis
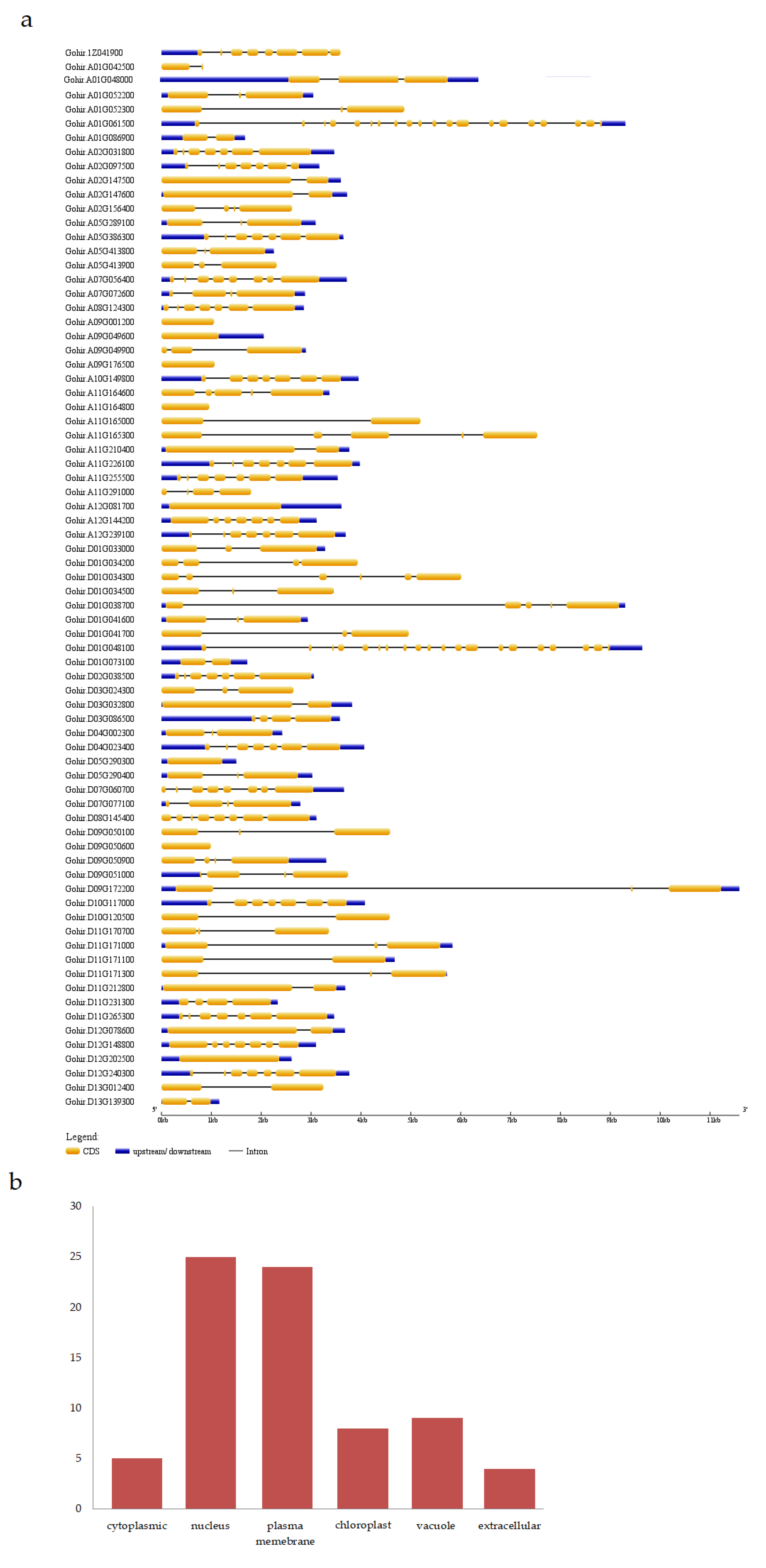
2.4. Subcellular Localization and Structure Analysis
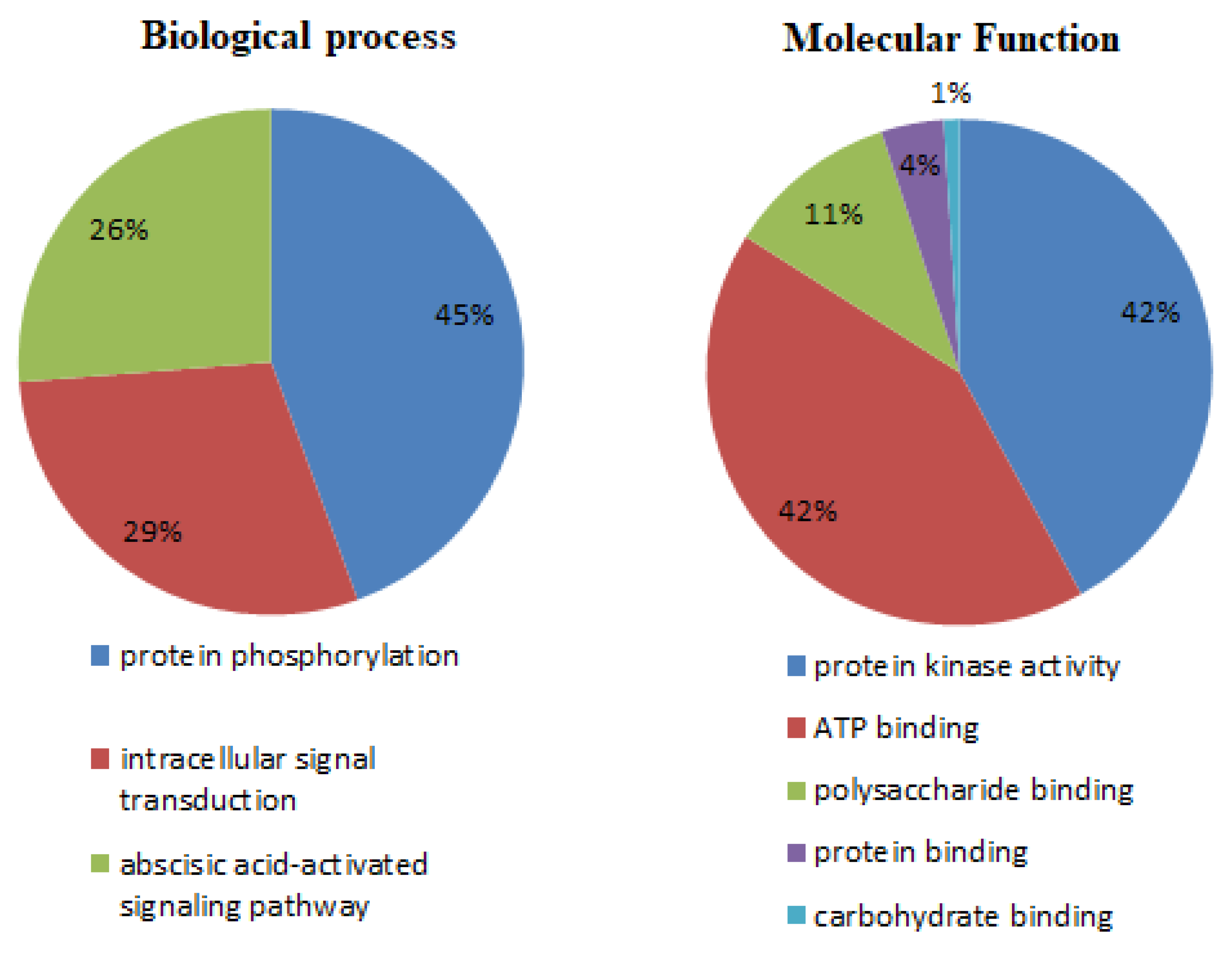
2.5. Gene Ontology (GO) Annotation
2.6. Syntenic Analysis and Duplication of Genes
2.7. Prediction of miRNA Target and Transcription Factor Binding Sites (TFBS) Analysis
2.8. Plant Material and Hydroponic Culture
2.9. RNA Sequence and RT-qPCR Analysis
3. Results
3.1. Genetic Linkage Map Features
3.2. Assessment of Collinearity of the Genetic and Physical Map of G. hirsutum
3.3. Phylogenetic Analysis
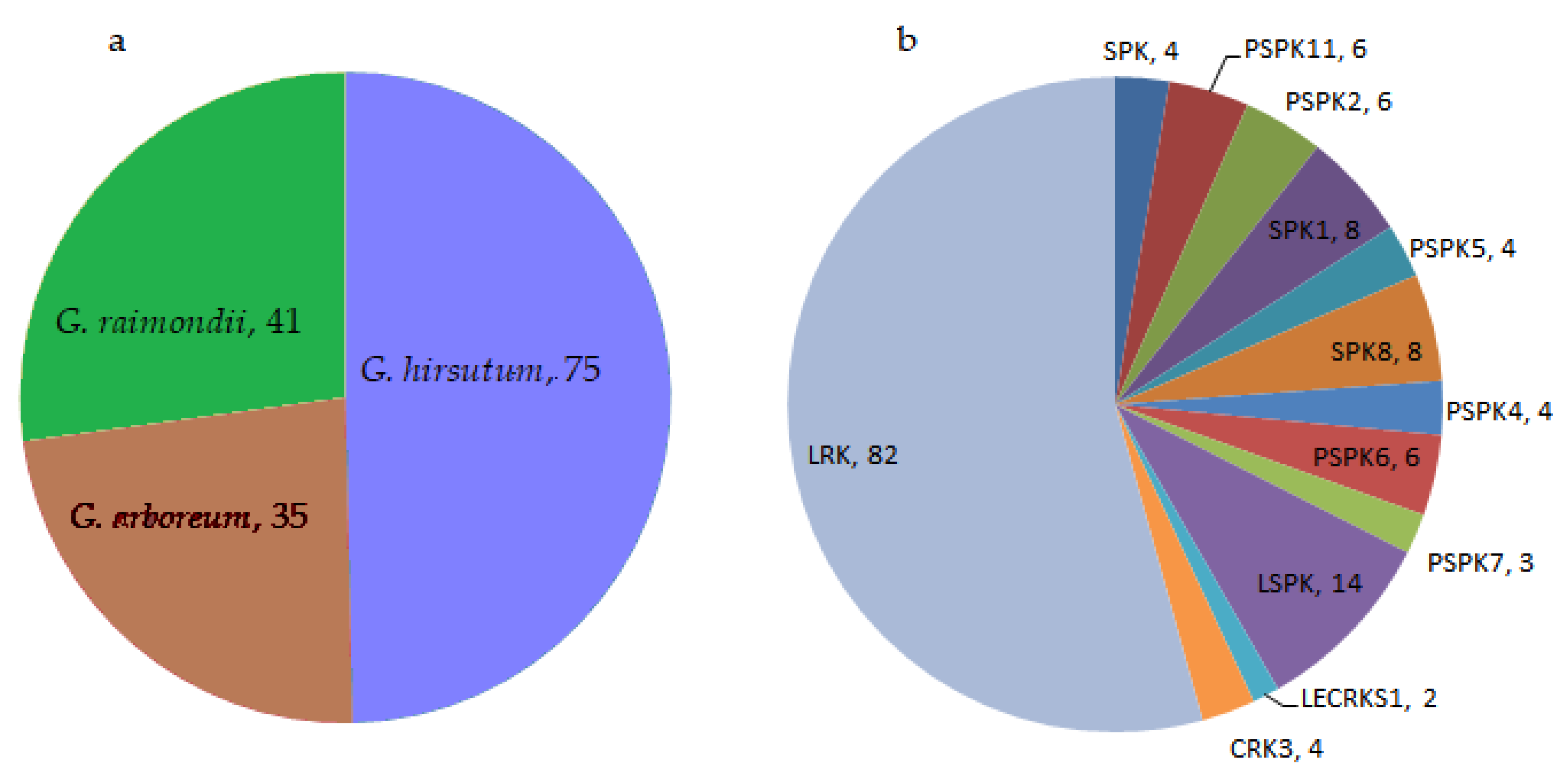
3.4. Chromosomal Distribution of Pkinase Genes
3.5. Structural Analysis and Localization of Genes
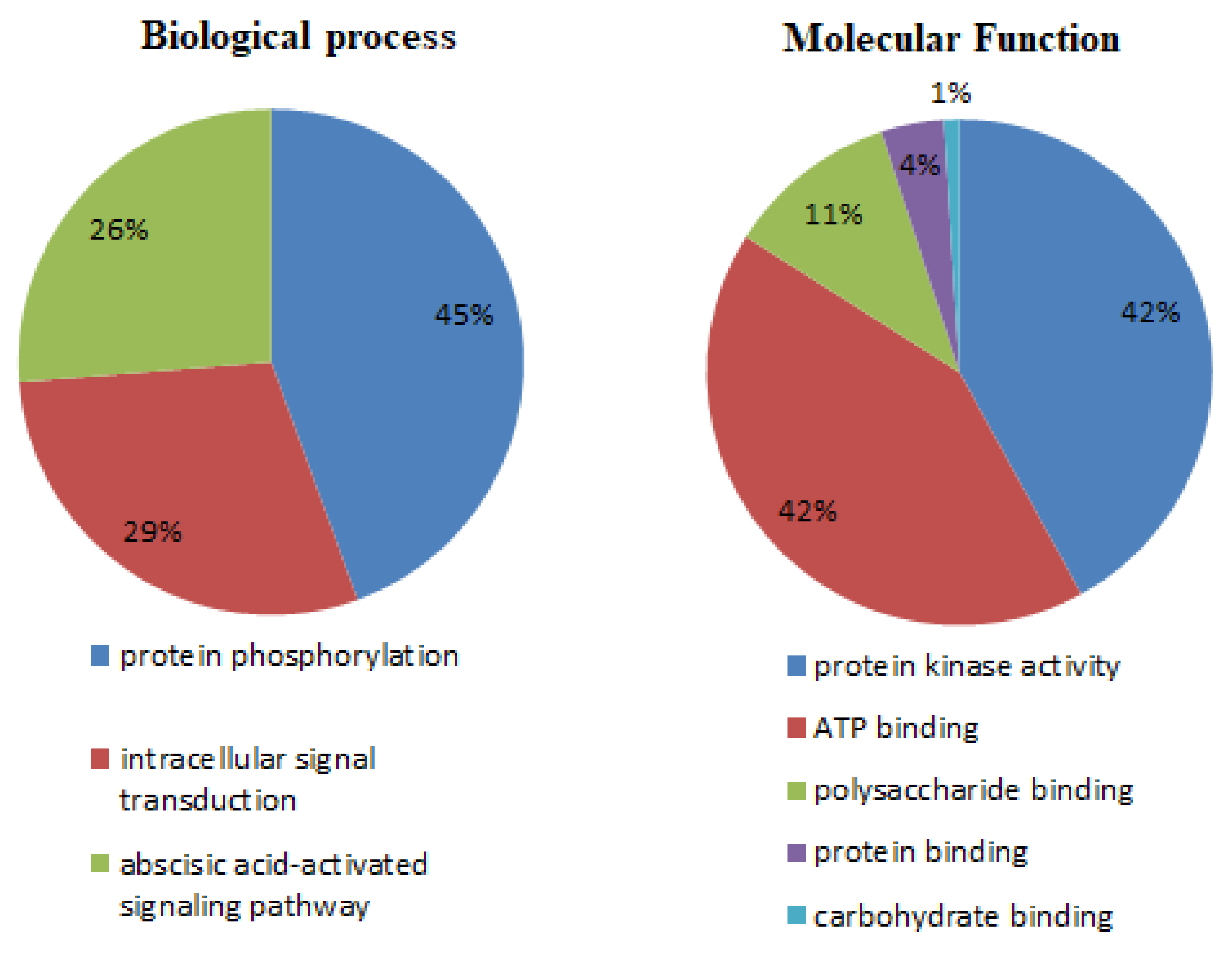
3.6. Gene Annotation by GO Analysis
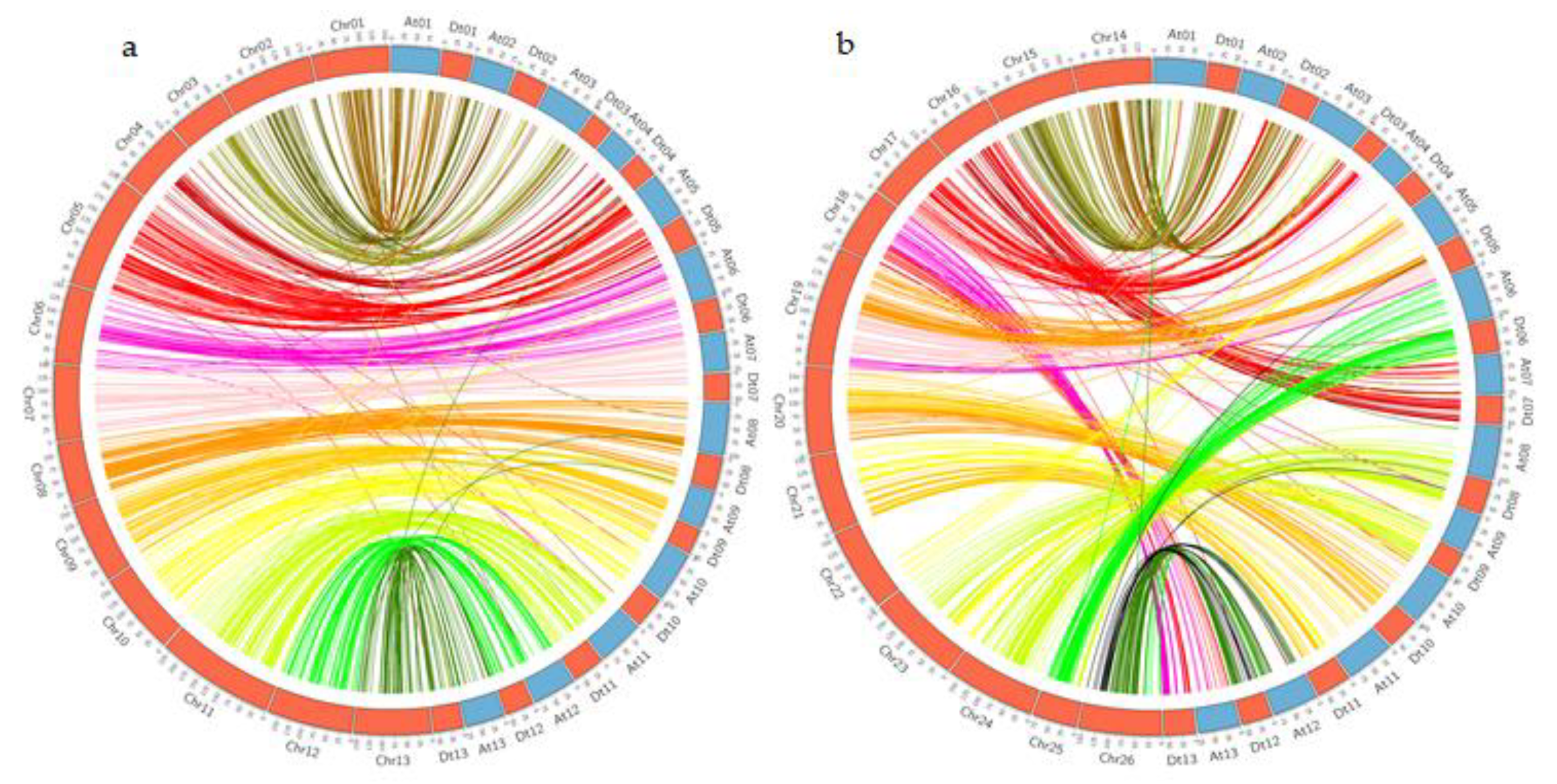
3.7. Duplication and Syntenic Analysis of Genes
3.8. Prediction of Transcription Factor Binding Sites
3.9. miRNA Target Analysis of Genes
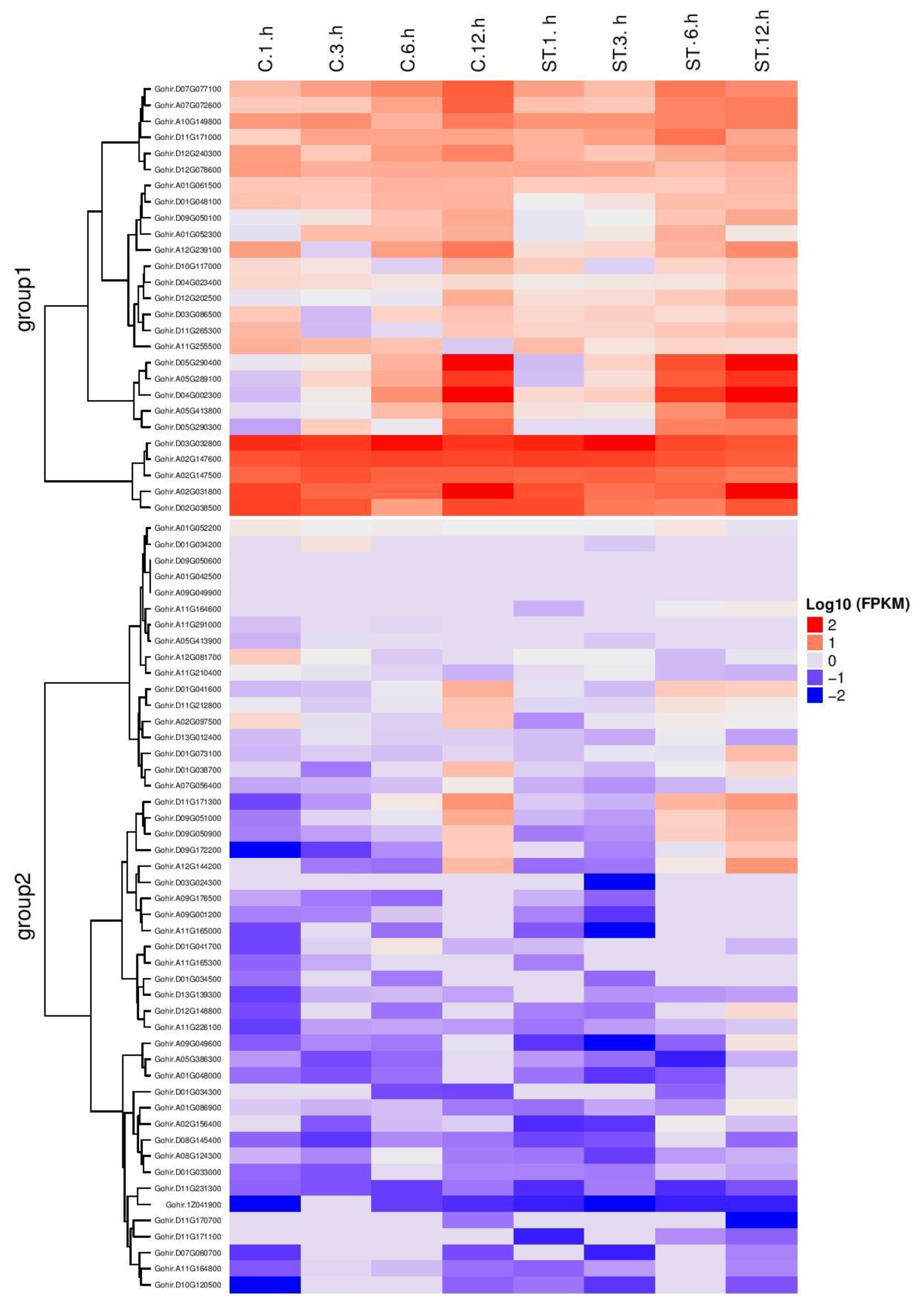
3.10. RNA Sequence Data of Salt Tolerant Genes
3.11. RT-qPCR Analysis of the Candidate Genes under Salt Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine Triphosphate |
| BP | Biological Process |
| CDS | Coding Sequence |
| CF | Cellular Function |
| CIRCOS | Circular Genome Data Visualization |
| GO | Gene Ontology |
| GSDS | Gene Structure Display Server |
| MF | Molecular Function |
| MYA | Million Years Ago |
| NaCl | Sodium Chloride |
| NCBI | National Centre for Biotechnology Information |
| SSR | Simple Sequence Repeat |
References
- Cepal, N. The Outlook for Agriculture and Rural Development in the Americas: A Perspective on Latin America and the Caribbean 2017–2018; FAO, IICA: San Jose, Costa Rica, 2017. [Google Scholar]
- Gallagher, J.P.; Grover, C.E.; Rex, K.; Moran, M.; Wendel, J.F. A new species of cotton from Wake Atoll, Gossypium stephensii (Malvaceae). Syst. Bot. 2017, 42, 115–123. [Google Scholar] [CrossRef]
- Wendel, J.F.; Grover, C.E. Taxonomy and evolution of the cotton genus, Gossypium. Cotton 2015, 25–44. [Google Scholar] [CrossRef]
- Kunbo, W.; WENDEL, J.F.; Jinping, H. Designations for individual genomes and chromosomes in Gossypium. J. Cotton Res. 2018, 1, 3. [Google Scholar]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Khan, M.K.R.; Zhou, Z.; Wang, X.; Cai, X.; Ilyas, M.K.; Wang, C.; Wang, Y.; Li, Y.; Liu, F. A high-density SSR genetic map constructed from a F2 population of Gossypium hirsutum and Gossypium darwinii. Gene 2015, 574, 273–286. [Google Scholar] [CrossRef]
- Maxted, N.; Kell, S.; Toledo, Á.; Dulloo, E.; Heywood, V.; Hodgkin, T.; Hunter, D.; Guarino, L.; Jarvis, A.; Ford-Lloyd, B. A global approach to crop wild relative conservation: Securing the gene pool for food and agriculture. Kew Bull. 2010, 65, 561–576. [Google Scholar] [CrossRef]
- Atwell, B.J.; Wang, H.; Scafaro, A.P. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 2014, 215, 48–58. [Google Scholar] [CrossRef]
- Roy, S.J.; Negrão, S.; Tester, M. Salt resistant crop plants. Curr. Opin. Biotechnol. 2014, 26, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Deinlein, U.; Stephan, A.B.; Horie, T.; Luo, W.; Xu, G.; Schroeder, J.I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, X.; Bressan, R.A.; Hasegawa, P.M.; Pardo, J.M. Ion homeostasis in NaCl stress environments. Plant Physiol. 1995, 109, 735. [Google Scholar] [CrossRef] [PubMed]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Cho, Y.-G. Plant hormones in salt stress tolerance. J. Plant Biol. 2015, 58, 147–155. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013, 32, 959–970. [Google Scholar] [CrossRef]
- Lu, N.; Roldan, M.; Dixon, R.A. Characterization of two TT2-type MYB transcription factors regulating proanthocyanidin biosynthesis in tetraploid cotton, Gossypium hirsutum. Planta 2017, 246, 323–335. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Zhu, J.-K.; Liu, J.; Xiong, L. Genetic analysis of salt tolerance in Arabidopsis: Evidence for a critical role of potassium nutrition. Plant Cell 1998, 10, 1181–1191. [Google Scholar] [CrossRef]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.-S.; Guo, Y.; Dietrich, M.A.; Schumaker, K.S.; Zhu, J.-K. Regulation of SOS1, a plasma membrane Na+/H+ exchanger in Arabidopsis thaliana, by SOS2 and SOS3. Proc. Natl. Acad. Sci. USA 2002, 99, 8436–8441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, G.; Wang, Y.; Zhou, Z.; Meng, Y.; Chen, B. Effect of soil salinity on physiological characteristics of functional leaves of cotton plants. J. Plant Res. 2013, 126, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Liang, C.; Meng, Z.; Malik, W.; Zhu, T.; Zong, X.; Guo, S.; Zhang, R. Progress in genome sequencing will accelerate molecular breeding in cotton (Gossypium spp.). 3 Biotech 2016, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xu, Y.; Lu, P.; Wang, X.; Li, Z.; Cai, X.; Zhou, Z.; Wang, Y.; Zhang, Z.; Lin, Z. Salt stress responsiveness of a wild cotton species (Gossypium klotzschianum) based on transcriptomic analysis. PLoS ONE 2017, 12, e0178313. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Cao, J.; Ni, L.; Zhu, Y.; Zhang, A.; Tan, M.; Jiang, M. ZmCPK11 is involved in abscisic acid-induced antioxidant defence and functions upstream of ZmMPK5 in abscisic acid signalling in maize. J. Exp. Bot. 2012, 64, 871–884. [Google Scholar] [CrossRef]
- Vivek, P.J.; Tuteja, N.; Soniya, E.V. CDPK1 from ginger promotes salinity and drought stress tolerance without yield penalty by improving growth and photosynthesis in Nicotiana tabacum. PLoS ONE 2013, 8, e76392. [Google Scholar] [CrossRef]
- Magwanga, R.; Lu, P.; Kirungu, J.; Diouf, L.; Dong, Q.; Hu, Y.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Z. GBS mapping and analysis of genes conserved between Gossypium tomentosum and Gossypium hirsutum cotton cultivars that respond to drought stress at the seedling stage of the BC2F2 generation. Int. J. Mol. Sci. 2018, 19, 1614. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV Wagening 2006. [Google Scholar]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Ditta, A.; Zhou, Z.; Cai, X.; Wang, X.; Okubazghi, K.; Shehzad, M.; Xu, Y.; Hou, Y.; Sajid Iqbal, M.; Khan, M. Assessment of genetic diversity, population structure, and evolutionary relationship of uncharacterized genes in a novel germplasm collection of diploid and allotetraploid Gossypium accessions using EST and genomic SSR markers. Int. J. Mol. Sci. 2018, 19, 2401. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Copley, R.R.; Doerks, T.; Ponting, C.P.; Bork, P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000, 28, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2014, 31, 1296–1297. [Google Scholar] [CrossRef]
- Voorrips, R. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Heredity 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J.; Li, D.; Zhang, Z.; Liu, F.; Zhou, X.; Wang, T.; Ling, Y.; Su, Z. PMRD: Plant microRNA database. Nucleic Acids Res. 2009, 38, D806–D813. [Google Scholar] [CrossRef] [PubMed]
- Boguski, M.S.; Lowe, T.M.; Tolstoshev, C.M. dbEST—Database for “expressed sequence tags”. Nat. Genet. 1993, 4, 332. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Higo, H. PLACE: A database of plant cis-acting regulatory DNA elements. Nucleic Acids Res. 1998, 26, 358–359. [Google Scholar] [CrossRef]
- Oluoch, G.; Zheng, J.; Wang, X.; Khan, M.K.R.; Zhou, Z.; Cai, X.; Wang, C.; Wang, Y.; Li, X.; Wang, H. QTL mapping for salt tolerance at seedling stage in the interspecific cross of Gossypium tomentosum with Gossypium hirsutum. Euphytica 2016, 209, 223–235. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Arnon, D.I. The water-culture method for growing plants without soil. Circ. Calif. Agric. Exp. Stn. 1950, 347, 32. [Google Scholar]
- Gregory, T.R. Understanding evolutionary trees. Evol. Educ. Outreach 2008, 1, 121. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Zheng, H.-J.; Hu, Y.; Lu, G.; Yang, C.-Q.; Chen, J.-D.; Chen, J.-J.; Chen, D.-Y.; Zhang, L. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef]
- Hu, R.; Qi, G.; Kong, Y.; Kong, D.; Gao, Q.; Zhou, G. Comprehensive analysis of NAC domain transcription factor gene family in Populus trichocarpa. BMC Plant Biol. 2010, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Dong, Q.; Hu, Y.; Zhou, Z.; Cai, X.; Wang, X.; Hou, Y.; Wang, K. Cotton late embryogenesis abundant (LEA2) genes promote root growth and confer drought stress tolerance in transgenic Arabidopsis thaliana. Genes Genomes Genet. 2018, 8, 2781–2803. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kong, H.; Sun, Y.; Zhang, X.; Zhang, W.; Altman, N.; DePamphilis, C.W.; Ma, H. Genome-wide analysis of the cyclin family in Arabidopsis and comparative phylogenetic analysis of plant cyclin-like proteins. Plant Physiol. 2004, 135, 1084–1099. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.; Wang, X.; Cai, X.; Zhou, Z.; Zhang, Z.; Salih, H.; Wang, K. Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in upland cotton. BMC Genet. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Magwanga, R.O.; Cai, X.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Jin, D.; Guo, X.; Wei, Y. Deep transcriptome analysis reveals reactive oxygen species (ROS) network evolution, response to abiotic stress, and regulation of fiber development in cotton. Int. J. Mol. Sci. 2019, 20, 1863. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-C.; Lee, T.-Y.; Huang, H.-D.; Huang, H.-Y.; Pan, R.-L. PlantPAN: Plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genom. 2008, 9, 561. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic-and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef]
- Boss, I.W.; Renne, R. Viral miRNAs: Tools for immune evasion. Curr. Opin. Microbiol. 2010, 13, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 2014, 66, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehzad, M.; Ditta, A.; Cai, X.; Zhou, Z.; Wang, X.; Xu, Y.; Hou, Y.; Yasir, M.; Khan, M.; Wang, K. Genome wide characterization, evolution and expression analysis of FBA gene family under salt stress in Gossypium species. Biologia 2019, 1–14. [Google Scholar] [CrossRef]
- Juturu, V.N.; Mekala, G.K.; Kirti, P. Current status of tissue culture and genetic transformation research in cotton (Gossypium spp.). Plant Cell Tissue Organ Cult. (PCTOC) 2015, 120, 813–839. [Google Scholar] [CrossRef]
- Kirungu, J.; Deng, Y.; Cai, X.; Magwanga, R.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Wang, K.; Liu, F. Simple sequence repeat (SSR) genetic linkage map of D genome diploid cotton derived from an interspecific cross between Gossypium davidsonii and Gossypium klotzschianum. Int. J. Mol. Sci. 2018, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, Y.; Chen, D.; Liu, D.; Hu, M.; Dong, J.; Zhang, X.; Song, L.; Shen, F. The catalase gene family in cotton: Genome-wide characterization and bioinformatics analysis. Cells 2019, 8, 86. [Google Scholar] [CrossRef]
- Hundertmark, M.; Hincha, D.K. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.; Lu, P.; Kirungu, J.; Cai, X.; Zhou, Z.; Wang, X.; Diouf, L.; Xu, Y.; Hou, Y.; Hu, Y. Whole genome analysis of cyclin dependent kinase (CDK) gene family in cotton and functional evaluation of the role of CDKF4 Gene in drought and salt stress tolerance in plants. Int. J. Mol. Sci. 2018, 19, 2625. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Lehti-Shiu, M.D.; Thomashow, M.; Shiu, S.-H. Evolution of stress-regulated gene expression in duplicate genes of Arabidopsis thaliana. PLoS Genet. 2009, 5, e1000581. [Google Scholar] [CrossRef] [PubMed]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef]
- Blanc, G.; Hokamp, K.; Wolfe, K.H. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Vandepoele, K.; Raes, J.; De Veylder, L.; Rouzé, P.; Rombauts, S.; Inzé, D. Genome-wide analysis of core cell cycle genes in Arabidopsis. Plant Cell 2002, 14, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Dossa, K.; Diouf, D.; Cissé, N. Genome-wide investigation of Hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front. Plant Sci. 2016, 7, 1522. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Baker, R.J. Subgenome-specific markers in allopolyploid cotton Gossypium hirsutum: Implications for evolutionary analysis of polyploids. Gene 2003, 306, 99–103. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Xu, F.-C.; Guo, D.-D.; Zhao, J.-R.; Liu, J.; Guo, Y.-W.; Singh, P.K.; Ma, X.-N.; Long, L.; Botella, J.R. Calcium-dependent protein kinases in cotton: Insights into early plant responses to salt stress. BMC Plant Biol. 2018, 18, 15. [Google Scholar] [CrossRef]
- Grant, D.; Cregan, P.; Shoemaker, R.C. Genome organization in dicots: Genome duplication in Arabidopsis and synteny between soybean and Arabidopsis. Proc. Natl. Acad. Sci. USA 2000, 97, 4168–4173. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Wang, X.; Fu, L.; Sun, J.; Zheng, W.; Li, Z. Identification of the trehalose-6-phosphate synthase gene family in winter wheat and expression analysis under conditions of freezing stress. J. Genet. 2015, 94, 55–65. [Google Scholar] [CrossRef]
- Initiative, A. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796. [Google Scholar] [CrossRef]
- Jain, M.; Nijhawan, A.; Arora, R.; Agarwal, P.; Ray, S.; Sharma, P.; Kapoor, S.; Tyagi, A.K.; Khurana, J.P. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007, 143, 1467–1483. [Google Scholar] [CrossRef]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide analysis, evolutionary expansion, and expression of early auxin-responsive SAUR gene family in rice (Oryza sativa). Genomics 2006, 88, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Culiat, C.T.; Chow, V.T.; Pervaiz, S. Functional and evolutionary analyses on expressed intronless genes in the mouse genome. FEBS Lett. 2006, 580, 1472–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.-H.; Willmann, M.R.; Chen, H.-C.; Sheen, J. Calcium signaling through protein kinases. The Arabidopsis calcium-dependent protein kinase gene family. Plant Physiol. 2002, 129, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Lv, W.; Jiang, S.; Zhang, D.; Cai, G.; Pan, J.; Li, D. Genome-wide identification and expression analysis of calcium-dependent protein kinase in maize. BMC Genom. 2013, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.G.O. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Kulik, A.; Wawer, I.; Krzywińska, E.; Bucholc, M.; Dobrowolska, G. SnRK2 protein kinases—Key regulators of plant response to abiotic stresses. Omics J. Integr. Biol. 2011, 15, 859–872. [Google Scholar] [CrossRef]
- Ghelis, T.; Bolbach, G.; Clodic, G.; Habricot, Y.; Miginiac, E.; Sotta, B.; Jeannette, E. Protein tyrosine kinases and protein tyrosine phosphatases are involved in abscisic acid-dependent processes in Arabidopsis seeds and suspension cells. Plant Physiol. 2008, 148, 1668–1680. [Google Scholar] [CrossRef]
- Kim, J.-S.; Mizoi, J.; Yoshida, T.; Fujita, Y.; Nakajima, J.; Ohori, T.; Todaka, D.; Nakashima, K.; Hirayama, T.; Shinozaki, K. An ABRE promoter sequence is involved in osmotic stress-responsive expression of the DREB2A gene, which encodes a transcription factor regulating drought-inducible genes in Arabidopsis. Plant Cell Physiol. 2011, 52, 2136–2146. [Google Scholar] [CrossRef]
- Li, C.; Ng, C.K.-Y.; Fan, L.-M. MYB transcription factors, active players in abiotic stress signaling. Environ. Exp. Bot. 2015, 114, 80–91. [Google Scholar] [CrossRef]
- Nakamichi, N.; Takao, S.; Kudo, T.; Kiba, T.; Wang, Y.; Kinoshita, T.; Sakakibara, H. Improvement of Arabidopsis biomass and cold, drought and salinity stress tolerance by modified circadian clock-associated PSEUDO-RESPONSE REGULATORs. Plant Cell Physiol. 2016, 57, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Zhao, J.-H.; Guo, H.-S. Trans-kingdom RNA silencing in plant–fungal pathogen interactions. Mol. Plant 2018, 11, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, D.; Zhang, X.; Chen, D.; Cheng, Y.; Shen, F. Plant microRNAs in cross-kingdom regulation of gene expression. Int. J. Mol. Sci. 2018, 19, 2007. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Burd, S.; Lers, A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015, 84, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W.; Chen, J.; Liu, J.; Xia, M.; Shen, F. Identification of miRNAs and their targets in cotton inoculated with Verticillium dahliae by high-throughput sequencing and degradome analysis. Int. J. Mol. Sci. 2015, 16, 14749–14768. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [Green Version]
- Marone, D.; Russo, M.; Laidò, G.; De Leonardis, A.; Mastrangelo, A. Plant nucleotide binding site–leucine-rich repeat (NBS-LRR) genes: Active guardians in host defense responses. Int. J. Mol. Sci. 2013, 14, 7302–7326. [Google Scholar] [CrossRef] [PubMed]
- Chiou, T.-J.; Aung, K.; Lin, S.-I.; Wu, C.-C.; Chiang, S.-F.; Su, C.-l. Regulation of phosphate homeostasis by microRNA in Arabidopsis. Plant Cell 2006, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.-S.P. Sensing the environment: Key roles of membrane-localized kinases in plant perception and response to abiotic stress. J. Exp. Bot. 2013, 64, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo Barrios, L.d.; Merchán Ignacio, F.; Laporte, P.; Thompson, R.D.; Clarke, J.H.; Sousa Martín, C.; Crespi, M.D. A novel plant leucine-rich repeat receptor kinase regulates the response of Medicago truncatula roots to salt stress. Plant Cell 2009, 21, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sheng, P.; Tan, J.; Chen, X.; Lu, G.; Ma, W.; Heng, Y.; Lin, Q.; Zhu, S.; Wang, J. Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the drought and salt tolerance transcription factor to regulate drought sensitivity in rice. J. Exp. Bot. 2014, 66, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-G.; Kim, D.S.; Jang, C.S. Comparative analysis of evolutionary dynamics of genes encoding leucine-rich repeat receptor-like kinase between rice and Arabidopsis. Genetica 2011, 139, 1023. [Google Scholar] [CrossRef] [PubMed]
- Chae, L.; Sudat, S.; Dudoit, S.; Zhu, T.; Luan, S. Diverse transcriptional programs associated with environmental stress and hormones in the Arabidopsis receptor-like kinase gene family. Mol. Plant 2009, 2, 84–107. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chu, X.; Li, Y.; Wang, C.; Guo, X. Cotton GhMKK1 induces the tolerance of salt and drought stress, and mediates defence responses to pathogen infection in transgenic Nicotiana benthamiana. PLoS ONE 2013, 8, e68503. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Lu, W.; Meng, F.; Wu, C.-A.; Guo, X. Cotton GhMKK5 affects disease resistance, induces HR-like cell death, and reduces the tolerance to salt and drought stress in transgenic Nicotiana benthamiana. J. Exp. Bot. 2012, 63, 3935–3951. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gossypium genome | Chromosome No. | Total Number of Genes | Gossypium genome | Chromosome No. | Total Number of Genes |
|---|---|---|---|---|---|
| (AD) Genome | At1 | 6 | A03 | 2 | |
| At2 | 5 | A04 | 3 | ||
| At5 | 4 | A05 | 1 | ||
| At7 | 2 | A07 | 2 | ||
| At8 | 1 | A08 | 1 | ||
| At9 | 4 | A09 | 5 | ||
| At10 | 1 | A10 | 1 | ||
| At11 | 8 | A11 | 7 | ||
| At12 | 3 | A12 | 3 | ||
| Dt14 | 1 | Scaffolds | 1 | ||
| Dt15 | 9 | Total | 35 | ||
| Dt16 | 2 | (D) Genome | D01 | 2 | |
| Dt17 | 3 | D02 | 10 | ||
| Dt18 | 2 | D03 | 4 | ||
| Dt19 | 2 | D04 | 1 | ||
| Dt20 | 2 | D05 | 1 | ||
| Dt21 | 7 | D06 | 6 | ||
| Dt22 | 2 | D07 | 6 | ||
| Dt23 | 5 | D08 | 4 | ||
| Dt24 | 1 | D09 | 2 | ||
| Dt26 | 4 | D11 | 1 | ||
| Scaffolds | 1 | D12 | 2 | ||
| Total | 75 | D13 | 2 | ||
| (A) Genome | A01 | 7 | Total | 41 | |
| A02 | 2 |
| Factor Name | Signal Sequence | Function | No. of Genes |
|---|---|---|---|
| Box 4 | ATTAAT | Involved in light responsiveness | 40 |
| TATC-box | TATCCCA | Involved in gibberellin-responsiveness | 38 |
| MYC | CAATTG | Role in cell proliferation | 50 |
| MYBS | CAACTG | Abiotic stress tolerance | 55 |
| CAAT-box | CAAT | Binding factors | 60 |
| AT-rich element | ATAGAAATCAA | AT-rich DNA binding protein (ATBP-1) | 49 |
| G-Box | CACGTT | In network regulating flag leaf senescence process | 40 |
| TATA-box | TATA | Core promoter element | 55 |
| ARE | AAACCA | Essential for the anaerobic induction | 41 |
| MBS | CAACTG | Involved in drought-inducibility | 53 |
| ABRE | CGCACGTGTC | Involved in the abscisic acid responsiveness | 54 |
| Myb-binding site | CAACAG | Plant Myb-binding site | 50 |
| TGACG-motif | TGACG | MeJA responsiveness | 40 |
| HD-Zip 1 | CAAT(A/T) ATG | Differentiation of palisade mesophyll cell | 20 |
| TGA-element | AACGAC | Auxin-responsive element | 35 |
| CCAAT-box | CAACGG | Plant MYBHv1 binding site | 39 |
| TCA-element | CCATCTTTTT | Salicylic acid responsiveness | 52 |
| P-box | CCTTTTG | Gibberellin-responsive element | 28 |
| DRE | GCCGAC | Stress responsiveness | 25 |
| 3-AF1 binding site | TAAGAGAGGAA | Light responsive element | 4 |
| O2-site | GATGATGTGG | Involved in zinc metabolism regulation | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shehzad, M.; Zhou, Z.; Ditta, A.; Cai, X.; Khan, M.; Xu, Y.; Hou, Y.; Peng, R.; Hao, F.; Shafeeq-ur-rahman; et al. Genome-Wide Mining and Identification of Protein Kinase Gene Family Impacts Salinity Stress Tolerance in Highly Dense Genetic Map Developed from Interspecific Cross between G. hirsutum L. and G. darwinii G. Watt. Agronomy 2019, 9, 560. https://doi.org/10.3390/agronomy9090560
Shehzad M, Zhou Z, Ditta A, Cai X, Khan M, Xu Y, Hou Y, Peng R, Hao F, Shafeeq-ur-rahman, et al. Genome-Wide Mining and Identification of Protein Kinase Gene Family Impacts Salinity Stress Tolerance in Highly Dense Genetic Map Developed from Interspecific Cross between G. hirsutum L. and G. darwinii G. Watt. Agronomy. 2019; 9(9):560. https://doi.org/10.3390/agronomy9090560
Chicago/Turabian StyleShehzad, Muhammad, Zhongli Zhou, Allah Ditta, Xiaoyan Cai, Majid Khan, Yanchao Xu, Yuqing Hou, Renhai Peng, Fushun Hao, Shafeeq-ur-rahman, and et al. 2019. "Genome-Wide Mining and Identification of Protein Kinase Gene Family Impacts Salinity Stress Tolerance in Highly Dense Genetic Map Developed from Interspecific Cross between G. hirsutum L. and G. darwinii G. Watt" Agronomy 9, no. 9: 560. https://doi.org/10.3390/agronomy9090560