Genetic Variation in Soybean at the Maturity Locus E4 Is Involved in Adaptation to Long Days at High Latitudes

Abstract
:
1. Introduction
2. Results
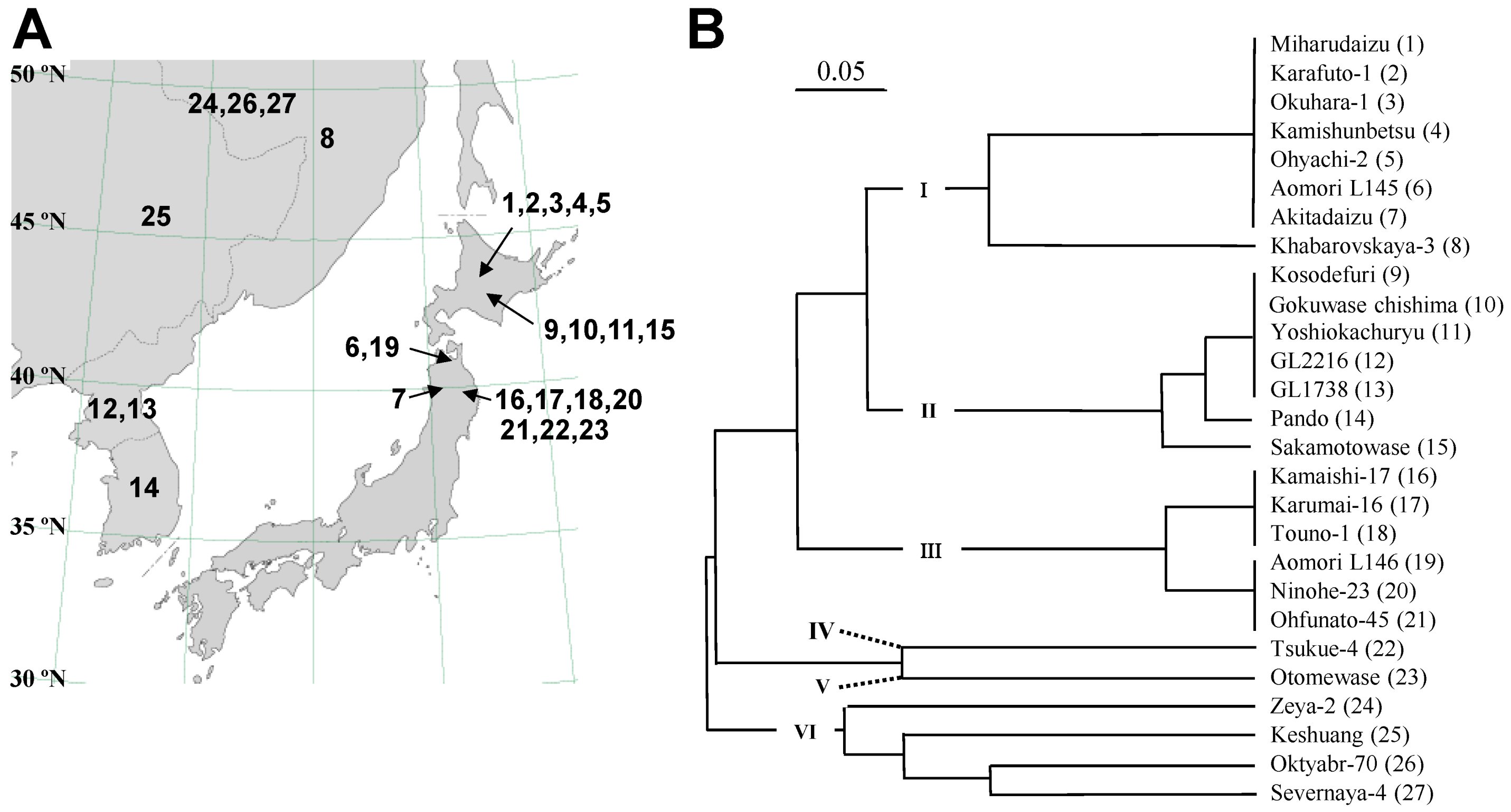
2.1. Classification of Photoperiod-Insensitive Soybean Accessions
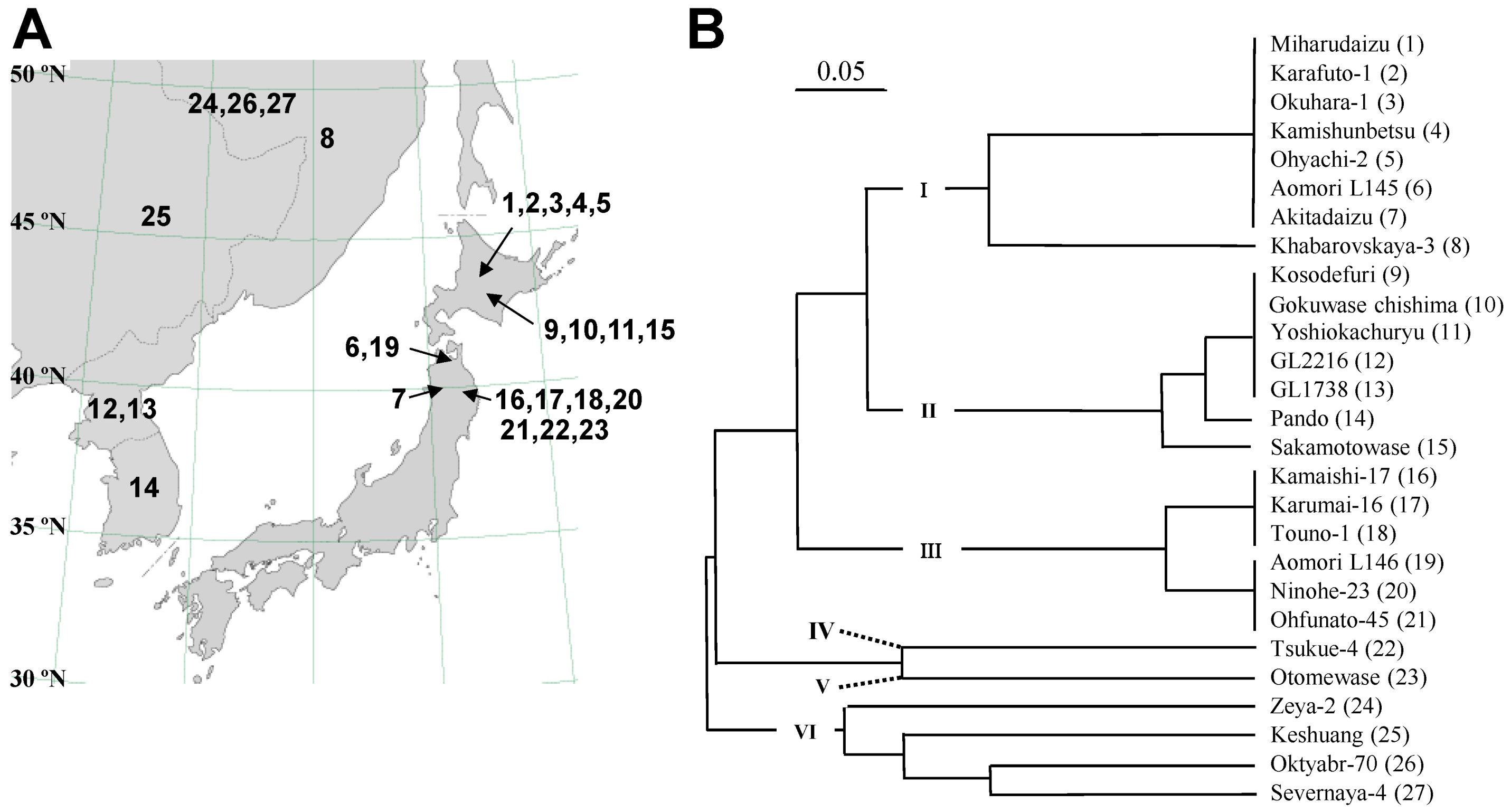
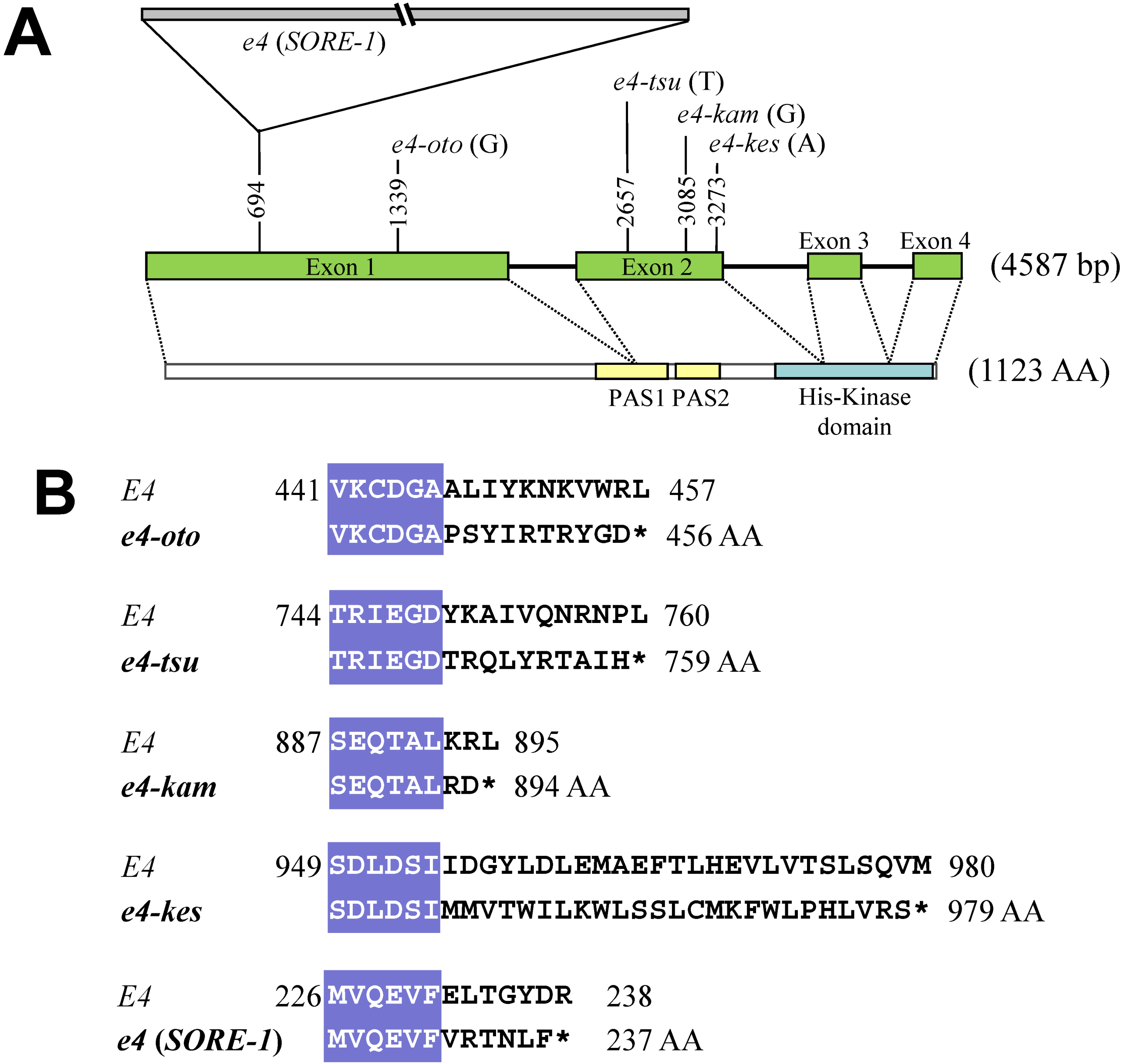
2.2. Sequence and DNA Marker Analyses of E4
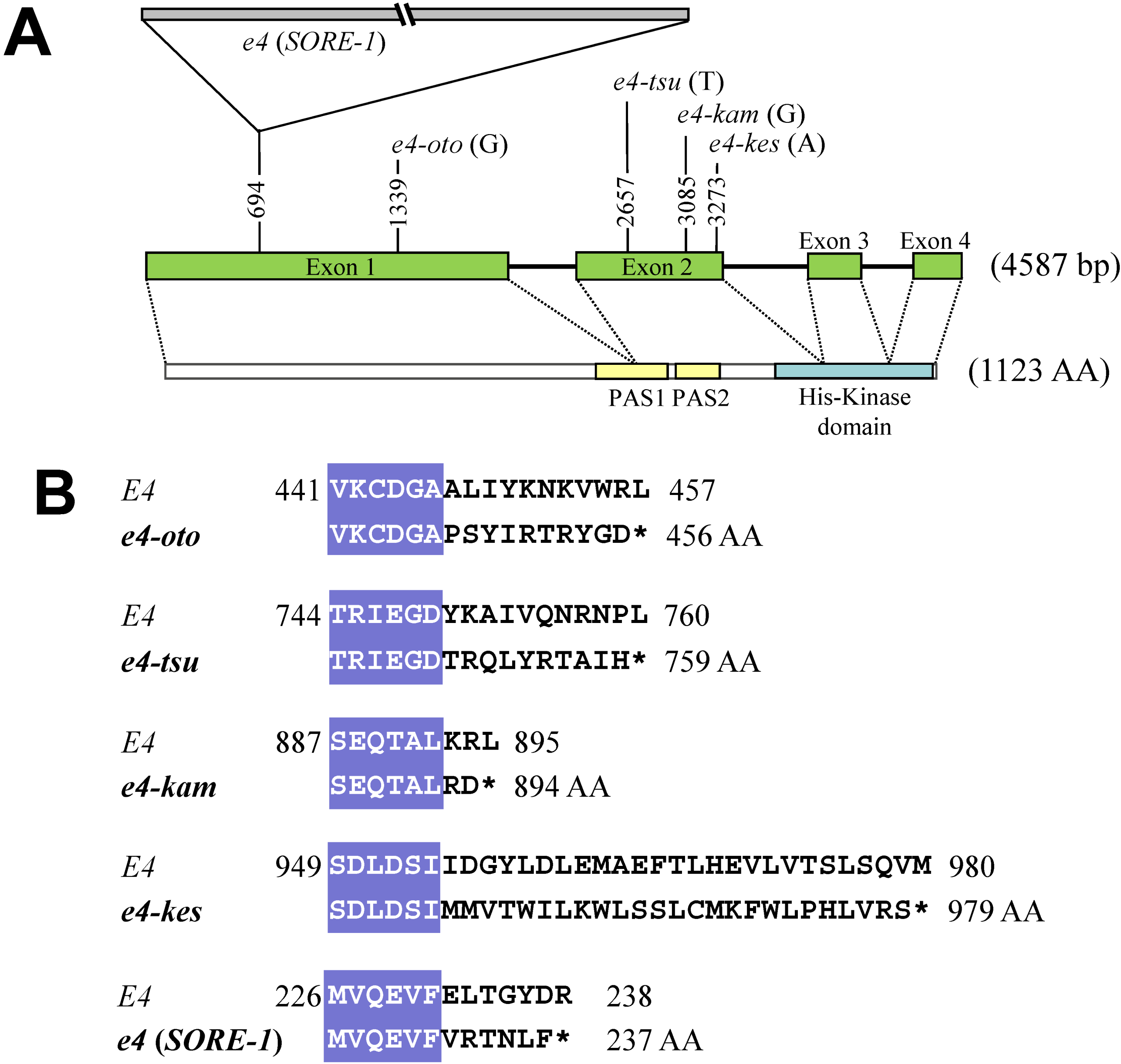
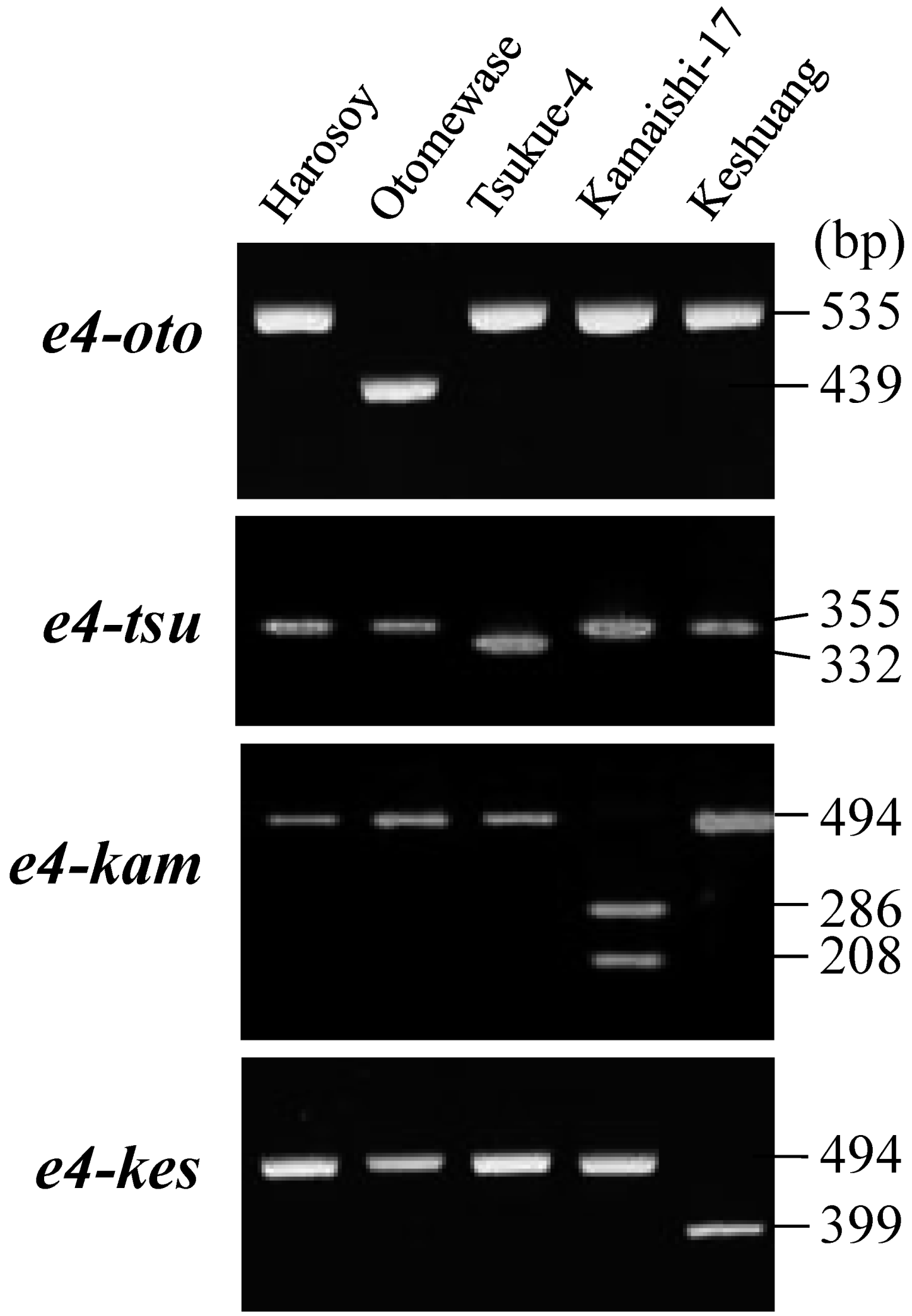
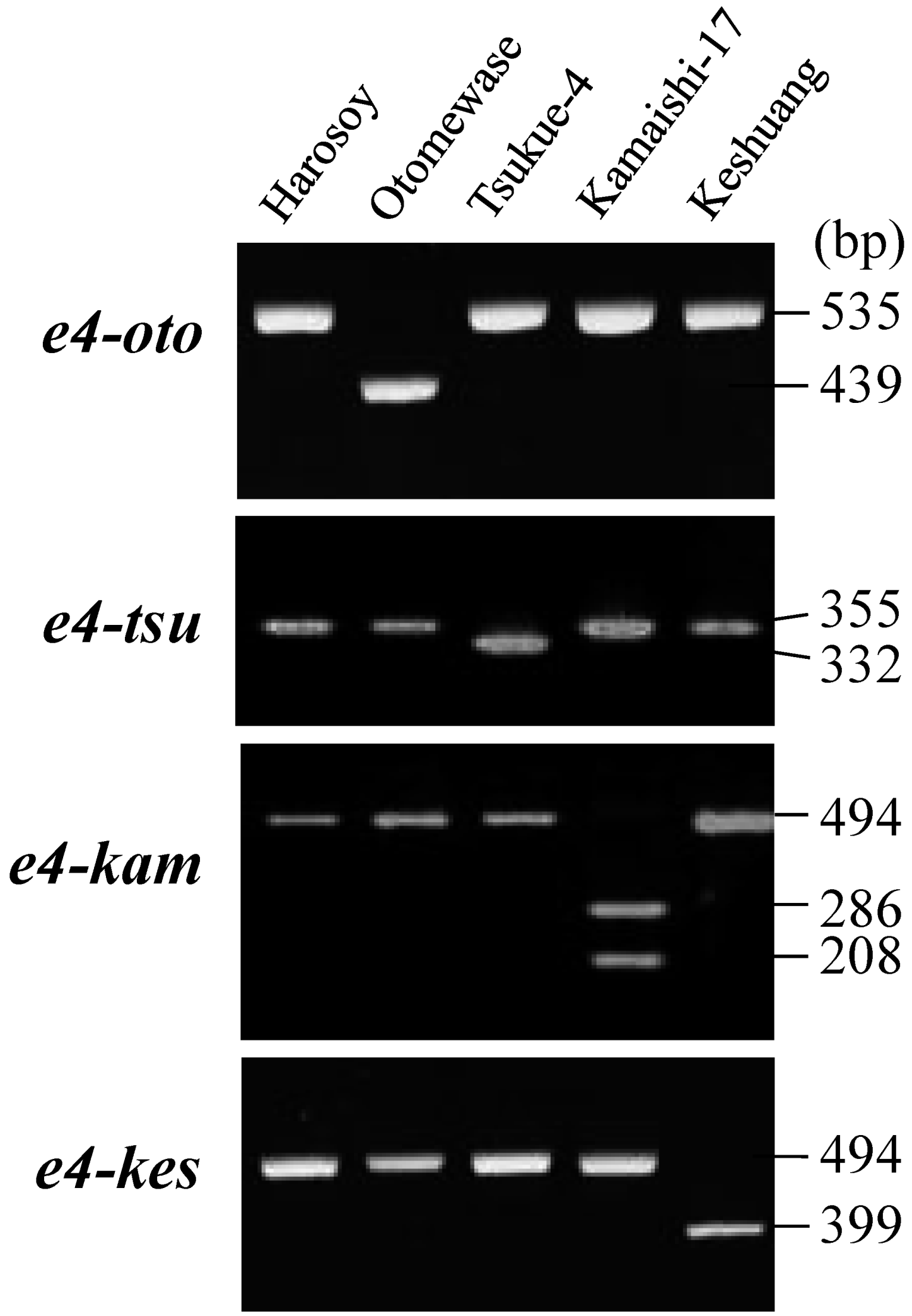
2.3. Survey of Genetic Variation Using Allele-Specific DNA Markers
2.4. Comparison of Nucleotide Diversity between E4 and GmphyA1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | S | Hap | π (s) (× 10−3) | θ (s) (× 10−3) | π (a) (× 10−3) | θ (a) (× 10−3) | |
|---|---|---|---|---|---|---|---|
| GmphyA1 | |||||||
| Cultivated soybean | 35 | 17 | 6 | 1.35 | 1.03 | 0.02 | 0.09 |
| Wild soybean | 24 | 18 | 12 | 1.31 | 1.40 | 0.03 | 0.10 |
| Combined | 59 | 20 | 15 | 1.35 | 1.36 | 0.03 | 0.16 |
| E4 (GmphyA2) | |||||||
| Cultivated soybean | 52 | 18 | 7 | 0.13 | 0.66 | 0.13 | 0.34 |
| Wild soybean | 25 | 39 | 13 | 2.11 | 2.01 | 0.56 | 0.61 |
| Combined | 77 | 44 | 19 | 1.12 | 1.65 | 0.39 | 0.70 |
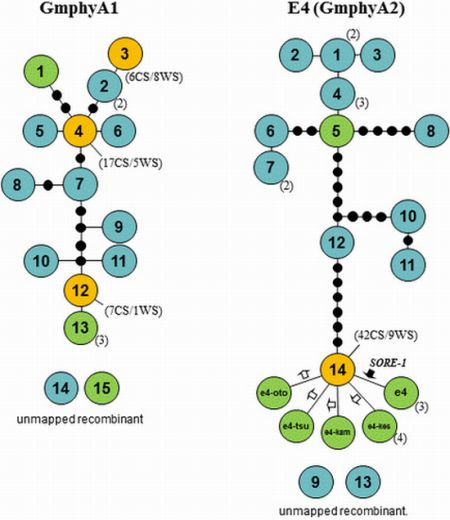
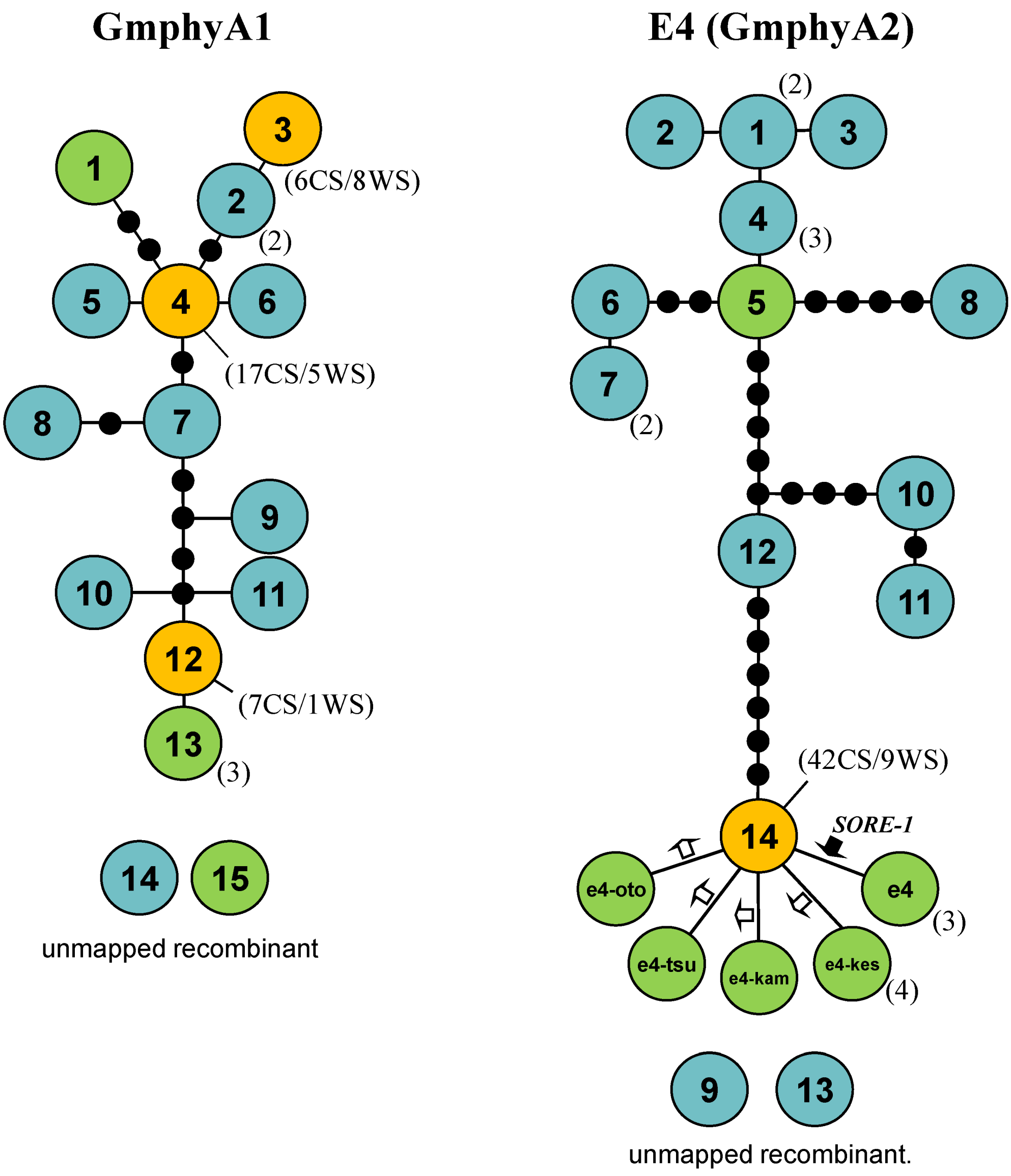
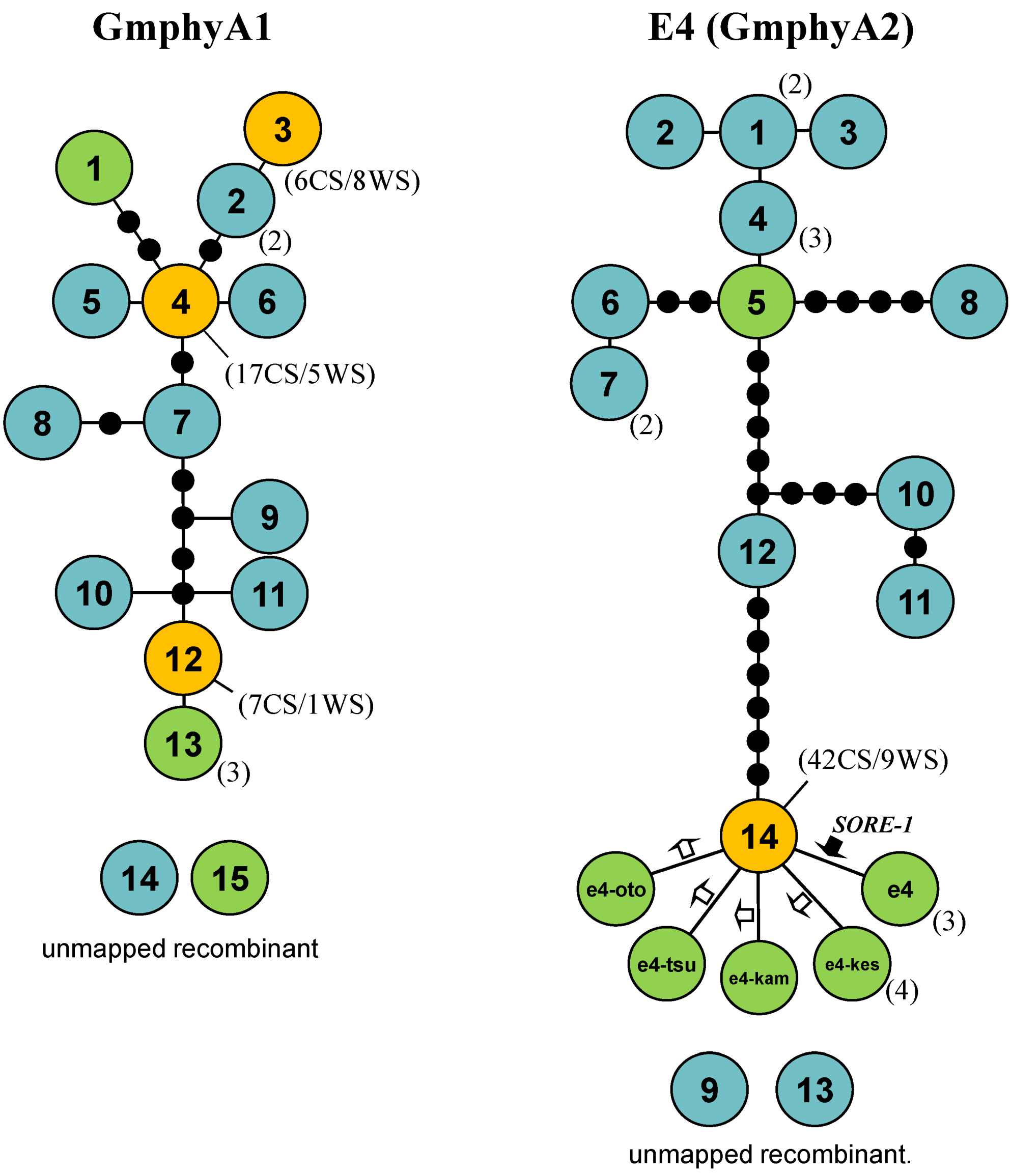
2.5. Haplotype Networks
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Methods
4.2.1. Classification of Photoperiod-Insensitive Accessions by Isozymes and SSRs
4.2.2. Sequence Analyses
4.2.3. Analysis of the Distribution of Loss-of-Function Alleles Using DNA Markers
4.2.4. Statistical Analyses
5. Conclusions
Acknowledgments
Accession Numbers
Supplementary Files
References
- Ehrenreich, I.M.; Hanzawa, Y.; Chou, L.; Roe, J.L.; Kover, P.X.; Purugganan, M.D. Candidate gene association mapping of Arabidopsis flowering time. Genetics 2009, 183, 325–335. [Google Scholar]
- Alonso-Blanco, C.; Aarts, M.G.M.; Bentsink, L.; Keurentjes, J.J.B.; Reymond, M.; Vreugdenhil, D.; Koornneef, M. What has natural variation taught us about plant development, physiology, and adaptation? Plant Cell 2009, 21, 1877–1896. [Google Scholar]
- Johanson, U.; West, J.; Lister, C.; Michaels, S.; Amashino, R.M.; Dean, C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 2000, 290, 344–347. [Google Scholar]
- Gazzani, S.; Gendall, A.R.; Lister, C.; Dean, C. Analysis of the molecular basis of flowering time variation in Arabidopsis accessions. Plant Physiol. 2003, 132, 1107–1114. [Google Scholar] [CrossRef]
- Michaels, S.D.; He, Y.; Scortecci, K.C.; Amasino, R.M. Attenuation of FLOWERING LOCUS C activity as a mechanism for the evolution of summer-annual flowering behavior in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 10102–10107. [Google Scholar] [CrossRef]
- Shindo, C.; Aranzana, M.J.; Lister, C.; Baxter, C.;. Nicholls, C.; Nordborg, M.; Dean, C. Role of FRIGIDA and FLOWERING LOCUS C in determining variation in flowering time of Arabidopsis. Plant Physiol. 2005, 138, 1163–1173. [Google Scholar] [CrossRef]
- Shindo, C.; Bernasconi, G.; Hardtke, C.S. Natural genetic variation in Arabidopsis: Tools, traits and prospects for evolutionary ecology. Ann. Bot. 2007, 99, 1043–1054. [Google Scholar] [CrossRef]
- Takahashi, Y.; Teshima, K.M.; Yokoi, S.; Innan, H.; Shimamoto, K. Variation in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc. Natl. Acad. Sci. USA 2009, 106, 4555–4560. [Google Scholar]
- Buzzell, R.I. Inheritance of a soybean flowering response to fluorescent-daylength conditions. Can. J. Genet. Cytol. 1971, 13, 703–707. [Google Scholar]
- Buzzell, R.I.; Voldeng, H.D. Inheritance of insensitivity to long day length. Soybean Genet. Newsl. 1980, 7, 26–29. [Google Scholar]
- Saindon, G.; Voldeng, H.D.; Beversdorf, W.D.; Buzzell, R.I. Genetic control of long daylength response in soybean. Crop Sci. 1989, 29, 1436–1439. [Google Scholar]
- Cober, E.R.; Tanner, J.W.; Voldeng, H.D. Genetic control of photoperiod response in early-maturing near-isogenic soybean lines. Crop Sci. 1996, 36, 601–605. [Google Scholar]
- Cober, E.R.; Tanner, J.W.; Voldeng, H.D. Soybean photoperiod-sensitivity loci respond differentially to light quality. Crop Sci. 1996, 36, 606–610. [Google Scholar] [CrossRef]
- Cober, E.R.; Voldeng, H.D. A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T. Crop Sci 2001, 41, 698–701. [Google Scholar] [CrossRef]
- Cober, E.R.; Voldeng, H.D. Low R:FR light quality delays flowering of E7E7 soybean lines. Crop Sci. 2001, 41, 1823–1826. [Google Scholar]
- Watanabe, S.; Harada, K.; Abe, J. Genetic and molecular bases of photoperiod responses of flowering in soybean. Breed Sci. 2012, 61, 531–543. [Google Scholar] [CrossRef]
- Liu, B.; Kanazawa, A.; Matsumura, H.; Takahashi, R.; Harada, K.; Abe, J. Genetic redundancy in soybean photoresponses associated with duplication of phytochrome A gene. Genetics 2008, 180, 996–1007. [Google Scholar]
- Watanabe, S.; Hideshima, R.; Xia, Z.; Tsubokura, Y.; Sato, S.; Nakamoto, Y.; Yamanaka, N.; Takahashi, R.; Ishimoto, M.; Anai, T.; et al. Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 2009, 182, 1251–1262. [Google Scholar]
- Xia, Z.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lü, S.; et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar]
- Abe, J.; Xu, D.H.; Miyano, A.; Komatsu, K.; Kanazawa, A.; Shimamoto, Y. Photoperiod-insensitive Japanese soybean landraces differ at two maturity loci. Crop Sci. 2003, 43, 1300–1304. [Google Scholar]
- Liu, B.; Abe, J. QTL mapping for photoperiod-insensitivity of a Japanese soybean landrace Sakamotowase. J. Hered. 2009, 101, 251–256. [Google Scholar]
- Kanazawa, A.; Liu, B.; Kong, F.; Arase, S.; Abe, J. Adaptive evolution involving gene duplication and insertion of a novel Ty1/copia-like retrotransposon in soybean. J. Mol. Evol. 2009, 69, 164–175. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar]
- Watterson, G. On the number of segregating sites in genetical models without recombination. Theor. Pop. Biol. 1975, 7, 188–193. [Google Scholar]
- Cannon, S.B.; Shoemaker, R.C. Evolutionary and comparative analyses of the soybean genome. Breed Sci. 2012, 61, 437–444. [Google Scholar] [CrossRef]
- Schlueter, J.A.; Scheffler, B.E.; Schlueter, S.D.; Shoemaker, R.C. Sequence conservation of homeologous bacterial artificial chromosomes and transcription of homeologous genes in soybean (Glycine max L. Merr.). Genetics 2006, 174, 1017–1028. [Google Scholar]
- Lin, J.Y.; Stupar, R.M.; Hans, C.; Hyten, D.L.; Jackson, S.A. Structural and functional divergence of a 1-Mb duplicated region in the soybean (Glycine max) genome and comparison to an orthologous region from Phaseolus vulgaris. Plant Cell 2010, 22, 2545–2561. [Google Scholar] [CrossRef]
- Hecht, V.; Foucher, F.; Ferrándiz, C.; Macknight, R.; Navarro, C.; Morin, J.; Vardy, M.E.; Ellis, N.; Beltran, J.P.; Rameau, C.; Weller, J.L. Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol. 2005, 137, 1420–1434. [Google Scholar] [CrossRef]
- Casal, J.J.; Sanchez, R.A.; Yanovsky, M.J. The function of phytochrome A. Plant Cell Environ. 1997, 20, 813–819. [Google Scholar]
- Franklin, K.A.; Allen, T.; Whitelam, G.C. Phytochrome A is an irradiance-dependent red light sensor. Plant J. 2007, 50, 108–117. [Google Scholar] [CrossRef]
- Franklin, K.A.; Whitelam, G.C. Phytochrome A function in red light sensing. Plant Signal. Behav. 2007, 2, 383–385. [Google Scholar]
- Weller, J.L.; Murfet, I.C.; Reid, J.B. Pea mutants with reduced sensitivity to far-red light define an important role for phytochrome A in day-length detection. Plant Physiol. 1997, 114, 1225–1236. [Google Scholar]
- Weller, J.L.; Beauchamp, N.; Kerckhoffs, H.J.; Platten, D.; Reid, J.B. Interaction of phytochrome A and B in the control of de-etiolation and flowering in pea. Plant J. 2001, 26, 283–294. [Google Scholar] [CrossRef]
- Takano, M.; Kanegae, H.; Shinomura, T.; Miyano, A.; Hirochika, H.; Furuya, M. Isolation and characterization of rice phytochrome A mutants. Plant Cell 2001, 13, 521–534. [Google Scholar]
- Takano, M.; Inagaki, N.; Xie, X.; Yuzurihara, N.; Hihara, F.; Ishizuka, T.; Yano, M.; Nishimura, M.; Miyano, A.; Hirochika, H.; et al. Distinct and cooperative functions of phytochromes A, B, and C in the control of deetiolation and flowering in rice. Plant Cell 2005, 17, 3311–3325. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, X.; Lee, R.; Li, Y.; Specht, J.E.; Nelson, R.L.; McClean, P.E.; Qiu, L.; Ma, J. Artificial selection for determinate growth habit in soybean. Proc. Natl. Acad. Sci. USA 2010, 107, 8563–8568. [Google Scholar]
- Liu, B.; Watanabe, S.; Uchiyama, T.; Kong, F.; Kanazawa, A.; Xia, Z.; Nagamatsu, A.; Arai, M.; Yamada, T.; Kitamura, K.; et al. The soybean stem growth habit gene Dt1 is an ortholog of Arabidopsis TERMINAL FLOWER 1. Plant Physiol. 2010, 153, 198–210. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N. An “electronic fluorescent pictograph” browser for exploring and analyzing large-Scale biological data sets. PloS One 2007, 8, e718. [Google Scholar]
- Available online: http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi (accessed on 10 December 2012).
- Abe, J.; Ohara, M.; Shimamoto, Y. New electrophoretic mobility variants observed in wild soybean (Glycine soja) distributed in Japan and Korea. Soybean Genet. Newsl. 1992, 19, 63–72. [Google Scholar]
- Abe, J.; Xu, D.H.; Suzuki, Y.; Kanazawa, A.; Shimamoto, Y. Soybean germplasm pools in Asia revealed by nuclear SSRs. Theor. Appl. Genet. 2003, 106, 445–453. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.57c; University of Washington Press: Seattle WA, USA, 1997. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tsubokura, Y.; Matsumura, H.; Xu, M.; Liu, B.; Nakashima, H.; Anai, T.; Kong, F.; Yuan, X.; Kanamori, H.; Katayose, Y.; et al. Genetic Variation in Soybean at the Maturity Locus E4 Is Involved in Adaptation to Long Days at High Latitudes. Agronomy 2013, 3, 117-134. https://doi.org/10.3390/agronomy3010117
Tsubokura Y, Matsumura H, Xu M, Liu B, Nakashima H, Anai T, Kong F, Yuan X, Kanamori H, Katayose Y, et al. Genetic Variation in Soybean at the Maturity Locus E4 Is Involved in Adaptation to Long Days at High Latitudes. Agronomy. 2013; 3(1):117-134. https://doi.org/10.3390/agronomy3010117
Chicago/Turabian StyleTsubokura, Yasutaka, Hisakazu Matsumura, Meilan Xu, Baohui Liu, Hiroko Nakashima, Toyoaki Anai, Fanjiang Kong, Xiaohui Yuan, Hiroyuki Kanamori, Yuichi Katayose, and et al. 2013. "Genetic Variation in Soybean at the Maturity Locus E4 Is Involved in Adaptation to Long Days at High Latitudes" Agronomy 3, no. 1: 117-134. https://doi.org/10.3390/agronomy3010117