Pea (Pisum sativum L.) in the Genomic Era

 ,
, 
 ,
,
Abstract
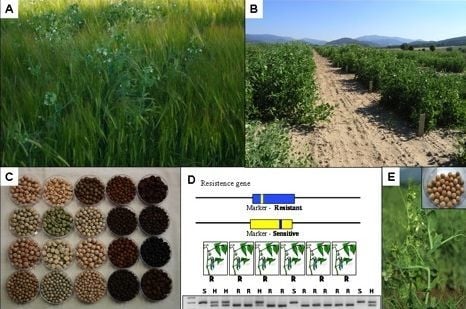
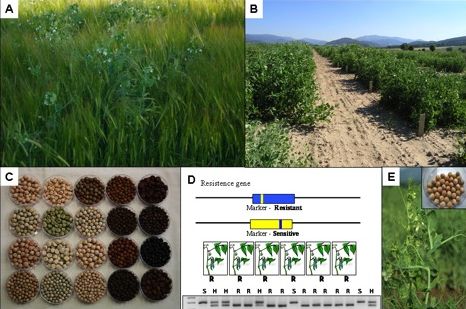
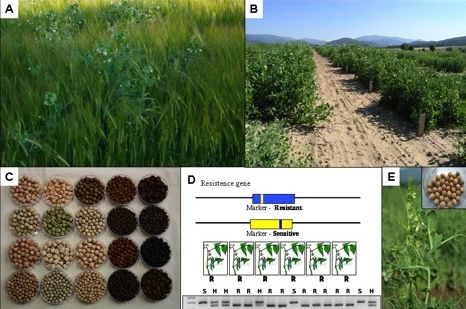
:
1. Introduction
2. Origin of Pea
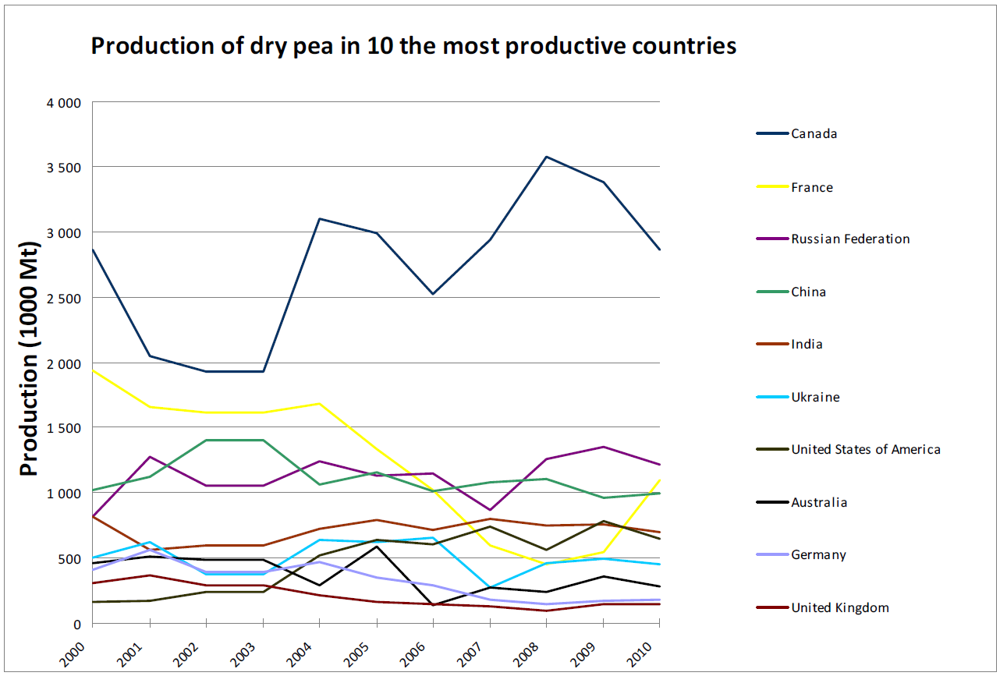
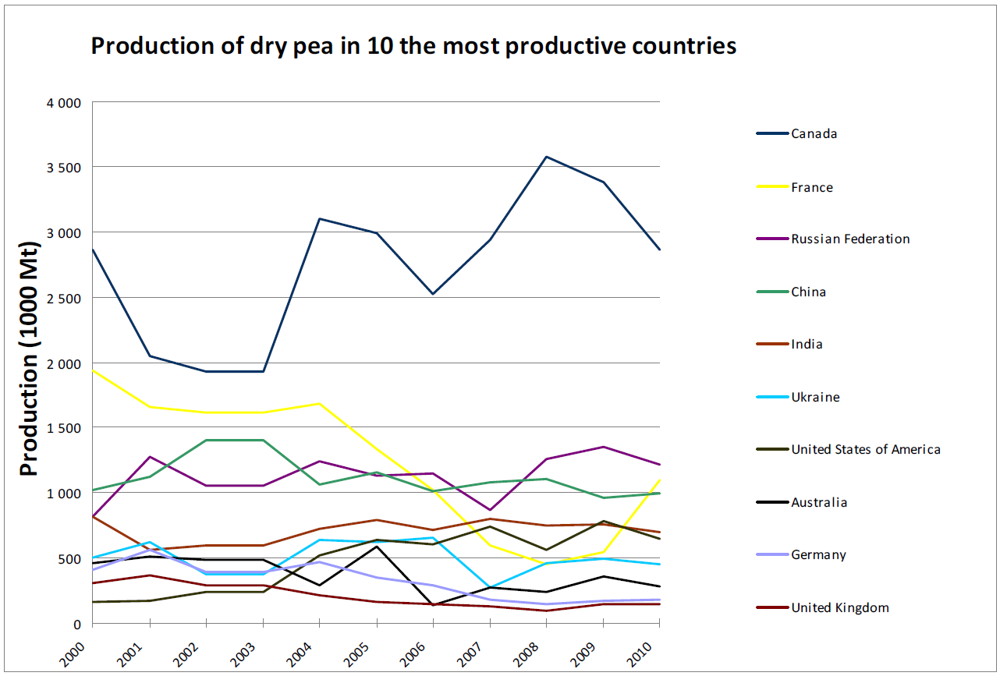
3. Global Pea Cultivation
4. Assessment and Conservation of Pea Diversity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Country | Institute | Number of Accessions | Web site | Genotyped | Phenotyped | Core |
|---|---|---|---|---|---|---|---|
| VIR | Russia | N.I. Vavilov Research Institute of Plant Industry, St. Petersburg | 6,790 | http://www.vir.nw.ru/data | not | not | |
| USDA | USA | Plant Germplasm Introduction and Testing Research Station, Pullman | 5,400 | http://www.ars-grin.gov | yes | yes | formed |
| BAR | Italy | Istituto del Germoplasma, Bari | 4,297 | http://www.ba.cnr.it/areagg34/germoplasma | not | not | |
| SAD | Bulgaria | Institute of Plant Introduction and Genetic Resources, Sadovo | 2,787 | http://www.genebank.hit.bg | not | not | |
| NGB | Sweden | NordGen, Nordic Genetic Resource Centre, Alnarp | 2,724 | http://www.ngb.se/sesto | not | not | |
| CGN | The Netherlands | Centre for Genetic Resources, Wageningen | 1,008 | http://www.cgn.wur.nl/pgr/ | not | not | |
| ATFC | Australia | Australian Temperate Field Crop Collection, Horsham | 6,567 | http://www2.dpi.qld.gov.au/extra/asp/AusPGRIS | yes | yes | formed |
| ICARDA | Syria | International Center for Agricultural Research in the Dry Areas, Aleppo | 6,105 | http://www.icarda.cgiar.org | not | not | |
| GAT | Germany | Leibniz Institute of Plant Genetics and Crop Plant Research, Gaterleben | 5,336 | http://fox-serv.ipk-gatersleben.de/ | not | not | |
| ICAR | China | Institute of Crop Sciences, CAAS China | 3,837 | http://icgr.caas.net.cn/cgris | partly | not | |
| JIC | UK | John Innes Centre, Norwich | 3,557 | http://www.jic.ac.uk/GERMPLAS/pisum | yes | yes | formed |
| WTD | Polland | Plant Breeding and Acclimatization Institute Blonie, Radzikow | 2,899 | http://www.ihar.edu.pl/gene_bank/ | not | not | |
| INRA | France | INRA CRG Légumineuse à grosses graines, Dijon | 1,891 | http://195.220.91.17/legumbase/ | partly | yes | formed |
| UKR | Ukraine | Yurjev Institute of Plant Breeding, Kharkov | 1,671 | http://www.bionet.nsc.ru | not | not | |
| CZE | Czech Republic | AGRITEC, Research, Breeding and Services Ltd., Sumperk | 1,284 | http://genbank.vurv.cz/genetic/resources | yes | yes | formed |
| HUN | Hungary | Institute for Agrobotany, Tapioszele | 1,188 | http://www.rcat.hu | not | not |
5. Pea Mutant Collections
6. Use of Pea Genetic Diversity in Trait Identification
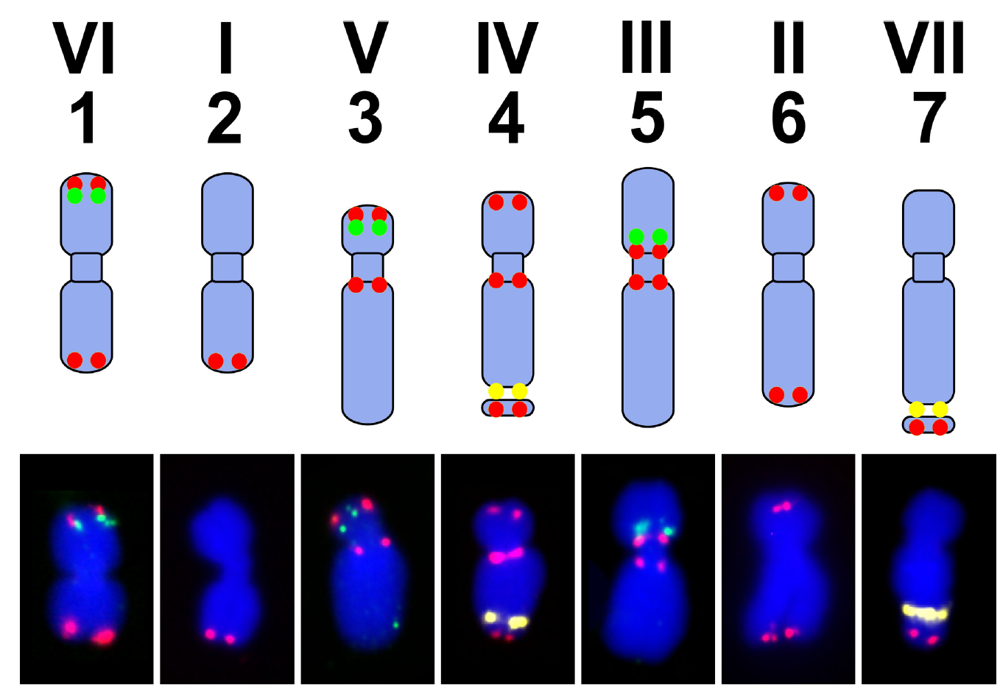
7. Pea Genome, Karyotype and Genetic Maps
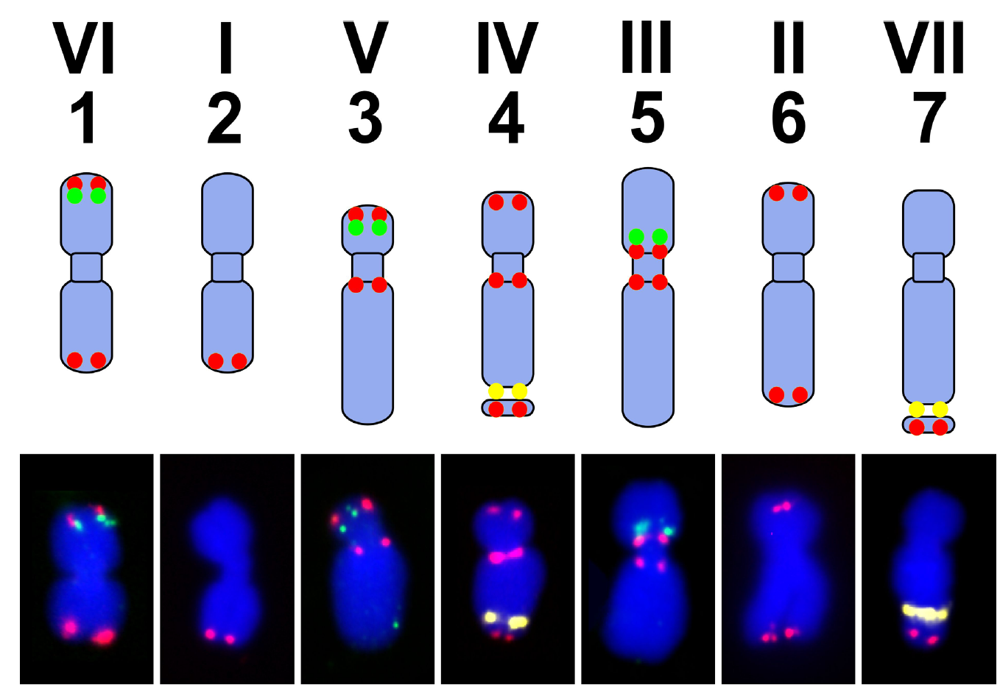
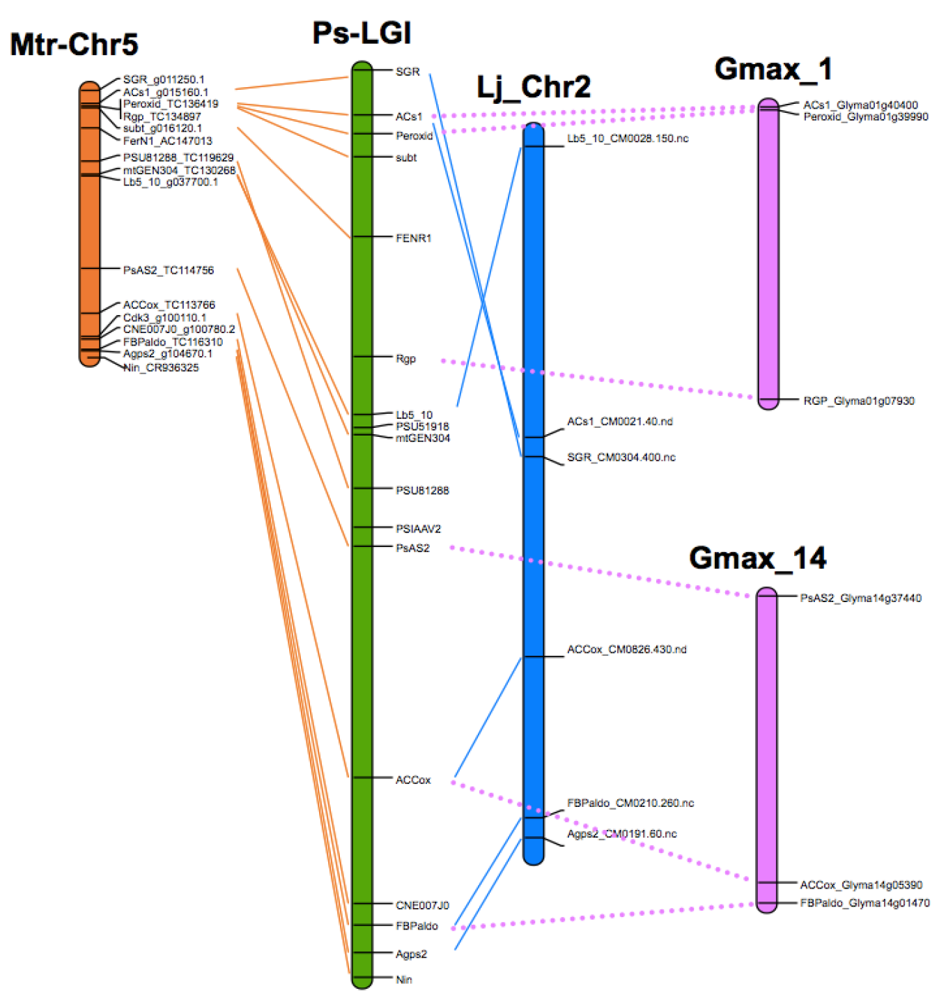
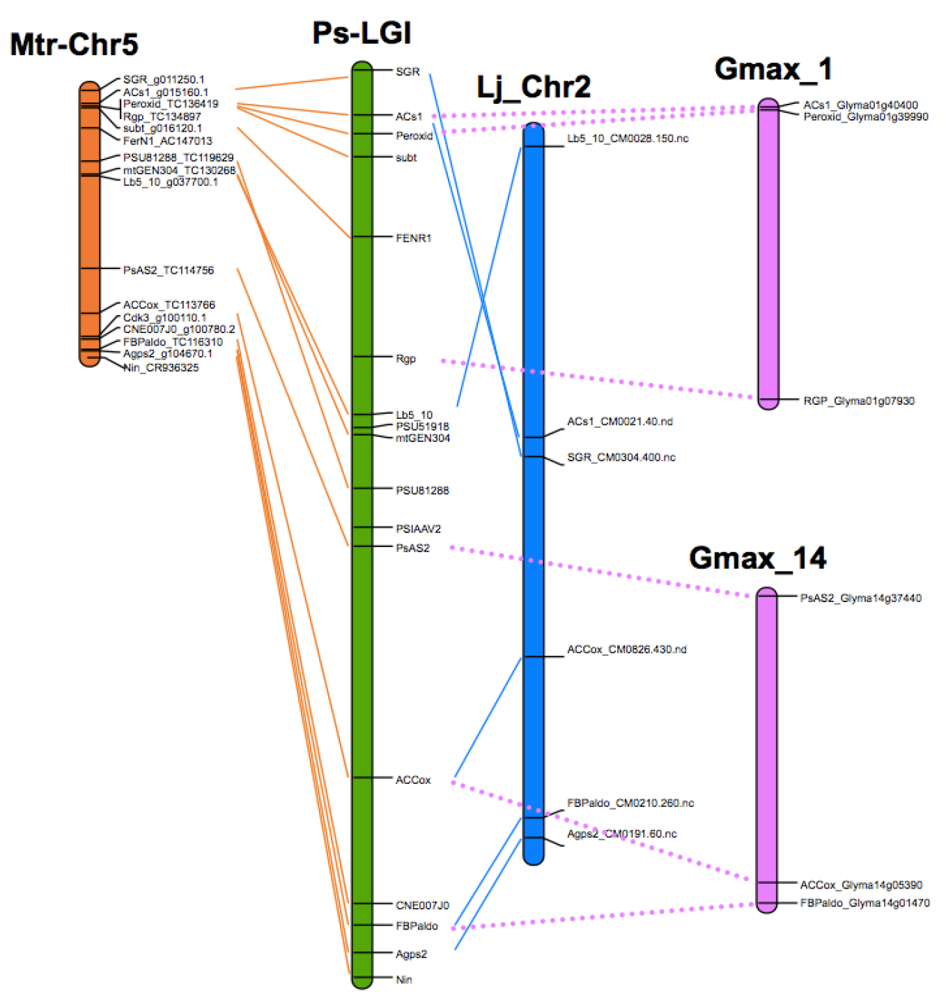
8. Translational Genomics—From Model Legumes to Pea
| P. sativum | M. truncatula | L. japonicus |
|---|---|---|
| I | 5 | 2 |
| II | 1 | 5 |
| III | 2, 3 | 1, 3 |
| IV | 8 | 4 |
| V | 7 | 1 |
| VI | 2, 6 | 6 |
| VII | 4 | 3, 4 |
| International Legume Database & Information Service (ILDIS) | http://www.ildis.org/ |
| Legume Information System (LIS) | http://www.comparative-legumes.org/ |
| Phytozome -Soybean Gbrowser | http://www.phytozome.net/cgi-bin/gbrowse/soybean/ |
| Medicago truncatula HapMap Project | http://www.medicagohapmap.org/cgi-bin/gbrowse/mthapmap/ |
| Legume phylo-informatics database | http://www.public.asu.edu/~mfwojci/legumephylo_dBase.html |
| Cool Season Food Legume Genome Database | http://www.gabcsfl.org/ |
| INRA Dijon Legume genetic and genomic resources | http://www.thelegumeportal.net |
| A bioinformatics gateway towards integrative legume biology | http://www.legoo.org/ |
| UTILLdb : URGV TILLING pea database | http://urgv.evry.inra.fr/UTILLdb |
| INRA LegumeBase | http://195.220.91.17/legumbase/index.php?mode=0&id= |
| Legume proteomes | http://iant.toulouse.inra.fr/plants/legumes/cgi/legumes.cgi |
| KnowPulse | http://knowpulse2.usask.ca |
| LegumeIP | http://plantgrn.noble.org/LegumeIP |
9. Transcriptome and Proteome Analysis
10. Molecular Markers
10.1. Molecular Marker Development and Application
10.2. Transition to Gene-based Markers EST-SSRs and SNPs
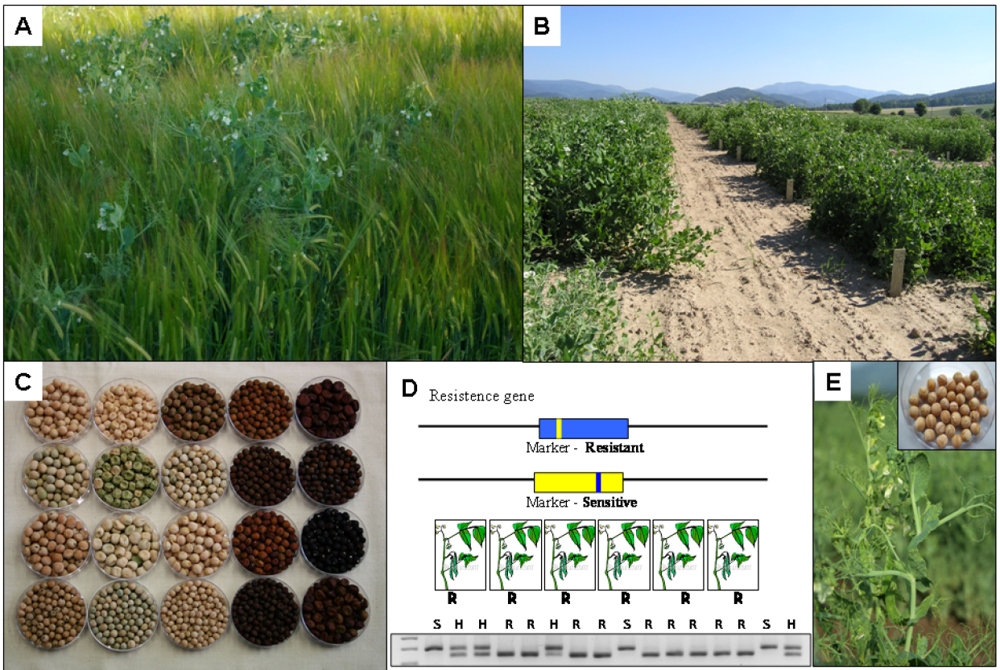
10.3. Markers for Disease and Pest Resistance
10.4. Markers for Flowering Time
10.5. Markers for Agronomic Traits (QTLs and Major Genes)
11. Genomic Analysis of Pea
12. Application of Genomics and Phenomics in Pea Breeding: Status and Outlook
13. Conclusions
Acknowledgements
References
- Lock, M.; Mackinder, B.; Schrirer, B.; Lewis, G. Legumes of the World; 2005; Royal Botanical Gardens: Kew, UK. [Google Scholar]
- Vance, C.P.; Graham, P.H. Legumes: Importance and constraints to greater use. Plant Physiol. 2003, 131, 872–877. [Google Scholar] [CrossRef] [PubMed]
- FAOSTAT. 2011. Available online: http://faostat.fao.org (accessed on 11 January 2012).
- Regnier, J.M.; Duparque, M.; Peyronnet, C.; Grosjean, F.; Bastianelli, D. Feeding value of pea (Pisum sativum, L.)—1 Chemical composition of different categories of pea. Anim. Sci. 1998, 67, 609–619. [Google Scholar] [CrossRef]
- Phillips, D.A. Efficiency of symbiotic nitrogen fixation in legumes. Ann. Rev. Plant Physiol. 1980, 31, 29–49. [Google Scholar] [CrossRef]
- Sumner, L.W.; Dixon, R.A. Legume natural products: Understanding and manipulating complex pathways for human and animal health. Plant Physiol. 2003, 131, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Hellens, R.P.; Coyne, C.J.; Timmerman-Vaughan, G.M.; Hofer, J.I.; Ellis, T.H.N. Mendel, 150 years on. Trends Plant Sci. 2011, 16, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.J.; Reid, J.B. Mendel’s genes: Toward a full molecular characterization. Genetics 2011, 189, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mumm, R.H.; Moose, S.P. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.J. From Near East centre of origin the prized pea migrates throughout world. Diversity 1995, 11, 118–119. [Google Scholar]
- Hopf, M.; Zohary, D. Domestication of Plants in the Old World; 2000; Oxford University Press: Oxford, UK. [Google Scholar]
- Ambrose, M.J.; Maxted, N.; Coyne, C.J.; Ford, R.; Redden, R.J.; Kosterin, O.; Corander, J.; Flavell, A.J.; Kenicer, G.; Smýkal, P.; et al. Phylogeny, phylogeography and genetic diversity of the Pisum genus. Plant Genet. Res. 2011, 9, 4–18. [Google Scholar] [CrossRef]
- Makasheva, R.K. Gorokh (Pea). In Kulturnaya Flora SSR; Ed., Korovina, O.N., Eds.; 1979; Kolos Publishing: Leningrad, Russia. [Google Scholar]
- Ambrose, N.; Maxted, N. Peas (Pisum L.) Chapter 10. Plant Genetic Resources of Legumes in the Mediterranean; Eds., Bennett, S.J., Maxted, N., Eds.; 2000; Kluwer Academic Publishers: Dordrecht, The Netherlands. [Google Scholar]
- Ford-Lloyd, B.; Jarvis, A.; Guarino, L.; Hunter, D.; Hodgkin, T.; Heywood, V.; Dulloo, E.; Toledo, A.; Kell, S.; Maxted, N. A global approach to crop wild relative conservation: Securing the gene pool for food and agriculture. Kew Bull. 2010, 65, 561–576. [Google Scholar] [CrossRef]
- Ellis, T.H.N. Pisum. In Wild Crop Relatives: Genomic and Breeding Resources; Ed., Kole, C., Eds.; 2011; Springer-Verlag: Berlin-Heidelberg, Germany. [Google Scholar]
- Flavell, A.J.; Ellis, T.H.N.; Ambrose, M.J.; Marshall, D.; Smýkal, P.; Shaw, P.; Grzebyta, J.; Vershinin, A.; Jing, R. The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Flavell, A.; Dumet, D.; Duc, G.; Debouck, D.; Smýkal, P.; Berger, J.; Ellis, N.; Ambrose, M.; Dwivedi, S.L.; Upadhyaya, H.D.; et al. Legume genetic resources: Management, diversity assessment, and utilization in crop improvement. Euphytica 2011, 180, 27–47. [Google Scholar] [CrossRef]
- Miles, C.A.; Furman, B.J.; Ambrose, M.J.; Redden, R.J.; McGee, R.J.; Coyne, C.J. Genetic Adjustment to Changing Climates: Pea. In Crop Adaptation to Climate Change; Eds., Hall, A.E., Lotze-Campen, H., Hatfield, J.L., Redden, R.J., Yadav, S.S., Eds.; 2011; Wiley Blackwell: Chichester, UK. [Google Scholar]
- Ford, R.; Ades, P.; Yan, L.; Gu, J.; Liu, X.; Xu, Y.; Liu, J.; Guan, J.; Wang, S.; Liu, Q.; Redden, R.; Zong, X. Analysis of a diverse global Pisum sp. collection and comparison to a Chinese local P. sativum collection with microsatellite markers. Theor. Appl. Genet. 2009, 118, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.J.; Timmerman-Vaughan, G.M.; Kisha, T.; Watt, C.A.; McGee, R.J.; Hu, J.; Brown, A.F.; Kwon, S.J. Population Genetic Sub-structure within the USDA ARS Pisum Core Collection and Its Potential as a Platform for Association Mapping. In Proceedings of the Plant , 13–17 January 2012; San Diego, CA, USA.
- Burstin, J.; Lallemand, J.; Lejeune-Hénaut, J.; Weinachter, C.; Potier, J.; Deniot, G.; Lainé, A.L.; Arnau, G.; Aubert, G.; Baranger, A.G. Genetic diversity within Pisum sativum using protein- and PCR-based markers. Theor. Appl. Genet. 2004, 108, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, A.; Tullu, A.; Warkentin, T.D.; Zhang, C.; Tar’an, B. Genetic diversity among varieties and wild species accessions of pea (Pisum sativum L.) based on molecular markers, and morphological and physiological characters. Genome 2005, 48, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.W.J.; Brouwer, J.B.; Itman, C.; Le Roux, K.; Ford, R. Diversity analysis and genotyping in Pisum with sequence tagged microsatellite site (STMS) markers. Euphytica 2002, 124, 397–405. [Google Scholar] [CrossRef]
- Griga, M.; Flavell, A.J.; Jarkovský, J.; Corander, J.; Hýbl, M.; Smýkal, P. Genetic diversity and population structure of pea (Pisum sativum L.) varieties derived from combined retrotransposon, microsatellite and morphological marker analysis. Theor. Appl. Genet. 2008, 117, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Flavell, A.J.; Ellis, T.H.N.; Knox, M.R.; Ambrose, M.J.; Kiss, G.; Seres, A.; Johnson, R.; Jing, R. Gene-based sequence diversity analysis of field pea (Pisum). Genetics 2007, 177, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.J.; Vershinin, A.V.; Knox, M.R.; Poyser, S.J.; Ellis, T.H.N. Polymorphism of insertion sites of Ty1-copia class retrotransposons and its use for linkage and diversity analysis in pea. Mol. Gen. Genet. 1998, 260, 9–19. [Google Scholar] [PubMed]
- Isaac, P.; Ellis, T.H.N.; Russell, J.; Jing, R.; Booth, A.; Bolshakov, V.N.; Flavell, A.J. A microarray–based high throughput molecular marker genotyping method: The tagged microarray marker (TAM) approach. Nucl. Acids Res. 2003, 31. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flavell, A.J.; Ellis, T.H.N.; Ambrose, M.; Vershinin, A.V.; Lee, J.M.; Knox, M.R.; Jing, R.C. Insertional polymorphism and antiquity of PDR1 retrotransposon insertions in Pisum species. Genetics 2005, 171, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Smýkal, P. Development of an efficient retrotransposon-based fingerprinting method for rapid pea variety identification. J. Appl. Genet. 2006, 47, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Melchinger, A.E.; Reif, J.C. Genetical and mathematical properties of similarity and dissimilarity coefficients applied in plant breeding and seed bank management. Crop Sci. 2005, 45, 1–7. [Google Scholar] [CrossRef]
- Rannala, B.; Beaumont, M.A. The Bayesian revolution in genetics. Nat. Rev. Genet. 2004, 5, 251–261. [Google Scholar] [PubMed]
- Sillanpää, M.J.; Waldmann, P.; Corander, J. Bayesian analysis of genetic differentiation between populations. Genetics 2003, 164, 367–374. [Google Scholar]
- Griga, M.; Hýbl, M.; Procházka, S.; Hanáček, P.; Dočkalová, Z.; Smýkal, P.; Cieslarová, J. Molecular evidence of genetic diversity changes in pea (Pisum sativum L.) germplasm after long-term maintenance. Genet. Resour. Crop Evol. 2011, 58, 439–451. [Google Scholar] [CrossRef]
- Larrianzar, E.; Imran, A.; Gonzalez, E.M.; Hafeez, F.Y.; Arrese-Igor, C.; Shivakumar, B.G.; Kantar, F. Efficient Biological Nitrogen Fixation under Warming Climates. In Climate Change and Management of Cool Season Grain legume Crops; Eds., Patil, A., Redden, R., McNeil, D., Yadav, S., Eds.; 2010; Springer: Dordrecht, The Netherland. [Google Scholar] [Green Version]
- Slattery, J.; Croser, J.; Ford, R.; Leonforte, T.; Redden, B. Pea (Pisum sativum L.). In Genetic Resources, Chromosome Engineering, and Crop Improvement: Series II—Grain Legumes; Ed., Singh, R., Eds.; 2004; Department of Crop Sciences,University of Illinois: Champaign, IL, USA. [Google Scholar] [Green Version]
- Dixit, G.P.; Tanveer, H.; Choudhury, P.R. Identification and detection of genetic relatedness among important varieties of pea (Pisum sativum L.) grown in India. Genetica 2007, 130, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.E.; Lotze-Campen, H.; Hatfield, J.L.; Redden, R.J.; Yadav, S.S. Crop Adaptation to Climate Change; 2011; Wiley Blackwell: Chichester, UK. [Google Scholar] [Green Version]
- Redden, R.J.; Rose, I.A.; Guan, J.; Yan, Q.; Liu, Y.; Feng, Q.; Yuan, M.; Wu, K.; Liu, Y.; He, C.; et al. Collecting and surveying landraces of pea (Pisum sativum) and faba bean (Vicia faba) in Qinghai province of China. Plant Genet. Res. Newsl. 2008, 156, 1–10. [Google Scholar]
- Paull, J.; Redden, R.J.; Leonforte, T.; Rose, I.A.; Enneking, D.; Lichi, L.; Wang, L.; Zong, X.; Yuhua, H.; Bao, S. Collection of pea (Pisum sativum) and faba bean (Vicia faba) germplasm in Yunnan. Plant Genet. Res. Newsl. 2008, 156, 11–22. [Google Scholar]
- Kneller, M.; Scheldman, X.; Jarvis, A.; Ehsan, E.; Snook, L.K. Crop germplasm Diversity: The Role of Gene Bank Collections in Facilitating Adaptation to Climate Change. In Crop Adaptation to Climate Change; Eds., Hall, A.E., Lotze-Campen, H., Hatfield, J.L., Redden, R.J., Yadav, S.S., Eds.; 2011; Wiley Blackwell: Chichester, UK. [Google Scholar] [Green Version]
- Yan, G.; Khan, T.; Hardie, D.C.; Byrne, O.M. Genetic analysis of pod and seed resistance to pea weevil in a Pisum sativum x P. fulvum interspecific cross. Aust. J. Agric. Res. 2008, 59, 854–862. [Google Scholar] [CrossRef]
- Rubiales, D.; Moreno, M.T.; Torres, A.M.; Fondevilla, S. Identification of a new gene for resistance to powdery mildew in Pisum fulvum, a wild relative of pea. Breed Sci. 2007, 57, 181–184. [Google Scholar] [CrossRef]
- De Vilmorin, P. Fixite des Races de Froments. IVe Conference Internationale de Genetique—Paris 1911. Comptes-rendus et Rapport Edite s par Philippe de Vilmorin; Masson: Paris, France, 1913; pp. 312–316. [Green Version]
- Blixt, S. Mutation genetics in Pisum. Agric. Hort. Genet. 1972, 30, 1–293. [Google Scholar]
- Si, P.; Xu, X.; Nakagawa, H.; Islam, M.M.; Shah, T.; Toker, C.; Cagirgan, M.I.; Kharkwal, M.C. Legume Mutant Varieties for Food, Feed and Environmental Benefits. Proceedings of the 5th International Food Legumes Research Conference (IFLRC) 196. [Green Version]
- Guichard, C.; de Oliveira, Z.; Brunaud, V.; Aubert, G.; Savois, V.; Burstin, J.; Moussy, F.; Le Signor, C.; Schmidt, J.; Dalmais, M.; et al. UTILLdb, a Pisum sativum in silico forward and reverse genetics tool. Genome Biol. 2008, 9. [Google Scholar] [CrossRef]
- Messager, J.; Duc, G. A mutagenesis of pea (Pisum sativum L.) and the isolation of mutants for nodulation and nitrogen-fixation. Plant Sci. 1989, 60, 207–213. [Google Scholar] [CrossRef]
- Duc, G.; Huguet, T.; Sagan, M. Phenotypic characterization and classification of nodulation mutants of pea (Pisum sativum L.). Plant Sci. 1994, 100, 59–70. [Google Scholar] [CrossRef]
- Duc, G.; Sagan, M. Sym28 and Sym29, two new genes involved in regulation of nodulation in pea (Pisum sativum L.). Symbiosis 1996, 20, 229–245. [Google Scholar]
- Ambrose, M.; Tabata, S.; Sato, S.; Weng, L.; Yang, J.; Xu, S.; Wang, L.; Li, X.; Luo, Y.; Wang, Z.; et al. Genetic control of floral zygomorphy in pea (Pisum sativum L.). Proc. Natl. Acad. Sci. USA 2008, 105, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Le Signor, C.; Dalmais, M.; Dupin, A.; Weller, J.; Butcher, S.; Isaac, P.; Ambrose, M.; Moreau, C.; Turner, L.; Hofer, J.; et al. Tendril-less regulates tendril formation in pea leaves. Plant Cell 2009, 21, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.M.E.; Hofer, J.M.I.; Murray, S.R.; Frew, T.J.; Fiers, M.W.E.J.; Thomson, S.J.; Schwinn, K.E.; Lin-Wang, K.; Moreau, C.; Hellens, R.P.; et al. Identification of Mendel’s white flower character. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Duc, G.; Burstin, J.; Warkentin, T.; Hýbl, M.; Flavell, A.J.; Redden, R.; Ford, R.; Coyne, C.J.; Smýkal, P.; et al. Effort towards a world pea (Pisum sativum L.) germplasm core collection: The case for common markers and data compatibility. Pisum Genet. 2008, 40, 11–14. [Google Scholar]
- Buckler, E.S.; Gore, M.; Zhu, C. Status and prospects of association mapping in plants. Plant Genome 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, T.; Zhu, C.; Li, C.; Zhao, Y.; Feng, Q.; Zhao, Q.; Sang, T.; Wei, X.; Huang, X.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, A.J.; Morgante, M.; Scott Tingey, S.; Smith, O.S.H.; Dolan, M.; Jung, M.; Caldwell, K.S.; Ching, A. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet 2002, 3. [Google Scholar] [CrossRef]
- Keller, B.; Yahiaouia, N.; Mackay, M.; Street, K.; Bhullar, N.K. Unlocking wheat genetic resources for the molecular identification of previously undescribed functional alleles at the Pm3 resistance locus. Proc. Natl. Acad. Sci. USA 2009, 106, 9519–9524. [Google Scholar] [CrossRef] [PubMed]
- Flavell, A.J.; van Hintum, T.J.L.; Dicks, J.; Ambrose, M.J.; Ellis, T.H.N.; Marshall, D.; Davenport, G.F.; Lee, M.J. GERMINATE. A Generic database for integrating genotypic and phenotypic information for plant genetic resource collections. Plant Physiol. 2005, 139, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Mackay, M.; Sinnott, Q.; Emberland, G.; Bohning, M.; Kinard, G.; Franko, T.; Bretting, P.; Ayala-Silva, T.; Hummer, K.; Postman, J.; et al. GRIN-GLOBAL: An international project to develop a global plant genebank information management system. ISHS Acta Hort. 2010, 859, 49–55. [Google Scholar]
- Stougaard, J. Genetics and genomics of root symbiosis. Curr. Opin. Plant Biol. 2001, 4, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Greilhuber, J.; Dolezel, J. Greilhuber, J. Nuclear genome size. Are we getting closer? Cytometry 2010, 77, 635–642. [Google Scholar] [PubMed]
- Bernardi, G.; Montero, L.M.; Matassi, G.; Salinas, J. Compositional compartmentalization and compositional patterns in the nuclear genomes of plants. Nucl. Acids Res. 1988, 16, 4269–4285. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.F.; Cuellar, R.E.; Murray, M.G. DNA sequence organization in the pea genome. Biochemistry 1978, 17, 5781–5790. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.F.; Peters, D.L.; Murray, M.G. Ancient repeated sequences in the pea and mung bean genomes and implications for genome evolution. J. Mol. Evol. 1981, 17, 31–42. [Google Scholar] [CrossRef]
- Smith, D.B.; Smith, J.B.; Bennett, M.D.; Flavell, R.B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 1974, 12, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Neumann, P.; Novák, P. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinforma 2010, 11. [Google Scholar] [CrossRef]
- Navrátilová, A.; Neumann, P.; Macas, J. Repetitive DNA in the pea (Pisum sativum L.) genome: Comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC Genomics 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Pozárková, D.; Neumann, P. Highly abundant pea LTR retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol. Biol. 2003, 53, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Macas, J. Ogre elements—A distinct group of plant Ty3/gypsy-like retrotransposons. Gene 2007, 390, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Neumann, P.; Steinbauerová, V. Experimental evidence for splicing of intron-containing transcripts of plant LTR retrotransposon Ogre. Mol. Genet. Genomics 2008, 280, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Navrátilová, A.; Koblízková, A.; Neumann, P. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 2006, 173, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Navrátilová, A.; Koblízková, A.; Macas, J. Hypervariable 3´UTR region of plant LTR-retrotransposons as a source of novel satellite repeats. Gene 2009, 448, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Cerff, R.; Liaud, M.F.; Zhang, D.X.; Chavanne, F. Structure and evolution of Cyclops: A novel giant retrotransposon of the Ty3/Gypsy family highly amplified in pea and other legume species. Plant Mol. Biol. 1998, 37, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.H.; Vershinin, A.V. Heterogeneity of the internal structure of PDR1, a family of Ty1/copia-like retrotransposons in pea. Mol. Gen. Genet. 1999, 262, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Ellis, N.T.H.; Ambrose, M.J.; Knox, M.R.; Allnutt, T.R.; Vershinin, A.V. Transposable elements reveal the impact of introgression, rather than transposition, in Pisum diversity, evolution, and domestication. Mol. Biol. Evol. 2003, 20, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Koblízková, A.; Pozárková, D.; Neumann, P. PIGY: A new plant envelope-class LTR retrotransposon. Mol. Genet. Genomics 2005, 273, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Griga, M.; Macas, J.; Ford, R.; Kalendar, R.; Smýkal, P. Evolutionary conserved lineage of Angela-family retrotransposons as a genome-wide microsatellite repeat dispersal agent. Heredity 2009, 103, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Pozárková, D.; Neumann, P.; Macas, J. Zaba: A novel miniature transposable element present in genomes of legume plants. Mol. Genet. Genomics 2003, 269, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Koblízková, A.; Macas, J. Characterization of Stowaway MITEs in pea (Pisum sativum L.) and identification of their potential master elements. Genome 2005, 48, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Dolezel, J.; Widmer, A.; Hobza, R.; Hribova, E.; Kejnovsky, E.; Koblizkova, A.; Navratilova, A.; Neumann, P. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 2. [Google Scholar] [CrossRef]
- Macas, J.; Neumann, P.; Navrátilová, A. Long-range organization of plant satellite repeats investigated using strand-specific FISH. Cytogenet. Genome Res. 2005, 109, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Nouzová, M.; Neumann, P. Molecular and cytogenetic analysis of repetitive DNA in pea (Pisum sativum L.). Genome 2001, 44, 716–728. [Google Scholar] [PubMed]
- Macas, J.; Dolezel, J.; Vrána, J.; Pozárková, D.; Neumann, P. Chromosome sorting and PCR-based physical mapping in pea (Pisum sativum L.). Chromosome Res. 2002, 10, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.H.; Parker, J.S.; Hall, K.J. The relationship between genetic and cytogenetic maps of pea. I. Standard and translocation karyotypes. Genome 1997, 40, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Miravalle, R.J.; Lamm, R. A translocation tester set in Pisum. Hereditas 1959, 45, 417–440. [Google Scholar]
- Lamm, R. Transpositions in Pisum. Pisum Newsl. 1977, 9, 28–29. [Google Scholar]
- Folkeson, D. Assignment of linkage segments to the satellite chromosomes 4 and 7 in Pisum sativum. Hereditas 1990, 112, 257–263. [Google Scholar]
- Poyser, S.J.; Ellis, T.H.N. An integrated and comparative view of pea genetic and cytogenetic maps. New Phytol. 2002, 153, 17–25. [Google Scholar] [CrossRef]
- Schubert, I.; Kühne, M.; Fuchs, J. Assignment of linkage groups to pea chromosomes after karyotyping and gene mapping by fluorescent in situ hybridization. Chromosoma 1998, 107, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Lejeune-Hénault, I.; Coyne, C.J.; Baranger, A.; Rameau, C.; Aubert, G.; Pilet-Nayel, M.L.; Dubreuil, P.; Morin, J.; McPhee, K.E.; Loridon, K.; et al. Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor. Appl. Genet. 2005, 111, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Ellis, T.H.; Timmerman-Vaughan, G.M.; Swiecicki, W.K.; Weeden, N.F. The current pea linkage map. Pisum Genet. 1996, 28, 1–4. [Google Scholar]
- McPhee, K. Pea. In Pulses, Sugar and Tuber Crops. Genome Mapping and Molecular Breeding in Plants; Ed., Kole, C., Eds.; 2007; Volume 3, pp. 33–47. Springer-Verlag: Berlin, Germany. [Google Scholar]
- Burstin, J.; Huguet, T.; Lejeune-Hénaut, I.; Rameau, C.; Petit, A.; Quillet, M.C.; Loridon, K.; Jacquin, F.; Morin, J.; Aubert, G. Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume. Medicago truncatula. Theor. Appl. Genet. 2006, 112, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Marx, G.; Weeden, N.F. Further genetic analysis and linkage relationships of isozyme loci in the pea: Confirmation of the diploid nature of the genome. J. Hered. 1987, 78, 153–159. [Google Scholar]
- Swiêcicki, W.K.; Wolko, B.; Irzykowska, L. The genetic linkage map of pea (Pisum sativum L.) based on molecular, biochemical and morphological markers. Pisum Genet. 2001, 33, 13–18. [Google Scholar]
- Rameau, C.; Ellis, N.; Haurogne, K.; Laucou, V. Genetic mapping in pea. I. RAPD-based linkage map of Pisum sativum. Theor. Appl. Genet 1998, 97, 905–915. [Google Scholar] [CrossRef]
- Timmerman-Vaughan, G.M.; Frew, T.J.; McCallum, J.A.; Gilpin, B.J. A linkage map of the pea (Pisum sativum L.) genome containing cloned sequences of known function and expressed sequence tags (ESTs). Theor. Appl. Genet. 1997, 95, 1289–1299. [Google Scholar] [CrossRef]
- Boone, W.E.; Weeden, N.F. Mapping the Rb locus on linkage group III using long PCR followed by endonuclease digestion. Pisum Genet. 1999, 31, 36. [Google Scholar]
- Weeden, N.F.; Frew, T.J.; Timmerman-Vaughan, G.M. Characterization and linkage mapping of R-gene analogous DNA sequences in pea (Pisum sativum L.). Theor. Appl. Genet. 2000, 101, 241–247. [Google Scholar] [CrossRef]
- Gostimsky, S.; Toshchakova, E.; Konovalov, F. A CAPS marker set for mapping in linkage group III of pea (Pisum sativum L.). Cell. Mol. Biol. Lett. 2005, 10, 163–171. [Google Scholar] [PubMed]
- Berdnikov, V.A.; Rozov, S.M.; Swiecicki, W.K.; Timmerman-Vaughan, G.M.; Ellis, T.H.N.; Weeden, N.F. A consensus linkage map for Pisum sativum. Pisum Genet. 1998, 30, 1–3. [Google Scholar]
- Aubert, G.; Burstin, J.; Lejeune-Hénaut, I.; Donnadieu, C.; Jacquin, F.; Marty, A.; Charrel, H.; Deulvot, C. Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Murat, F.; Rond, C.; Houtin, H.; Potier, J.; Bourgeois, M.; Chauveau, A.; Salse, J.; Nicolas, M.; Savois, V.; Bordat, A.; et al. Translational genomics in legumes allowed placing insilico 5460 unigenes on the pea functional map and identified candidate genes in Pisum sativum L. G3. G3. Genes-Genomes-Genetics 2011, 2, 93–103. [Google Scholar]
- Shultz, J.; Brumett, S.; Coyne, C.; Decarie, J. Additional pea EST-SSR markers for comparative mapping in pea (Pisum sativum L.). Plant Breed. 2012, 131, 222–226. [Google Scholar] [CrossRef]
- Ellis, T.H.N.; Knox, M.R. Excess heterozygosity contributes to genetic map expansion in pea recombinant inbred populations. Genetics 2002, 162, 861–873. [Google Scholar] [PubMed]
- Ellis, T.H.N.; Knox, M.R. Stability and inheritance of methylation states at PstI sites in Pisum. Mol. Genet. Genomics 2001, 265, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.; Young, N.D. Translating Medicago truncatula genomics to crop legumes. Curr. Opin. Plant Biol. 2009, 12, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Tabata, S.; Isobe, S.; Asamizu, E.; Nakamura, Y.; Sato, S. Genome sequencing and genome resources in model legumes. Plant Physiol. 2007, 144, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.R.; Hoisington, D.A.; Singh, N.K.; Close, T.J.; Varshney, R.K. Orphan legume crops enter the genomics era! Curr. Opin. Plant Biol. 2009, 12, 202–210. [Google Scholar] [CrossRef]
- Jackson, S.A.; May, G.D.; Cannon, S.B. Three sequenced legume genomes and many crop species: Rich opportunities for translational genomics. Plant Physiol. 2009, 151, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, N.I. The law of homologous series in variation. J. Genetic. 1922, 12, 47–89. [Google Scholar] [CrossRef]
- Ladizinsky, G.; Muehlbauer, F.J.; Weeden, N.F. Extensive conservation of linkage relationships between pea and lentil genetic maps. J. Hered. 1992, 83, 123–129. [Google Scholar]
- Muehlbauer, F.J.; Simon, C.J. Construction of a chickpea linkage map and its comparison with maps of pea and lentil. J. Hered. 1997, 88, 115–119. [Google Scholar] [CrossRef]
- Cook, D.R.; Doyle, J.; Luckow, M.A.; Choi, H.K. Development of nuclear gene-derived molecular markers linked to legume genetic maps. Mol. Genet. Genomics 2006, 276, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.B.; Ellis, T.H.N.; Kereszt, G.E.; Kevei, Z.A.; Jaka, J.; Taylor, S.A.; Seres, A.; Kalo, P. Comparative mapping between Medicago sativa and Pisum sativum. Mol. Genet. Genomics 2004, 272, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Thelen, J.J.; Qijian Song, Q.; Hyten, D.L.; David, L.; Nelson, W.; Mitros, T.; Ma, J.; Schlueter, J.; Cannon, S.B.; Schmutz, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.L.; Macknight, R.C.; Murfet, I.C.; Sussmilch, F.C.; Liew, L.C.; Knowles, C.L.; Ridge, S.; Vander Schoor, J.K.; Laurie, R.E.; Hecht, V. The pea GIGAS gene is a FLOWERING LOCUS T homolog necessary for graft-transmissible specification of flowering but not for responsiveness to photoperiod. Plant Cell 2011, 23, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Bisseling, T.; Lauber, E.; Ané, J.M.; Journet, E.P.; Duc, G.; Kulikova, O.; Chalhoub, B.; Geurts, R.; Bres, C.; Levy, J.; et al. A putative Ca2+ and Calmodulin-dependent protein kinase required for bacterial and fungal symbioses. Science 2004, 303, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, F.; Tabata, S.; Kaneko, T.; Duc, G.; Szczyglowski, K.; Genua, A.; Aubert, G.; Sato, S.; Madsen, L.H.; Krusell, L.; et al. Shoot control of root development and nodulation is mediated by a receptor-like kinase. Nature 2002, 420, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Aubert, G.; Otsubo, Y.; Oka-Kira, E.; Okamoto, S.; Grossmann, C.; Koch, E.B.V.; Fukuhara, I.; Sato, N.; Krusell, L.; et al. The Clavata2 genes of pea and Lotus japonicus affect autoregulation of nodulation. Plant J. 2008, 65, 861–871. [Google Scholar]
- Weber, H.; Saalbach, I.; Fernie, A.R.; Fait, A.; Weichert, H.; Muller, M.; Radchuk, R.; Kuster, H.; Weigelt, K. Increasing amino acid supply in pea embryos reveals specific interactions of N and C metabolism, and highlights the importance of mitochondrial metabolism. Plant J. 2008, 55, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Hosein, F.; Desel, C.; Emery, R.J.N.; Wasternack, C.; Miersch, O.; Fernie, A.R.; Fait, A.; Rutten, T.; Küster, H.; Weigelt, K.; et al. ADP-glucose pyrophosphorylase-deficient pea embryos reveal specific transcriptional and metabolic changes of carbon-nitrogen metabolism and stress responses. Plant Physiol. 2009, 149, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Weschke, W.; Fernie, A.R.; Nunes-Nesi, A.; Küster, H.; Emery, R.J.N.; Hajirezaei, M.R.; Meitzel, T.; Radchuk, R.; Häusler, R.E.; Riebeseel, E.; et al. The 2-oxoglutarate/malate translocator mediates amino acid and storage protein biosynthesis in pea embryos. Plant J. 2010, 61, 350–363. [Google Scholar] [PubMed]
- Weber, H.; Weschke, W.; Feil, R.; Lunn, J.E.; Geigenberger, P.; Vigeolas, H.; Weier, D.; Emery, N.J.R.; Radchuk, R. Sucrose non-fermenting kinase 1 (SnRK1) coordinates metabolic and hormonal signals during pea cotyledon growth and differentiation. Plant J. 2010, 61, 324–338. [Google Scholar] [PubMed]
- Rubiales, D.; Cubero, J.I.; Krajinski, F.; Küster, H.; Fondevilla, S. Identification of genes differentially expressed in a resistant reaction to Mycosphaerella pinodes in pea using microarray technology. BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.P.M.; Bornberg-Bauer, E.; Bräutigam, A.; Shrestha, R.P.; Franssen, S.U. Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, J.; Garin, J.; Macherel, D.; Rabilloud, T.; Luche, S.; Jourdain, A.; Jaquinod, M.; Louwagie, M.; Bardel, J. A survey of the plant mitochondrial proteome in relation to development. Proteomics 2002, 2, 880–898. [Google Scholar] [CrossRef] [PubMed]
- Wienkoop, S.; Erik, P.; Saalbach, G. Characterisation by proteomics of peribacteroid space and peribacteroid membrane preparations from pea (Pisum sativum) symbiosomes. Proteomics 2002, 2, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Sommerer, N.; Negroni, L.; Huart, M.; Gallardo, K.; Schiltz, S. Proteome reference maps of vegetative tissues in pea. An investigation of nitrogen mobilization from leaves during seed filling. Plant Physiol. 2004, 135, 2241–2260. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, M.; Jacquin, F.; Savois, V.; Sommerer, N.; Labas, V.; Henry, C.; Burstin, J. Dissecting the proteome of pea mature seeds reveals the phenotypic plasticity of seed protein composition. Proteomics 2009, 9, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Jorrín, J.V.; Rubiales, D.; Maldonado, A.M.; Lopez, J.A.; Camafeita, E.; Curto, M. A proteomic approach to study pea (Pisum sativum) responses to powdery mildew (Erysiphe pisi). Proteomics 2006, 6, S163–S174. [Google Scholar] [CrossRef] [PubMed]
- Delbreil, B.; Lejeune-Hénaut, I.; Hilbert, J.L.; Sellier, H.; Valot, B.; Goulas, E.; Bahrman, N.; Dumont, E. A proteomic approach to decipher chilling response from cold acclimation in pea (Pisum sativum L.). Plant Sci. 2011, 180, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Jorrín, J.V.; Rubiales, D.; Gaudot, E.D.; Amiour, N.; Castillejo, M.A. A proteomic approach to studying plant response to crenate broomrape (Orobanche crenata) in pea (Pisum sativum). Phytochemistry 2004, 65, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Jorrin, J.V.; Rubiales, D.; Fondevilla, S.; Curto, M.; Castillejo, M.A. Two-dimensional electrophoresis based proteomic analysis of the pea (Pisum sativum) in response to Mycosphaerella pinodes. J. Agric. Food Chem. 2010, 58, 12822–12832. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Marget, P.; Huart, M.; Quillien, L.; Aubert, G.; Belghazi, M.; Savois, V.; Cassecuelle, F.; Jacquin, F.; Bourgeois, M. A PQL (protein quantity loci) analysis of mature pea seed proteins identifies loci determining seed protein composition. Proteomics 2011, 9, 1581–1594. [Google Scholar]
- Mullineaux, P.M.; Chinoy, C.; Ambrose, M.; Arques, J.L.; Oehlschlager, S.; Godward, J.; Jones, S.A.; Harrison, M.; Donarski, J.A.; Charlton, A.; et al. Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics 2008, 4, 312–327. [Google Scholar] [CrossRef]
- Oehlschlager, S.; Mullineaux, P.; Ellis, N.; Bean, S.; Chisholm, J.; Holmes, S.; Allnutt, T.; Charlton, A. NMR profiling of transgenic peas. Plant Biotechnol. J. 2004, 2, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D. Mapping polygenes. Ann. Rev. Genet. 1993, 27, 205–233. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D. QTL mapping and quantitative disease resistance in plants. Ann. Rev. Phytopatol. 1996, 34, 479–501. [Google Scholar] [CrossRef]
- Hýbl, M.; Dostálová, R.; Horáček, J.; Smýkal, P. Variety discrimination in pea (Pisum sativum L.) by molecular, biochemical and morphological markers. J. Appl. Genet. 2008, 49, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Scholz, U.; Grosse, I.; Thiel, T.; Miehe, H.; Funke, T.; Lange, M.; Künne, C. CR-EST: A resource for crop ESTs. Nucleic Acids Res. 2005, 33, D619–D621. [Google Scholar] [PubMed]
- Bhalla, P.L.; Smyth, G.K.; Phipson, B.; Beveridge, C.A.; Singh, M.B.; Wong, C.E.; Liang, D. Molecular dissection of the pea shoot apical meristem. Exp. Bot. 2009, 60, 4201–4213. [Google Scholar] [CrossRef]
- Mitchell, S.E.; Buckler, E.S.; Kawamoto, K.; Poland, J.A.; Sun, Q.; Glaubitz, J.C.; Elshire, R.J. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Cockram, J.; White, J.; Zuluaga, D.L.; Smith, D.; Comadran, J.; Macaulay, M.; Luo, Z.; Kearsey, M.J.; Werner, P.; Harrap, D.; et al. Genome-wide association mapping to candidate polymorphism resolution in the unsequenced barley genome. Proc. Natl. Acad. Sci. USA 2010, 107, 21611–21616. [Google Scholar] [CrossRef] [PubMed]
- Graner, A.; Kilian, B.; Haseneyer, G.; van Eeuwijk, F.A.; Malosetti, M.; Sharma, R.; Pasam, R.K. Genome-wide association studies for agronomical traits in a world wide spring barley Collection. BMC Plant Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, M.E.; Jannink, J.L.; Lorenz, A.J.; Heffner, E.L. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Pfleger, F.L.; Kraft, J.M. Compendium of Pea Diseases and Pests; 2001; APS Press: St. Paul, MN, USA. [Google Scholar]
- Weeden, N.F.; Grey, W.; Hance, S.T. Indentification of tolerance to Fusarium solani in Pisum sativum ssp. elatius. Pisum Genet. 2004, 36, 9–13. [Google Scholar]
- Porter, L.; Weeden, N. The genetic basis of Fusarium root rot tolerance in the Afghanistan pea. Pisum Genet. 2007, 39, 35–36. [Google Scholar]
- Xue, A.G.; McLaren, D.L.; Gossen, B.D.; Strelkov, S.E.; Hwang, S.F.; Conner, R.L.; Chang, K.F.; Hwang, R.; Feng, J. Identification of microsatellite markers linked to quantitative trait loci controlling resistance to Fusarium root rot in field pea. Can. J. Plant Sci. 2011, 91, 199–204. [Google Scholar] [CrossRef]
- Coyne, C.J.; Inglis, D.A.; McPhee, K.E.; McClendon, M.T.; Keller, K.E.; Okubara, P.A. Y15_999Fw, a dominant SCAR marker linked to the Fusarium wilt race 1 (Fw) resistance gene in pea. Pisum Genet. 2005, 37, 32–35. [Google Scholar]
- Muehlbauer, F.J.; Grajal-Martin, M.J. Genomic location of the Fw gene for resistance to fusarium wilt race 1 in peas. J. Hered. 2002, 93, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.J.; McPhee, K.E.; Inglis, D.A.; McClendon, M.T. DNA markers for Fusarium wilt race 1 resistance gene in pea. J. Am. Soc. Hort. Sci. 2002, 127, 602–607. [Google Scholar]
- Muehlbauer, F.J.; Whitehead, S.J.; Inglis, D.A.; Coyne, C.J. Chromosomal location of Fwf in pea. Pisum Genet. 2000, 32, 20–22. [Google Scholar]
- Coyne, C.J.; Gunderson, B.; Inglis, D.A.; McPhee, K.E. Mapping a resistance gene for Fusarium wilt Race 2 on LG IV of pea (Pisum sativum L.). Plant Breed. 2012. [Google Scholar] [CrossRef]
- Matthews, P.; Lewis, B.G.; Darby, P. heritance and Expression of Resistance to Ascochyta pisi. In The Pea Crop; Eds., Dawkins, T.C., Heath, M.C., Hebblethwaite, P.D., Eds.; 1985; Butterworths: London, UK. [Google Scholar]
- Lewis, B.G.; Matthews, P.; Clulow, S.A. Genetical analysis of resistance to Mycosphaerella pinodes in pea seedlings. Euphytica 1991, 58, 183–189. [Google Scholar] [CrossRef]
- Falloon, K.; Murray, S.; Gilpin, M.; Butler, R.; Khan, T.; Russell, A.C.; Frew, T.J.; Timmerman-Vaughan, G.M. QTL mapping of partial resistance to field epidemics of Ascochyta blight of peas. Crop Sci. 2002, 42, 2100–2111. [Google Scholar] [CrossRef]
- Khan, T.; Russell, A.; Lakeman, M.B.; Johnston, P.; Falloon, K.; Gilpin, M.; Murray, S.; Butler, R.; Frew, T.J.; Timmerman-Vaughan, G.M. Validation of quantitative trait loci for Ascochyta blight resistance in pea (Pisum sativum L.), using populations from two crosses. Theor. Appl. Genet. 2004, 109, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- DeKoeyer, D.; Xue, A.; Bing, D.; Woods, S.; Blade, S.; Vandenberg, A.; Miranda, D.; Somers, D.J.; Warkentin, T.; Tar’an, B.; et al. Quantitative trait loci for lodging resistance, plant height and partial resistance to mycosphaerella blight in field pea (Pisum sativum L.). Theor. Appl. Genet 2003, 107, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Baranger, A.; Morin, G.; Deniot, G.; Frankewitz, A.; Prioul, S. Mapping of quantitative trait loci for partial resistance to Mycosphaerella pinodes in pea (Pisum sativum L.), at the seedling and adult plant stages. Theor. Appl. Genet. 2004, 108, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.M.; Cubero, J.I.; Vaz Patto, M.C.; Rubiales, D.; Satovic, Z.; Almeida, N.F.; Fondevilla, S. Identification of common genomic regions controlling resistance to Mycosphaerella pinodes, earliness and architectural traits in different pea genetic backgrounds. Euphytica 2011, 18, 43–52. [Google Scholar]
- Tazelaar, M.F.; van Norel, A.; Heringa, R.J. Resistance to powdery mildew (Erysiphe polygoni D.C.) in peas (Pisum sativum L.). Euphytica 1969, 18, 163–169. [Google Scholar]
- Harland, S.C. Inheritance of immunity to mildew in Peruvian forms of Pisum sativum. Heredity 1948, 2, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.D.; Penner, G.A.; Tiwari, K.R. Inheritance of powdery mildew resistance in pea. Can. J. Plant Sci. 1997, 77, 307–310. [Google Scholar] [CrossRef]
- Rubiales, D.; Moreno, M.T.; Carver, T.L.W.; Fondevilla, S. Macroscopic and histological characterisation of genes er1 and er2 for powdery mildew resistance in pea. Eur. J. Plant Pathol. 2006, 115, 309–321. [Google Scholar] [CrossRef]
- Devienne, D.; Cousin, R.; Belajouza, M.; Ranade, S.; Isaac, P.; Dirlewanger, E. Restriction fragment length polymorphism analysis of loci associated with disease resistance genes and developmental traits in Pisum sativum L. Theor. Appl. Genet. 1994, 88, 17–27. [Google Scholar]
- Weeden, N.F.; Frew, T.J.; Timmerman-Vaughan, G.M. Linkage analysis of er1, a recessive Pisum sativum gene for resistance to powdery mildew fungus (Erysiphe pisi D.C). Theor. Appl. Genet. 1994, 88, 1050–1055. [Google Scholar]
- Warkentin, T.D.; Penner, G.A.; Tiwari, K.R. Identification of coupling and repulsion phase RAPD markers for powdery mildew resistance gene er-1 inpea. Genome 1998, 41, 440–444. [Google Scholar]
- Leitão, J.M.; Farinho, M.; Tavares-Sousa, M.; Damaso, M.; Formiga, S.; Ribeiro, R.; Marques, C.; Pereira, G. Identification of DNA markers linked to an induced mutated gene conferring resistance to powdery mildew in pea (Pisum sativum L.). Euphytica 2010, 171, 327–335. [Google Scholar] [CrossRef]
- Tuvesson, S.; Issac, P.; Ceptilis, A.; Weibull, P.; Henriksson, T.; Dayteg, C.; von Post, R.; Eklund, M.; Ek, M. Microsatellite markers for powdery mildew resistance in pea (Pisum sativum L.). Hereditas 2005, 142, 86–91. [Google Scholar] [PubMed]
- Warkentin, T.D.; Penner, G.A.; Tiwari, K.R. Identification of AFLP markers for powdery mildew resistance gene er2 in pea. Pisum Genet. 1999, 31, 27–29. [Google Scholar]
- Ricciardi, L.; Lotti, C.; Bai, Z.; Visser, R.G.F.; Cillo, F.; Marcotrigiano, A.R.; Appiano, M.; Schiavulli, A.; Pavan, S. Pea powdery mildew er1 resistance is associated to loss-of-function mutations at a MLO homologous locus. Theor. Appl. Genet. 2011, 123, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Panstruga, R.; Bisseling, T.; Ivanov, S.; Reinstaedler, A.; Humphry, M. Durable broad-spectrum powdery mildew resistance in pea er1 plants is conferred by natural loss-of-function mutations in PsMLO1. Mol. Plant Pathol. 2011, 12, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Rathour, R.; Sharma, S.K.; Banayal, K.; Pathania, S.; Sharma, S.; Katoch, V. Molecular mapping of pea powdery mildew resistance gene er2 to pea linkage group III. Mol. Breed. 2010, 25, 229–237. [Google Scholar] [CrossRef]
- Torres, A.M.; Moreno, M.T.; Rubiales, D.; Fondevilla, S. Identification and validation of RAPD and SCAR markers linked to the gene Er3 conferring resistance to Erysiphe pisi DC in pea. Mol. Breed. 2008, 22, 193–200. [Google Scholar] [CrossRef]
- Srivastava, C.; Chand, R.; Kushwaha, C. Role of aeciospores in outbreaks of pea (Pisum sativum) rust (Uromyces fabae). Eur. J. Plant Path. 2006, 115, 323–330. [Google Scholar] [CrossRef]
- Torres, A.M.; Rubiales, D.; Satovic, Z.; Barilli, E. Mapping of quantitative trait loci controlling partial resistance against rust incited by Uromyces pisi (Pers.) Wint. in a Pisum fulvum L. intraspecific cross. Euphytica 2010, 175, 151–159. [Google Scholar] [CrossRef]
- Ram, R.S.; Katiyar, R.P. Genetics of rust resistance in pea. Indian J. Genet. 1987, 47, 46–48. [Google Scholar]
- Singh, B.D.; Chand, R.; Srivastava, C.P.; Sarode, S.B.; Kushwaha, C.; Yadav, K.; Vijayalakshmi, S. Identification of RAPD markers linked to the rust (Uromyces fabae) resistance gene in pea (Pisum sativum). Euphytica 2005, 144, 265–274. [Google Scholar] [CrossRef]
- Srivastava, C.P.; Chand, R.; Joshi, A.K.; Singh, B.D.; Singh, A.K.; Rai, R. Molecular mapping for resistance to pea rust caused by Uromyces fabae (Pers.) de-Bary. Theor. Appl. Genet. 2011, 123, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Muehlbauer, F.J.; Grau, C.R.; McGee, R.J.; Weeden, N.F. A gene influencing tolerance to common root rot is located on linkage group IV. Pisum Genet. 2000, 32, 53–55. [Google Scholar]
- Coyne, C.J.; Baranger, A.; Kraft, J.M.; McGee, R.J.; Muehlbauer, F.J.; Pilet-Nayel, M.L. Quantitative trait loci for partial resistance to Aphanomyces root rot in pea. Theor. Appl. Genet. 2002, 106, 28–39. [Google Scholar] [PubMed]
- Coyne, C.J.; Baranger, A.; Kraft, J.M.; McGee, R.J.; Muehlbauer, F.J.; Pilet-Nayel, M.L. Consistent QTL in pea for partial resistance to Aphanomyces euteiches isolates from United States and France. Phytopathology 2005, 95, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Mangin, P.; Klein, A.; Rivière, J.P.; Esnault, R.; L’Anthoëne, V.; Le Goff, I.; McGee, R.J.; Coyne, C.J.; Baranger, A.; Hamon, C.; et al. New consistent QTL in pea associated with partial resistance to Aphanomyces euteiches in multiple field and controlled environments from France and the United States. Theor. Appl. Genet. 2010, 123, 261–281. [Google Scholar]
- Taylor, J.D.; Ellis, N.; Hunter, P.J. Association of dominant loci for resistance to Pseudomonas syringae pv. pisi with linkage groups II, VI and VII of Pisum sativum. Theor. Appl. Genet. 2001, 103, 129–135. [Google Scholar] [CrossRef]
- Caminero, C.; Rubiales, D.; Fernández-Romero, M.D.; Satovic, Z.; Martın-Sanz, A.; Fondevilla, S. Identification of quantitative trait loci involved in resistance to Pseudomonas syringae pv. syringae in pea (Pisum sativum L.). Euphytica 2012. [Google Scholar] [CrossRef]
- Taylor, J.D.; Elvira-Recuenco, M. Resistance to bacterial blight (Pseudomonas syringae pv. pisi) in Spanish pea (Pisum sativum) landraces. Euphytica 2001, 118, 305–311. [Google Scholar] [CrossRef]
- Sillero, J.C.; Moreno, M.T.; Rubiales, D. Search for resistance to crenate broomrape (Orobanche crenata) in pea germplasm. Genet. Res. Crop Evol. 2005, 52, 853–861. [Google Scholar] [CrossRef]
- Fondevilla, S.; Rispail, N.; Sillero, J.; Castillejo, M.A.; Prats, E.; Pérez-de-Luque, A.; Fernández-Aparicio, M.; Rubiales, D. Breeding approaches for crenate broomrape (Orobanche crenata Forsk.) management in pea (Pisum sativum L.). Pest Manag. Sci. 2009, 65, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.M.; Cubero, J.I.; Rubiales, D.; Satovic, Z.; Román, B.; Valderrama, M.R. Locating genes associated with Orobanche crenata resistance in pea. Weed Res. 2004, 44, 323–328. [Google Scholar] [CrossRef]
- Rubiales, D.; Moreno, M.T.; Torres, A.M.; Emeran, A.A.; Satovic, Z.; Fernández-Aparicio, M.; Fondevilla, S. Identification of quantitative trait loci for specific mechanisms of resistance to Orobanche crenata Forsk. in pea (Pisum sativum L.). Mol. Breed. 2010, 25, 259–272. [Google Scholar] [CrossRef]
- Hardie, D.; Byrne, O. Incorporation of pea weevil resistance into a cultivar field peas. Grains Research and Development Corporation Project Report No UWA314SR, CLIMA,, University of Western Australia: Australia, 2004.
- Hampton, R.O.; Provvidenti, R. Chromosomal distribution of genes for resistance to seven potyviruses in Pisum sativum. Pisum Genet. 1991, 23, 26–28. [Google Scholar]
- Laursen, J.; Hjulsager, C.K.; Lund, O.S.; Johansen, E.I. Recessive resistance in Pisum sativum and Potyvirus pathotype resolved in a gene-for-cistron correspondence between host and virus. J. Virol. 2001, 75, 6609–6614. [Google Scholar] [CrossRef] [PubMed]
- Maule, A.; Ellis, N.; Thomas, C.; Eyers, S.; Gao, Z. Identification of markers tightly linked to sbm recessive genes for resistance to Pea seed-borne mosaic virus. Theor. Appl. Genet. 2004, 109, 488–494. [Google Scholar] [PubMed]
- Johansen, I.E.; Lund, O.S.; Hansen, K.R.; Tulinius, G.; Møller, I.S.; Bruun-Rasmussen, M. The same allele of translation initiation factor 4E mediates resistance against two Potyvirus spp. Pisum sativum. Mol. Plant Microbe Interact. 2007, 20, 1075–1082. [Google Scholar] [CrossRef]
- Uyeda, I.; Nakahara, K.S.; Abe, Y.; Andrade, M. The cyv-2 resistance to Clover yellow vein virus in pea is controlled by the eukaryotic initiation factor 4E. J. Gen. Plant Pathol. 2009, 75, 241–249. [Google Scholar] [CrossRef]
- Dostalová, R.; Navrátil, M.; Šafářová, D.; Smýkal, P. Marker assisted pea breeding: eIF4E allele specific markers to pea seed-borne mosaic virus (PSbMV) resistance. Mol. Breed. 2010, 26, 425–438. [Google Scholar] [CrossRef]
- Weeden, N.F.; Randhawa, H. Refinement of the position of En on LG III and identification of closely linked DNA markers. Pisum Genet. 2011. [Google Scholar]
- Providenti, R.; Weeden, N.F. A marker locus, Adh-1, for resistance to Pea Enation Mosaic Virus in Pisum sativum. J. Hered. 1988, 79, 128–131. [Google Scholar]
- Weeden, N.; Provvidenti, R.; Gu, W.K.; Yu, J. Identifying and mapping two DNA markers linked to the gene conferring resistance to Pea Enation Mosaic Virus. J. Am. Soc. Hort. Sci. 1995, 120, 730–733. [Google Scholar]
- Vander Schoor, J.K.; Knowles, C.L.; Wenden, B.; Sussmilch, F.C.; Liew, L.C.; Hecht, V.; Weller, J.L. Update on the genetic control of flowering in garden pea. J. Exp. Bot. 2009, 60, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Murfet, I.C. Flowering in Pisum. Hr, a gene for high response to photoperiod. Heredity 1973, 31, 157–164. [Google Scholar] [CrossRef]
- Murfet, I.C. Flowering in Pisum. Multiple alleles at the Lf locus. Heredity 1975, 35, 85–98. [Google Scholar] [CrossRef]
- Rameau, C.; Banfield, M.J.; Ellis, N.; Cadioux, S.; Courtiade, J.; Morin, J.; Foucher, F. DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER1/CENTRORADIALIS homologs that control two distinct phases of flowering initiation and development in pea. Plant Cell 2003, 15, 2742–2754. [Google Scholar] [CrossRef] [PubMed]
- Stempniak, J.J.; Boilleau, M.; Devaux, R.; Petit, A.; Morin, J.; Delbreil, B.; Fontaine, V.; Béthencourt, L.; Hanocq, E.; Lejeune-Hénaut, I.; et al. The flowering locus Hr colocalizes with a major QTL affecting winter frost tolerance in Pisum sativum L. Theor. Appl. Genet. 2008, 116, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.L.; Lambert, M.J.; Jones, S.E.; Liew, L.C.; Vander Schoor, J.K.; Knowles, C.L.; Hecht, V. Pea LATE BLOOMER1 is a GIGANTEA ortholog with roles in photoperiodic flowering, deetiolation, and transcriptional regulation of circadian clock gene homologs. Plant Physiol. 2007, 144, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Liew, L. Genetic Control of Photoperiod Responsiveness in Pea. Ph.D. Dissertation, University of Tasmania, Tasmania, Australia, 2011. [Google Scholar]
- Domoney, C.; Welham, T.; Duc, G.; Aubert, G.; Page, D. Combinatorial variation in coding and promoter sequences of genes at the Tri locus in Pisum sativum accounts for variation in trypsin inhibitor activity in seeds. Mol. Genet. Genomics 2002, 267, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Domoney, C.; Moreau, C.; Turner, L.; Welham, T.; Chinoy, C. The genetic control of seed quality traits: Effects of allelic variation at the Tri and Vc-2 genetic loci in Pisum sativum L. Euphytica 2011, 180, 107–122. [Google Scholar] [CrossRef]
- Timmerman-Vaughan, G.; Swiecicki, W.K. Molecular Marker Systems in Plant Breeding and Crop Improvement Biotechnology in Agriculture and Forestry,Localization of Important Traits: The Example Pea (Pisum sativum L.); Eds., Wenzel, G., Lorz, H., Eds.; 2005; Springer-Verlag: Berlin, Germany. [Google Scholar]
- Russell, A.C.; Weeden, N.F.; Frew, T.J.; McCallum, J.A.; Timmerman-Vaughan, G.M. Linkage mapping of quantitative trait loci controlling seed weight in pea (Pisum sativum L.). Theor. Appl. Genet. 1996, 93, 431–439. [Google Scholar] [CrossRef]
- Young, N.D.; Danesh, D.; Menancio-Hautea, D.I.; Fatokun, C.A. Evidence for orthologous seed weight genes in cowpea and mung bean based on RFLP mapping. Genetics 1992, 132, 841–846. [Google Scholar] [PubMed]
- Buss, G.R.; Saghai Maroof, M.A.; Maughan, P.J. Molecular-marker analysis of seed-weight: Genomic locations, gene action, and evidence for orthologous evolution among three legume species. Theor. Appl. Genet. 1996, 93, 574–579. [Google Scholar] [CrossRef]
- Wilson, D.; Russell, A.; McCallum, J.; Lakeman, M.; Murray, S.; Butler, R.; Frew, T.; Whitfield, C.; Mills, A.; Timmerman-Vaughan, G.M. Linkage mapping of QTL for seed yield, yield components, and developmental traits in pea. Crop Sci. 2005, 45, 1336–1344. [Google Scholar] [CrossRef]
- Bing, D.; Blade, S.; Vandenberg, A.; Miranda, D.; Somers, D.; Warkentin, T.; Tar’an, B. Quantitative trait loci for grain yield, seed protein content and days to maturity in field pea (Pisum sativum L.). Euphytica 2004, 136, 297–306. [Google Scholar] [CrossRef]
- Somers, D.J.; Vandenberg, A.; Bett, K.E.; Tullu, A.; Warkentin, T.D.; Tar’an, B.; Zhang, C. Selection for lodging resistance in field pea (Pisum sativum L.) using molecular markers. Crop Sci. 2006, 46, 321–329. [Google Scholar] [CrossRef]
- Wolko, B.; Irzykowska, L. Interval mapping of QTLs controlling yield-related traits and seed protein content in Pisum sativum. J. Appl. Genet. 2004, 45, 297–306. [Google Scholar] [PubMed]
- Duc, G.; Munier-Jolain, N.; Desprez, B.; Duchene, C.; Mangin, B.; Moessner, A.; Huart, M.; Marget, P.; Burstin, J. Developmental genes have pleiotropic effects on plant morphology and source capacity, eventually impacting on seed protein content and productivity in pea. Plant Physiol. 2007, 144, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Burstin, J.; Duc, G.; Marget, P.; Galmiche, F.; de Larambergue, H.; Fournier, S.; Rizvi, S.M.H.; Bourion, V. Genetic dissection of nitrogen nutrition in pea through a QTL approach of root, nodule, and shoot variability. Theor. Appl. Genet. 2010, 121, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Wolko, B.; Swiecicki, W.; Pniewski, T.; Kaczmarek, Z.; Gawłowska, M.; Bocianowski, J.; Krajewski, P. QTL for yield components and protein content: A multienvironment study of two pea (Pisum sativum L.) populations. Euphytica 2012, 183, 323–336. [Google Scholar] [CrossRef]
- Russell, A.; Frew, T.; Timmerman-Vaughan, G.; McCallum, J. Biochemical and genetic linkage analysis of green seed color in field pea. J. Am. Hort. Sci. 1997, 122, 218–225. [Google Scholar]
- Warkentin, T.D.; Vijayan, P.; Tar’an, B.; Bett, K.; Ubayasena, L. Genetic control and QTL mapping of cotyledon bleaching resistance in green field pea (Pisum sativum L.). Genome 2010, 53, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.D.; Tar’an, B.; Bett, K.; Ubayasena, L. Genetic control and identification of QTLs associated with visual quality traits of field pea (Pisum sativum L.). Genome 2011, 54, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, J.L.; Avia, K.; Frise, M.; Devaux, R.; Voedts, N.; Bodèle, S.; Sellier, H.; Vuylsteker, C.; Fontaine, V.; Dumont, E.; et al. Association of sugar content QTL and PQL with physiological traits relevant to frost damage resistance in pea under field and controlled conditions. Theor. Appl. Genet. 2009, 118, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Hedley, C.; Ellis, T.H.N.; Smith, A.M.; Bhattacharyya, M.K. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell 1990, 60, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hvoslef-Eide, A.K.; Ambrose, M.J.; Lee, Y.K.; Owusu, A.K. Growth, seed development and genetic analysis in wild type and Def mutant of Pisum sativum L. BMC Res. Notes 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, H.J.; Riesselmann, S.; Menke, M.; Lindemann, S.; Stackelberg, M. Identification of AFLP and STS markers closely linked to the def locus in pea. Theor. Appl. Genet. 2003, 106, 1293–1299. [Google Scholar] [PubMed]
- Weeden, N.F. Genetic changes accompanying the domestication of Pisum sativum: Is there a common genetic basis to the ‘domestication syndrome’ for legumes? Ann. Bot. 2007, 100, 1017–1025. [Google Scholar]
- Portais, J.C.; Danoun, S.; Matusova, R.; Letisse, F.; Pillot, J.P.; Dun, E.A.; Puech-Pagès, V.; Brewer, P.B.; Fermas, S.; Gomez-Roldan, V.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.B.; Davies, P.J.; Ross, J.J.; Lester, D.R. Mendel’s stem length gene (Le) encodes a gibberellin 3[beta]-hydroxylase. Plant Cell 1997, 9, 1435–1443. [Google Scholar] [PubMed]
- Olhoft, P.M.; Samac, D.A.; Somers, D.A. Recent advances in legume transformation. Plant Physiol. 2003, 131, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Ondrej, V.; Griga, M.; Smýkal, P.; Svabova, L. Agrobacterium-mediated transformation of Pisum sativum in vitro and in vivo. Biol. Plant. 2005, 49, 361–370. [Google Scholar] [CrossRef]
- Lund, O.S.; Johansen, I.E.; Nicolaisen, M.; MacFarlane, S.A.; Krath, B.N.; Constantin, G.D. Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J. 2004, 40, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, I.; Johansen, I.E.; Olsen, A.; Gronlund, M. Protocol: Using virus-induced gene silencing to study the arbuscular mycorrhizal symbiosis in Pisum sativum. Plant Methods 2010, 6. [Google Scholar] [CrossRef]
- Tamiru, M.; Mitsuoka, C.; Yoshida, K.; Matsumura, H.; Kanzaki, H.; Takagi, H.; Natsume, S.; Yoshida, K.; Kosugi, S.; Abe, A.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.R.; Keller, K.E.; Shultz, J.L.; Lightfoot, D.A.; Meksem, K.; Murray, S.; Timmerman-Vaughan, G.M.; Walling, J.G.; McClendon, M.T.; Coyne, C.J.; et al. Construction and characterization of two bacterial artificial chromosome libraries of pea (Pisum sativum L.) for the isolation of economically important genes. Genome 2007, 50, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Barth, S.; Milbourne, D.; Stefanović, S.; Perry, A.S.; Sémon, M.; Cusack, B.P.; Rice, D.W.; Aspinall, S.; Magee, A.M.; et al. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- The International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010, 8. [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smýkal, P.; Aubert, G.; Burstin, J.; Coyne, C.J.; Ellis, N.T.H.; Flavell, A.J.; Ford, R.; Hýbl, M.; Macas, J.; Neumann, P.; et al. Pea (Pisum sativum L.) in the Genomic Era. Agronomy 2012, 2, 74-115. https://doi.org/10.3390/agronomy2020074
Smýkal P, Aubert G, Burstin J, Coyne CJ, Ellis NTH, Flavell AJ, Ford R, Hýbl M, Macas J, Neumann P, et al. Pea (Pisum sativum L.) in the Genomic Era. Agronomy. 2012; 2(2):74-115. https://doi.org/10.3390/agronomy2020074
Chicago/Turabian StyleSmýkal, Petr, Gregoire Aubert, Judith Burstin, Clarice J. Coyne, Noel T. H. Ellis, Andrew J. Flavell, Rebecca Ford, Miroslav Hýbl, Jiří Macas, Pavel Neumann, and et al. 2012. "Pea (Pisum sativum L.) in the Genomic Era" Agronomy 2, no. 2: 74-115. https://doi.org/10.3390/agronomy2020074
APA StyleSmýkal, P., Aubert, G., Burstin, J., Coyne, C. J., Ellis, N. T. H., Flavell, A. J., Ford, R., Hýbl, M., Macas, J., Neumann, P., McPhee, K. E., Redden, R. J., Rubiales, D., Weller, J. L., & Warkentin, T. D. (2012). Pea (Pisum sativum L.) in the Genomic Era. Agronomy, 2(2), 74-115. https://doi.org/10.3390/agronomy2020074