Genome-Wide Mining and Characterization of SSR Markers for Gene Mapping and Gene Diversity in Gossypium barbadense L. and Gossypium darwinii G. Watt Accessions

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. PCR Primer Selection and Amplification
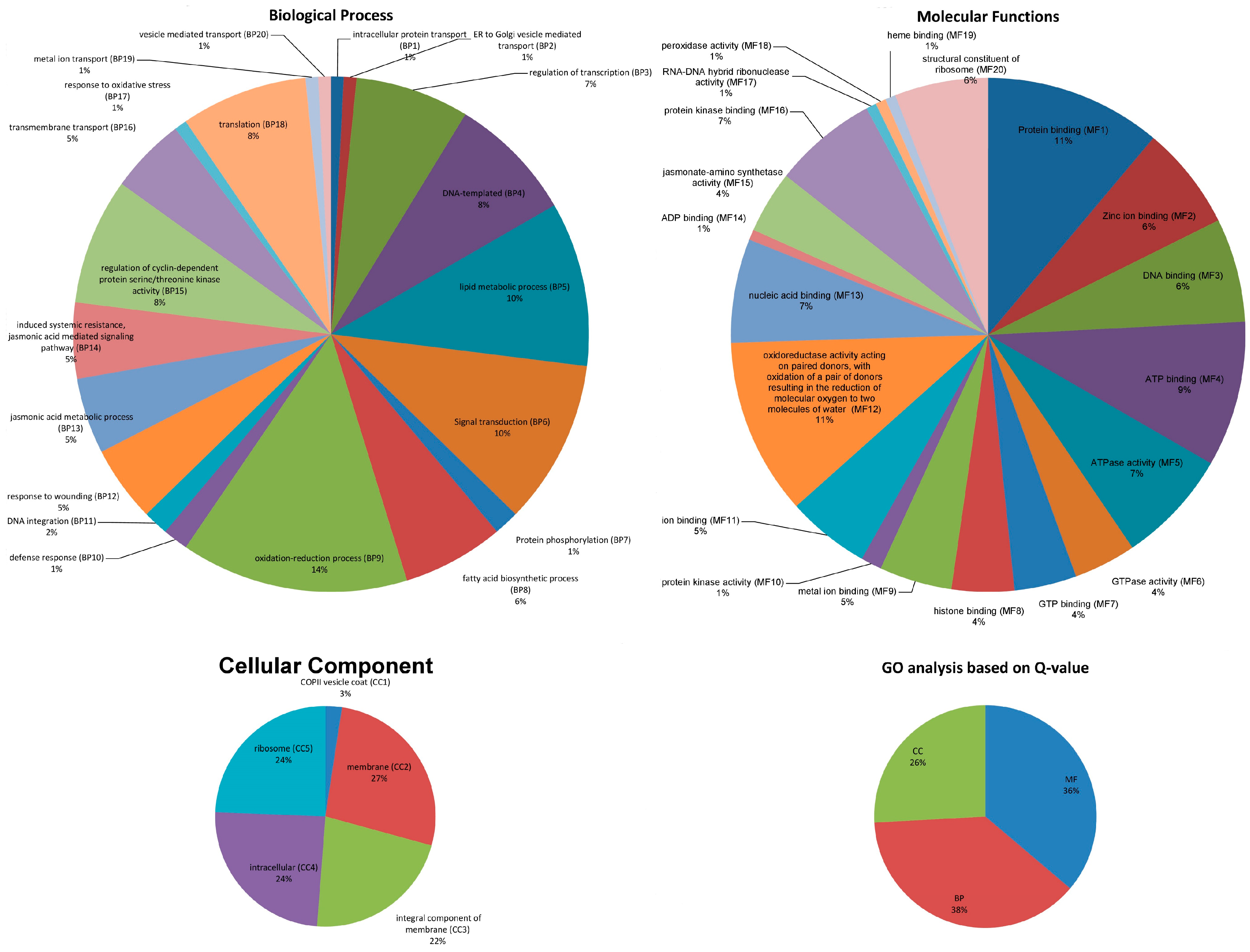
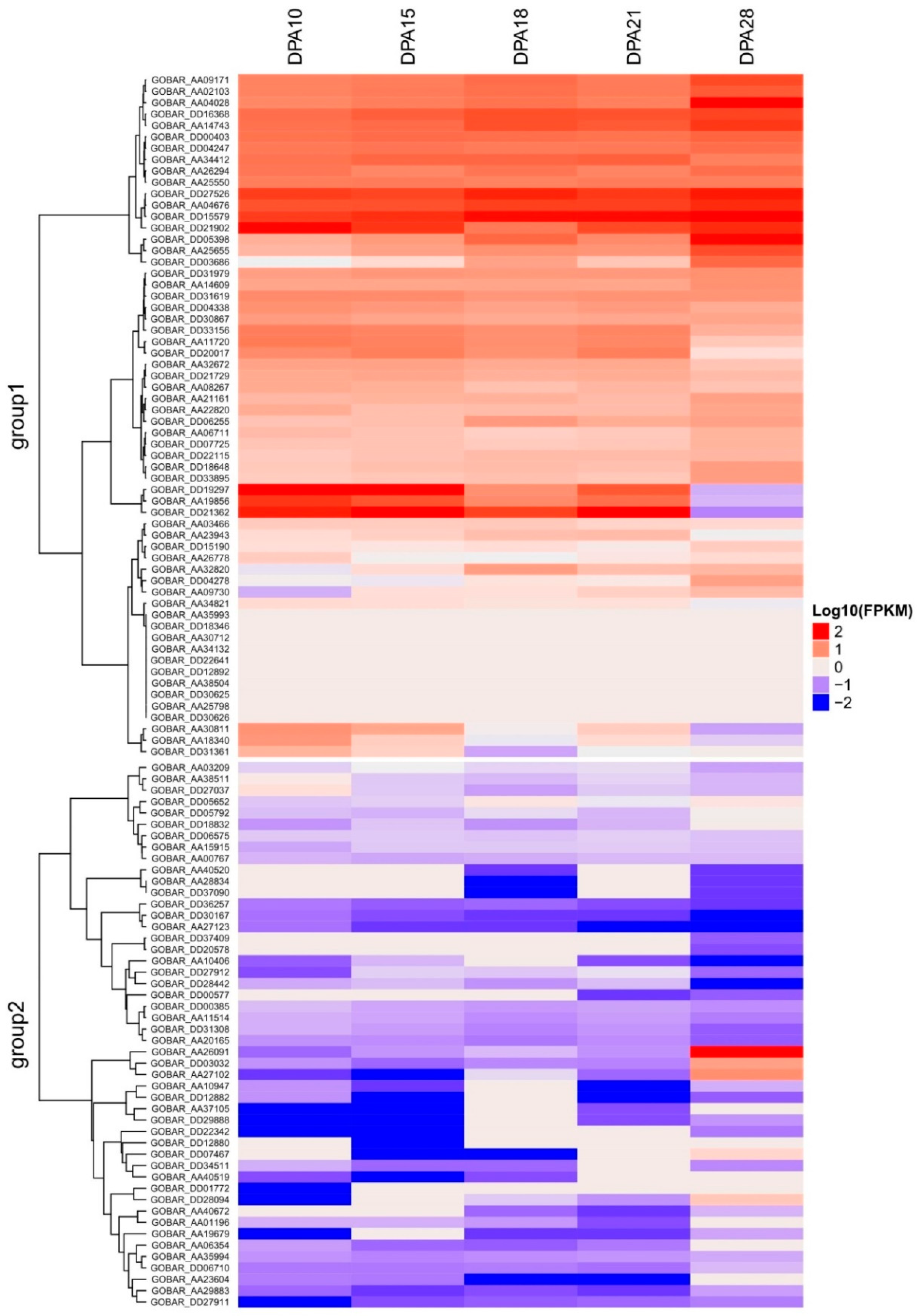
2.3. Identification of Genes with GO Functional Classification and Expression Analysis
2.4. Statistical Analysis
2.4.1. Polymorphism Analysis
2.4.2. Genetic Diversity Analysis
3. Results
3.1. Genome-Wide Mining and Characterization of Functional SSR Markers
3.2. GO Functional Annotations and Expression Analysis
3.3. Analysis of Genetic Diversity
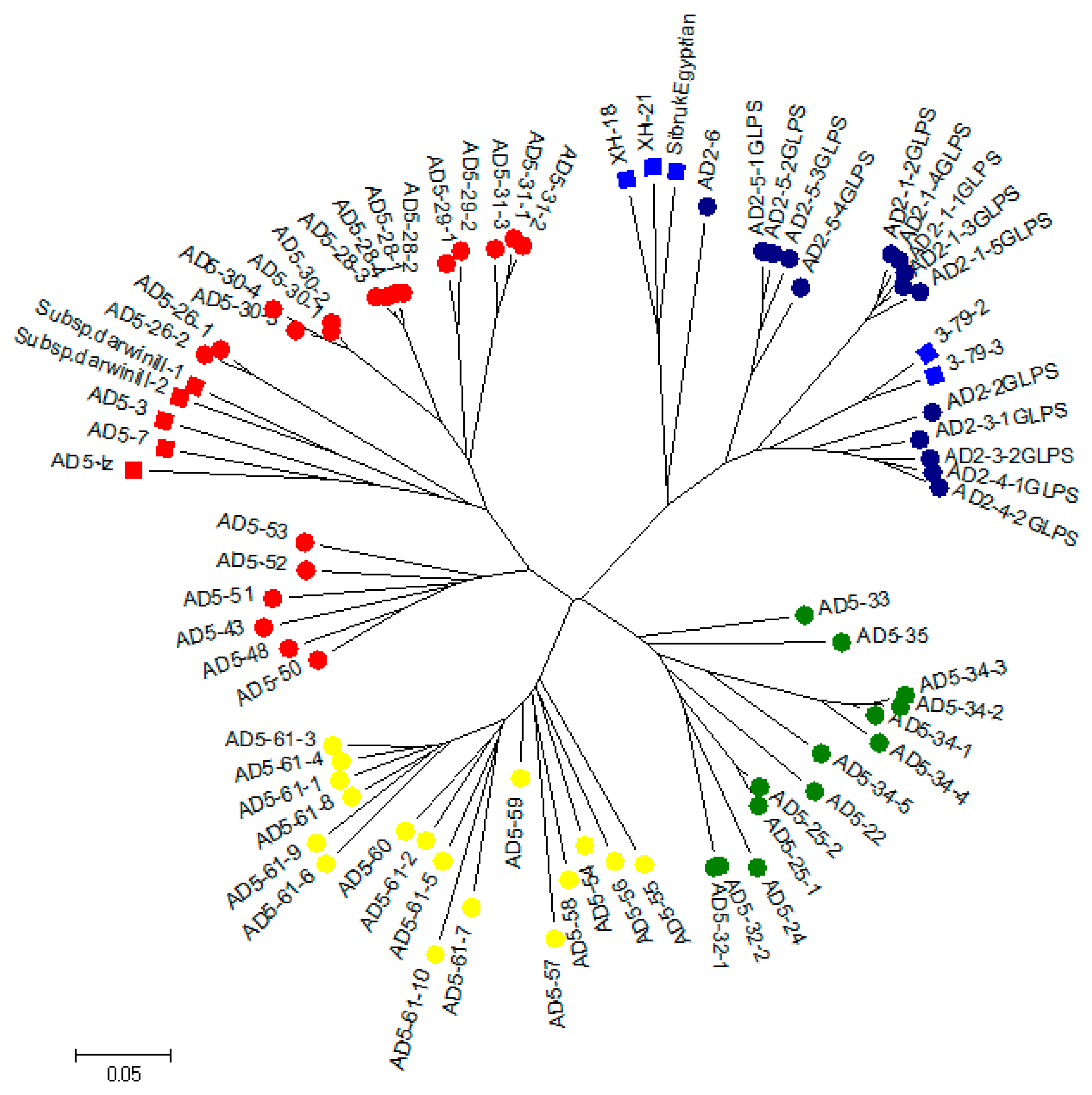
3.4. Phylogenetic Tree Analysis
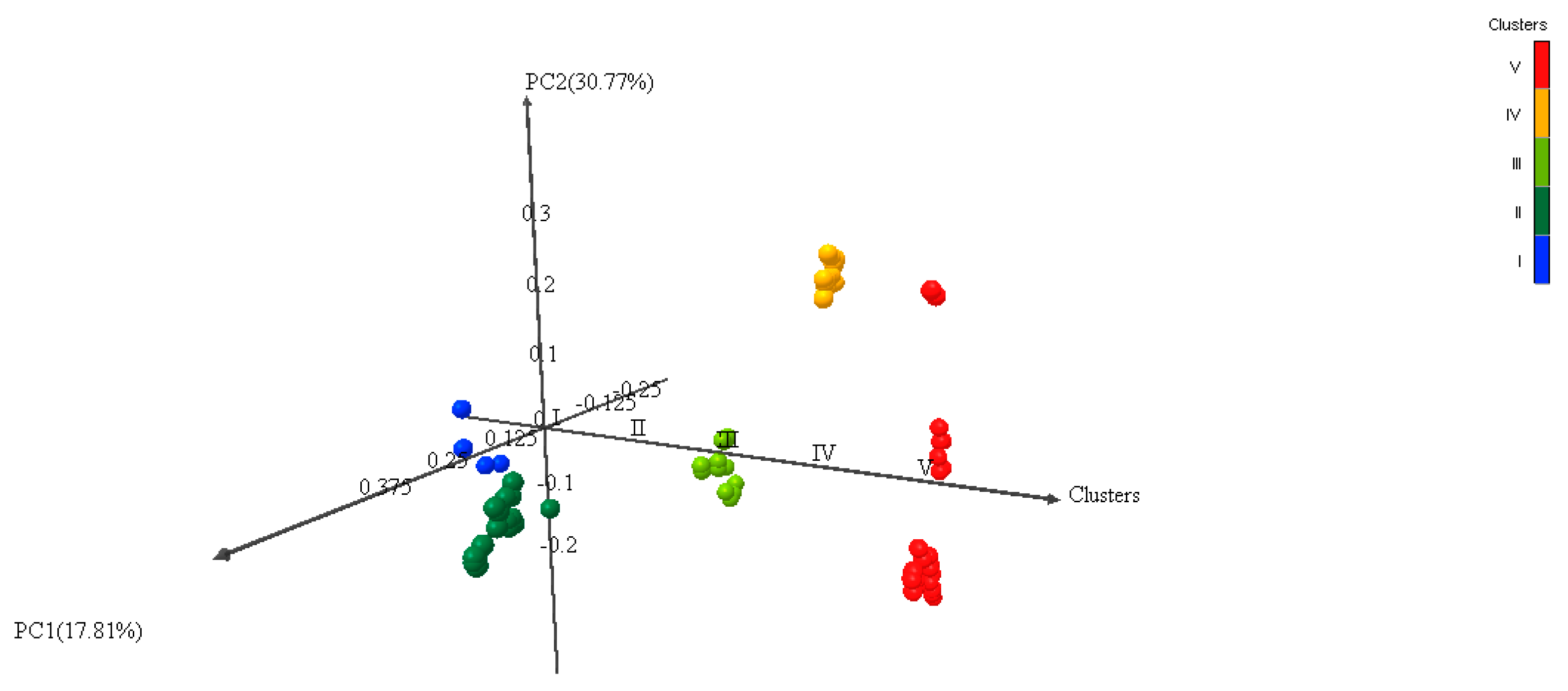
3.5. Multivariate Analysis of Genetic Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PIC | polymorphism information content |
| GD | genetic distance |
| Pop | population |
| AMOVA | analysis of molecular variance |
| PCoA | principal coordinate analysis |
| PCR | polymerase chain reaction |
| DNA | deoxyribonucleic acid |
| EST | expressed sequence tags |
| gSSR | genomic SSR |
| SSR | simple sequence repeats |
| DP | discrimination power |
| G | Gossypium |
| NAU | Nanjing Agriculture University |
References
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Grover, C.E. Taxonomy and evolution of the cotton genus, Gossypium. Cotton 2015, 57, 25–44. [Google Scholar]
- Zhang, J.; Lu, Y.; Yu, S. Cleaved aflp (caflp), a modified amplified fragment length polymorphism analysis for cotton. Theor. Appl. Genet. 2005, 111, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Hammer, K. Resolving the Challenge Posed by Agrobiodiversity and Plant Genetic Resouces—An Attempt; Kassel University Press: Kassel, Germany, 2004; 184p. [Google Scholar]
- Borlaug, N.E. Sixty-two years of fighting hunger: Personal recollections. Euphytica 2007, 157, 287–297. [Google Scholar] [CrossRef]
- Gerald, N.D.L.F.; Ursula, K.-F.; Thomas, L. Accelerating plant breeding. Cell 2013, 18, 667–672. [Google Scholar]
- Kadirvel, P.; Senthilvel, S.; Geethanjali, S.; Sujatha, M.; Varaprasad, K. Genetic markers, trait mapping and marker-assisted selection in plant breeding. In Plant Biology and Biotechnology; Springer India: New Delhi, India, 2015; pp. 65–88. [Google Scholar]
- Wang, Q.; Fang, L.; Chen, J.; Hu, Y.; Si, Z.; Wang, S.; Chang, L.; Guo, W.; Zhang, T. Genome-wide mining, characterization, and development of microsatellite markers in Gossypium species. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Cato, S.; Gardner, R.; Kent, J.; Richardson, T. A rapid pcr-based method for genetically mapping ests. Theor. Appl. Genet. 2001, 102, 296–306. [Google Scholar] [CrossRef]
- Liang, F.; Holt, I.; Pertea, G.; Karamycheva, S.; Salzberg, S.L.; Quackenbush, J. An optimized protocol for analysis of est sequences. Nucleic Acids Res. 2000, 28, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- John, Z.Y.; Fang, D.D.; Kohel, R.J.; Ulloa, M.; Hinze, L.L.; Percy, R.G.; Zhang, J.; Chee, P.; Scheffler, B.E.; Jones, D.C. Development of a core set of ssr markers for the characterization of Gossypium germplasm. Euphytica 2012, 187, 203–213. [Google Scholar]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of est-ssr markers and population genetic analyses for the desert biomass willow, salix psammophila. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-K.; Dake, T.M.; Singh, S.; Benscher, D.; Li, W.; Gill, B.; Sorrells, M.E. Development and mapping of est-derived simple sequence repeat markers for hexaploid wheat. Genome 2004, 47, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting est databases for the development and characterization of gene-derived ssr-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.A. Preliminary analysis of microsatellite markers derived from sugarcane expressed sequence tags (ests). Genet. Mol. Biol. 2001, 24, 155–159. [Google Scholar] [CrossRef]
- Feng, S.P.; Li, W.G.; Huang, H.S.; Wang, J.Y.; Wu, Y.T. Development, characterization and cross-species/genera transferability of est-ssr markers for rubber tree (Hevea brasiliensis). Mol. Breed. 2008, 23, 85–97. [Google Scholar] [CrossRef]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. Cottonfgd: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Guo, X.; Zhao, P.; Ruan, M.; Yu, X.; Zou, L.; Yang, Y.; Li, X.; Deng, D.; Xiao, J. Molecular diversity analysis, drought related marker-traits association mapping and discovery of excellent alleles for 100-day old plants by est-ssrs in cassava germplasms (Manihot esculenta Cranz). PLoS ONE 2017, 12, e0177456. [Google Scholar] [CrossRef] [PubMed]
- Percy, R.; Wendel, J.F. Allozyme evidence for the origin and diversification of Gossypium barbadense L. Theor. Appl. Genet. 1990, 79, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stewart, J. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Yu, J.; Jung, S.; Cheng, C.-H.; Ficklin, S.P.; Lee, T.; Zheng, P.; Jones, D.; Percy, R.G.; Main, D. Cottongen: A genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 2013, 42, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Okubazghi, K.W.; Li, X.-N.; Cai, X.-Y.; Wang, X.-X.; Chen, H.-D.; Zhou, Z.-L.; Wang, C.-Y.; Wang, Y.-H.; Fang, L.; Wang, K.-B. Genome-wide assessment of genetic diversity and fiber quality traits characterization in Gossypium hirsutum races. J. Integr. Agric. 2017, 16, 2402–2412. [Google Scholar] [CrossRef]
- Gallagher, J.P.; Grover, C.E.; Rex, K.; Moran, M.; Wendel, J.F. A new species of cotton from wake atoll, Gossypium stephensii (Malvaceae). Syst. Bot. 2017, 42, 115–123. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, W.; Zhang, T. Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L.× Gossypium barbadense L.) with a haploid population. Theor. Appl. Genet. 2002, 105, 1166–1174. [Google Scholar] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44. [Google Scholar] [CrossRef] [PubMed]
- Marconi, T.G.; Costa, E.A.; Miranda, H.R.; Mancini, M.C.; Cardoso-Silva, C.B.; Oliveira, K.M.; Pinto, L.R.; Mollinari, M.; Garcia, A.A.; Souza, A.P. Functional markers for gene mapping and genetic diversity studies in sugarcane. BMC Res. Notes 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Ditta, A.; Zhou, Z.; Cai, X.; Wang, X.; Okubazghi, K.W.; Shehzad, M.; Xu, Y.; Hou, Y.; Sajid Iqbal, M.; Khan, M.K.R.; et al. Assessment of genetic diversity, population structure, and evolutionary relationship of uncharacterized genes in a novel germplasm collection of diploid and allotetraploid Gossypium accessions using est and genomic ssr markers. Int. J. Mol. Sci. 2018, 19, 2401. [Google Scholar] [CrossRef] [PubMed]
- Priyam, A.; Woodcroft, B.J.; Rai, V.; Munagala, A.; Moghul, I.; Ter, F.; Gibbins, M.A.; Moon, H.; Leonard, G.; Rumpf, W. Sequenceserver: A modern graphical user interface for custom blast databases. Biorxiv 2015. [Google Scholar] [CrossRef]
- Kuhnert, P.; Venables, B.; Zocchi, S.S. An Introduction to R: Software for Statistical Modelling & Computing; CSIRO Mathematical and Information Sciences: Cleveland, Australia, 2005. [Google Scholar]
- Anderson, J.A.; Churchill, G.; Autrique, J.; Tanksley, S.; Sorrells, M. Optimizing parental selection for genetic linkage maps. Genome 1993, 36, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Tessier, C.; David, J.; This, P.; Boursiquot, J.-M.; Charrier, A. Optimization of the choice of molecular markers for varietal identification in vitis vinifera l. Theor. Appl. Genet. 1999, 98, 171–177. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. Powermarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.M.; Pinto, L.R.; Marconi, T.G.; Margarido, G.R.; Pastina, M.M.; Teixeira, L.H.M.; Figueira, A.V.; Ulian, E.C.; Garcia, A.A.F.; Souza, A.P. Functional integrated genetic linkage map based on est-markers for a sugarcane (Saccharum spp.) commercial cross. Mol. Breed. 2007, 20, 189–208. [Google Scholar] [CrossRef]
- Schloss, S.; Mitchell, S.; White, G.; Kukatla, R.; Bowers, J.; Paterson, A.; Kresovich, S. Characterization of rflp probe sequences for gene discovery and ssr development in sorghum bicolor (L.) moench. Theor. Appl. Genet. 2002, 105, 912–920. [Google Scholar] [PubMed]
- Hinze, L.L.; Gazave, E.; Gore, M.A.; Fang, D.D.; Scheffler, B.E.; Yu, J.Z.; Jones, D.C.; Frelichowski, J.; Percy, R.G. Genetic diversity of the two commercial tetraploid cotton species in the Gossypium diversity reference set. J. Hered. 2016, 107, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Juturu, V.N.; Mekala, G.K.; Kirti, P. Current status of tissue culture and genetic transformation research in cotton (Gossypium spp.). Plant Cell Tissue Organ Cult. (PCTOC) 2015, 120, 813–839. [Google Scholar] [CrossRef]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Ann. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Hulse-Kemp, A.M.; Ashrafi, H.; Zheng, X.; Wang, F.; Hoegenauer, K.A.; Maeda, A.B.; Yang, S.S.; Stoffel, K.; Matvienko, M.; Clemons, K. Development and bin mapping of gene-associated interspecific snps for cotton (Gossypium hirsutum L.) introgression breeding efforts. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Derkx, A.; Liu, D.C.; Buchner, P.; Hawkesford, M. Overexpression of a nac transcription factor delays leaf senescence and increases grain nitrogen concentration in wheat. Plant Biol. 2015, 17, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lin, R.; Feng, J.; Chen, W.; Qiu, D.; Xu, S. Tanac1 acts as a negative regulator of stripe rust resistance in wheat, enhances susceptibility to pseudomonas syringae, and promotes lateral root development in transgenic arabidopsis thaliana. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, Y.; Li, B.; Chang, J.; Chen, M.; Li, K.; Yang, G.; He, G. Tanac29, a nac transcription factor from wheat, enhances salt and drought tolerance in transgenic arabidopsis. BMC Plant Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.T.; de Almeida Engler, J.; Raes, J.; Magyar, Z.; De Groodt, R.; Inzé, D.; De Veylder, L. Molecular characterization of arabidopsis pho80-like proteins, a novel class of cdka; 1-interacting cyclins. Cell. Mol. Life Sci. 2004, 61, 1485–1497. [Google Scholar]
- Rudrabhatla, P.; Reddy, M.M.; Rajasekharan, R. Genome-wide analysis and experimentation of plant serine/threonine/tyrosine-specific protein kinases. Plant Mol. Biol. 2006, 60, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Ann. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Ablazov, A.; Tombuloglu, H. Genome-wide identification of the mildew resistance locus o (mlo) gene family in novel cereal model species brachypodium distachyon. Eur. J. Plant Pathol. 2016, 145, 239–253. [Google Scholar] [CrossRef]
- Zorzatto, C.; Machado, J.P.B.; Lopes, K.V.; Nascimento, K.J.; Pereira, W.A.; Brustolini, O.J.; Reis, P.A.; Calil, I.P.; Deguchi, M.; Sachetto-Martins, G. Nik1-mediated translation suppression functions as a plant antiviral immunity mechanism. Nature 2015, 520. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.E.; Lynch, T.; Peeters, J.; Snowden, C.; Finkelstein, R. A small plant-specific protein family of abi five binding proteins (afps) regulates stress response in germinating arabidopsis seeds and seedlings. Plant Mol. Biol. 2008, 67, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Verrier, P.J.; Bird, D.; Burla, B.; Dassa, E.; Forestier, C.; Geisler, M.; Klein, M.; Kolukisaoglu, U.; Lee, Y.; Martinoia, E. Plant abc proteins—A unified nomenclature and updated inventory. Trends Plant Sci. 2008, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Rancour, D.M.; Park, S.; Knight, S.D.; Bednarek, S.Y. Plant ubx-domain containing protein 1, pux1, regulates the oligomeric structure and activity of arabidopsis cdc48. J. Biol. Chem. 2004, 279, 54264–54274. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.-H.; Karlowski, W.M.; Pan, R.; Tzeng, Y.-H.; Mayer, K.F.; Li, W.-H. Comparative analysis of the receptor-like kinase family in arabidopsis and rice. Plant Cell 2004, 16, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Serna, L. The role of brassinosteroids and abscisic acid in stomatal development. Plant Sci. 2014, 225, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, M.; Jia, F.; Gong, J.; Palanga, K.K.; Shi, Y.; Li, J.; Shang, H.; Liu, A.; Chen, T.; Zhang, Z. Identification of stable quantitative trait loci (qtls) for fiber quality traits across multiple environments in Gossypium hirsutum recombinant inbred line population. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Bourgou, L.; Sawadogo, M.; Sanfo, D.; Lacape, J.-M. Ssr-based genetic diversity of traditional and perennial cotton (Gossypium spp.) populations collected in burkina faso. Genet. Resour. Crop Evol. 2017, 64, 1743–1759. [Google Scholar] [CrossRef]
- Ashraf, J.; Malik, W.; Iqbal, M.; Khan, A.; Qayyum, A.; Noor, E.; Abid, M.; Cheema, H.; Ahmad, M. Comparative analysis of genetic diversity among bt cotton genotypes using est-ssr, issr and morphological markers. J. Agric. Sci. Technol. 2016, 18, 517–531. [Google Scholar]
- Sethi, K.; Siwach, P.; Verma, S.K. Assessing genetic diversity among six populations of Gossypium arboreum L. Using microsatellites markers. Physiol. Mol. Biol. Plants 2015, 21, 531–539. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Species Name | Genome Type | Total No. | No. of Accessions | Place of Collection |
|---|---|---|---|---|---|
| 1 | G. barbadense | AD2 | 20 | 15 | Santa Cruz, Galapagos |
| 5 | Wild cotton germplasm nursery of China | ||||
| 2 | G. darwinii | AD5 | 59 | 13 | Santa Cruz, Galapagos |
| 25 | Isabella | ||||
| 16 | San Cristobal | ||||
| 5 | Wild cotton germplasm nursery of China |
| Source | df | SS | MS | Est. Var. | % |
|---|---|---|---|---|---|
| Among Pops | 4 | 1591.508 | 397.877 | 23.919 | 38% |
| Within Pops | 74 | 2915.137 | 39.394 | 39.394 | 62% |
| Total | 78 | 4506.646 | 63.313 | 100% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ditta, A.; Zhou, Z.; Cai, X.; Shehzad, M.; Wang, X.; Okubazghi, K.W.; Xu, Y.; Hou, Y.; Sajid Iqbal, M.; Khan, M.K.R.; et al. Genome-Wide Mining and Characterization of SSR Markers for Gene Mapping and Gene Diversity in Gossypium barbadense L. and Gossypium darwinii G. Watt Accessions. Agronomy 2018, 8, 181. https://doi.org/10.3390/agronomy8090181
Ditta A, Zhou Z, Cai X, Shehzad M, Wang X, Okubazghi KW, Xu Y, Hou Y, Sajid Iqbal M, Khan MKR, et al. Genome-Wide Mining and Characterization of SSR Markers for Gene Mapping and Gene Diversity in Gossypium barbadense L. and Gossypium darwinii G. Watt Accessions. Agronomy. 2018; 8(9):181. https://doi.org/10.3390/agronomy8090181
Chicago/Turabian StyleDitta, Allah, Zhongli Zhou, Xiaoyan Cai, Muhammad Shehzad, Xingxing Wang, Kiflom Weldu Okubazghi, Yanchao Xu, Yuqing Hou, Muhammad Sajid Iqbal, Muhammad Kashif Riaz Khan, and et al. 2018. "Genome-Wide Mining and Characterization of SSR Markers for Gene Mapping and Gene Diversity in Gossypium barbadense L. and Gossypium darwinii G. Watt Accessions" Agronomy 8, no. 9: 181. https://doi.org/10.3390/agronomy8090181
APA StyleDitta, A., Zhou, Z., Cai, X., Shehzad, M., Wang, X., Okubazghi, K. W., Xu, Y., Hou, Y., Sajid Iqbal, M., Khan, M. K. R., Wang, K., & Liu, F. (2018). Genome-Wide Mining and Characterization of SSR Markers for Gene Mapping and Gene Diversity in Gossypium barbadense L. and Gossypium darwinii G. Watt Accessions. Agronomy, 8(9), 181. https://doi.org/10.3390/agronomy8090181