1. Introduction
Since the twentieth century, different blending methods, types of reactions, and polymerization processes have been used to prepare two component polymers, attempting to combine or optimize properties, expand applications, or simply optimize the cost/properties relationship [
1,
2,
3]. It has been demonstrated that the contribution of each polymeric component is improved if phase separation occurs at the microscopic level, which implies that chemical blending offers better component interaction than physical blending [
4]. Additionally, the mechanical superiority of polymer blends containing a gradient in composition over materials with uniform composition in the polymer bulk (e.g., gradient interpenetrating polymer networks (gIPNs) versus regular IPNs obtained by a three step method using bulk polymerization) has been shown [
5,
6,
7]. Such superiority has been attributed to the synergic contribution of polymeric region(s) rich in component A as well as of region(s) rich in component B, in the spatial structure of the bulk polymer, which leads to the optimization of properties [
6]. Moreover, using the suspension process, forming gradients by comers/monomer diffusion in polymer beads followed by an in situ polymerization, better control in transport properties [
8,
9], or ionic exchange optimization [
10,
11] has been achieved in polymer blends. Nevertheless, for materials obtained by conventional or traditional free radical polymerization (FRP), if sequential homopolymerizations of A and B components are used in emulsion, a two-phase core-shell type polymer is usually obtained, where phase separation generates a weak interaction between the components, inhibiting the optimization of properties. However, by appropriate comonomer mixing taking advantage of their common difference in reactivities [
12], gradients in composition have been formed since a long time ago to produce for example a gradient in properties like refraction-index in copolymers [
13]. Basically, gradients in polymers may be formed in one or more constituents of a polymeric material, carrying out variations in space, structure, composition, or synthesis method [
14,
15,
16,
17]. With that perspective, taking into consideration that covalent bonding offers the strongest component interaction and that gradients in composition help for the synergic contribution of the attributes of each component, the attainment of convenient gradual changes in the composition spectrum of copolymer chains (weight composition distribution of chains; WCD) could improve the mechanical properties. One way to search for these characteristics using free radical copolymerization (FRC) is to prepare copolymers by means of a semicontinuous process varying the feed comonomer composition along the reaction. That is, in principle forcing the reaction, copolymer chains rich in A, as well as chains rich in B can be formed, regardless of the relative reactivities (for not extremely distant values). Additionally, with a gradual variation in feeding composition, the formation of chains with intermediate compositions of monomers A and B can also be attained; such chains may serve as compatibilizers to reduce the tendency to separate the polymer bulk into two phases. With that aim, using the styrene-2-ethylhexyl acrylate system with a seeded multistage semicontinuous emulsion process, promising results were reported years ago using linear feeding profiles for the comonomers with variations in global composition [
18]; besides, seed type or particle size may also be varied [
19]. If the cumulative composition of the copolymer chains is followed along the reaction and used to estimate the WCD of the copolymer bulk, the copolymer properties can be correlated to that composition spectrum. With this aim, the styrene/
n-butyl acrylate (S/BA) system with styrene seed was polymerized in emulsion, considering several feeding profiles for the 70/30
w/
w composition. For the copolymers obtained, significant changes in the toughening effect caused by the BA contribution were observed, while their Young’s modulus was sustained to a certain extent at room temperature. Those changes in mechanical behavior corresponded to variations in the copolymer composition spectrum [
20]. Based on those results, to demonstrate the full correspondence between the composition spectrum of the copolymer chains and the mechanical properties (dynamic and static) of a copolymer system, in this study several feeding profiles using the 50/50
w/
w S/BA as chemical system and a semicontinuous emulsion process were considered and the correspondent WCDs are estimated. The S/BA monomer pair is a system of interest for the combination of rigidity-elasticity properties with distant glass transition temperatures (–55 °C for BA and 100 °C for S correspond to the homopolymers), and shows moderate phase segregation [
21]. Their relative reactivities are 0.19 for BA and 0.72 for S [
22].
For the reactions, starting with a polystyrene (PS) seed, predetermined feeding stages are used with linear, parabolic, or linear V type (e.g., linear descendant-ascendant) profiles to change the instantaneous S/BA comonomer composition and orientate, since at the beginning of the reaction the composition changes in the new copolymer chains. With the feeding schemes used, the formation of chains rich in S units (looking for a high modulus) and chains rich in BA units (for high deformation capacity) is intended. In the end, this approach may be used as a method to tailor the properties by means of useful gradual changes in the WCD of the copolymer chains. To be used as references for mechanical properties, an equivalent statistical copolymer (SC) and a core-shell type polymer in two stages (T-S) were also prepared and tested.
2. Experimental Section Materials
Styrene and
n-butyl acrylate monomers (both from Sigma Aldrich, St, Louis, MO, USA, purity >99%) were disinhibited with ionic exchange resins; methyl ester hydroquinone resin was used for BA, and 4-tert-butylcathechol for S. As surfactant, sodium dodecylsulfate (SDS; Aldrich, St, Louis, MO, USA, purity >98%) was used as acquired. Potassium persulfate was used as initiator (KPS; Aldrich, purity >98%). Sodium bicarbonate (Arm and Hammer, Ewing, NJ, USA) was used to buffer pH changes during the reaction. Distilled water was used for every polymerization reaction. In all polymerizations, nitrogen gas was used to purge the reaction system. Twenty wt % of solid polymer (PS, T-S or copolymers) was obtained at the end of every reaction. The reactions were carried out at 70 °C in a 4 L glass reactor, stirring the system at 400 rpm; reaction times >12 h were used to maximize monomer(s) conversion (followed by gravimetry). The SC synthesis was carried out by adding quickly to the reactor the required amount of monomers (S/BA, 50/50
w/
w) at room temperature while stirring. Before the addition of the required amount of initiator, the whole system was heated up to start the batch reaction. For the other polymeric materials, a PS seed was obtained first by batch emulsion polymerization. The T-S polymer was obtained by adding to the reactor the required amount of PS seed and for the second stage the required amount of BA monomer (BA/PS, 50/50
w/
w) was quickly added to such a reactor while stirring; then, the whole system was heated up and a similar procedure to the one followed for the SC synthesis was carried out. The five forced composition copolymers (FCCs) were obtained by means of seeded semicontinuous emulsion processes; for the FCCs the global styrene content was also 50 wt %. At the start of each copolymerization reaction, the reactor was charged with a seed latex containing 50 g of PS and the amount of distilled water required to complete 1600 g of total load. Then, 10 sequential “comonomer feeding stages” of 12 min each one were implemented (except for the V type feeding profile where 20 stages of 6 min were used). At the beginning of each stage, an aqueous solution containing 45 g (except for the reaction of the V type feeding profile, where 22.5 g of water were used) of distilled water, and the required amounts of KPS, SDS, and NaHCO
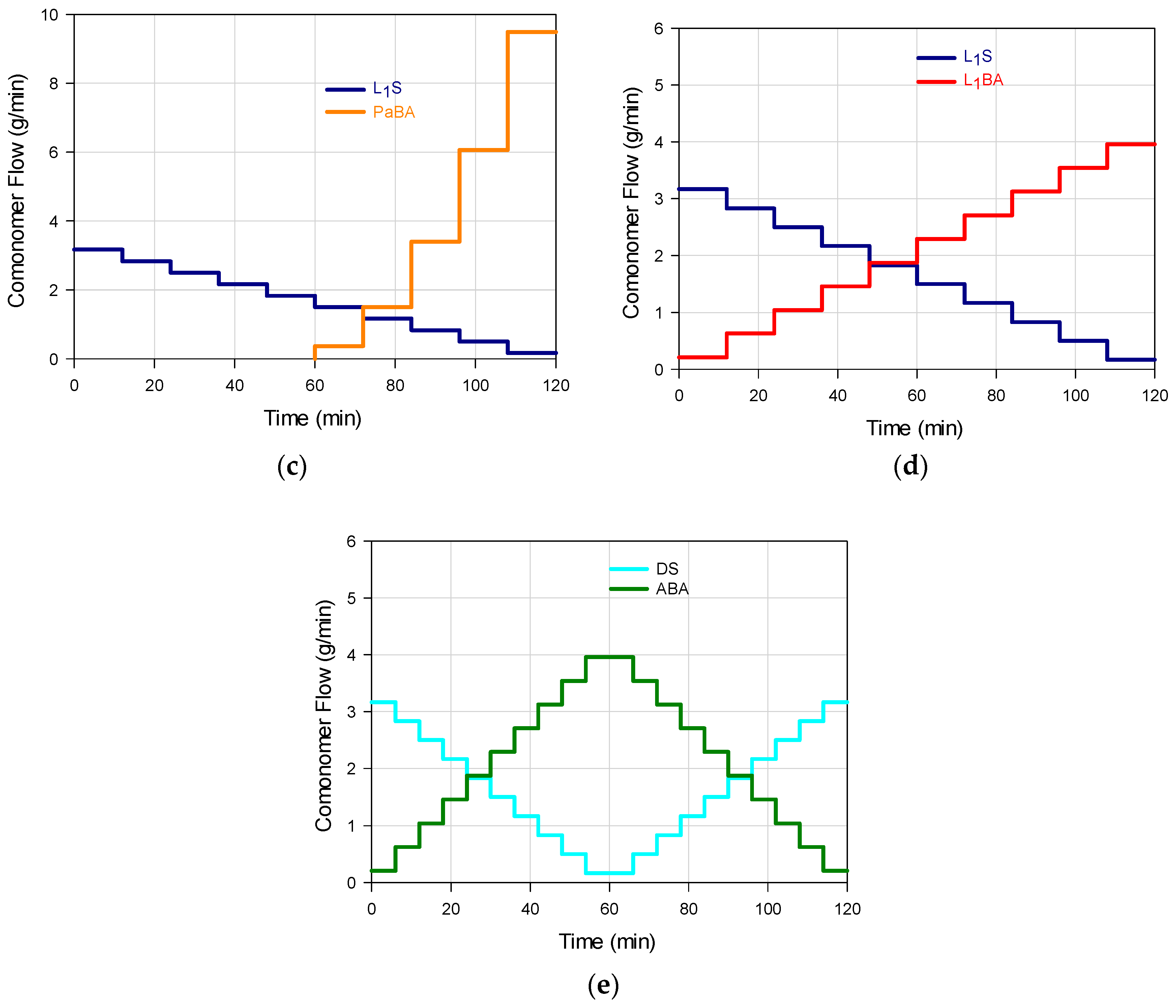
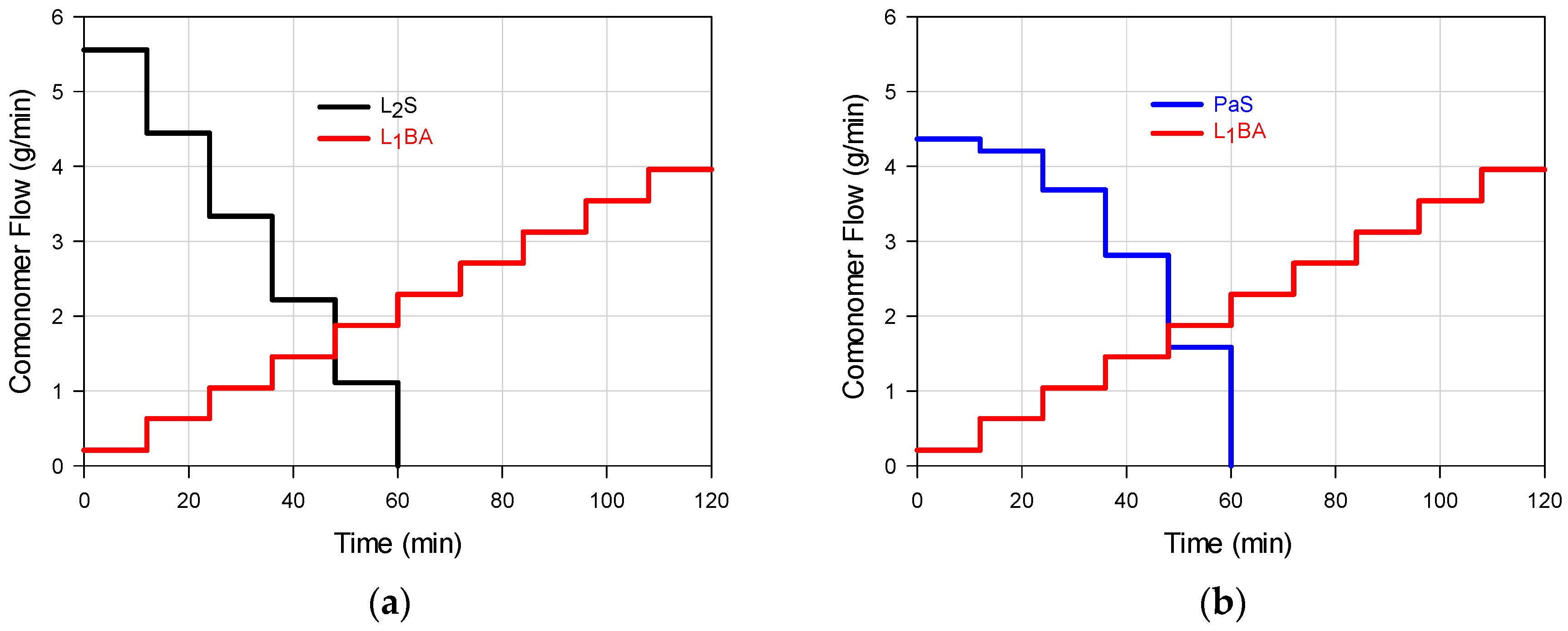
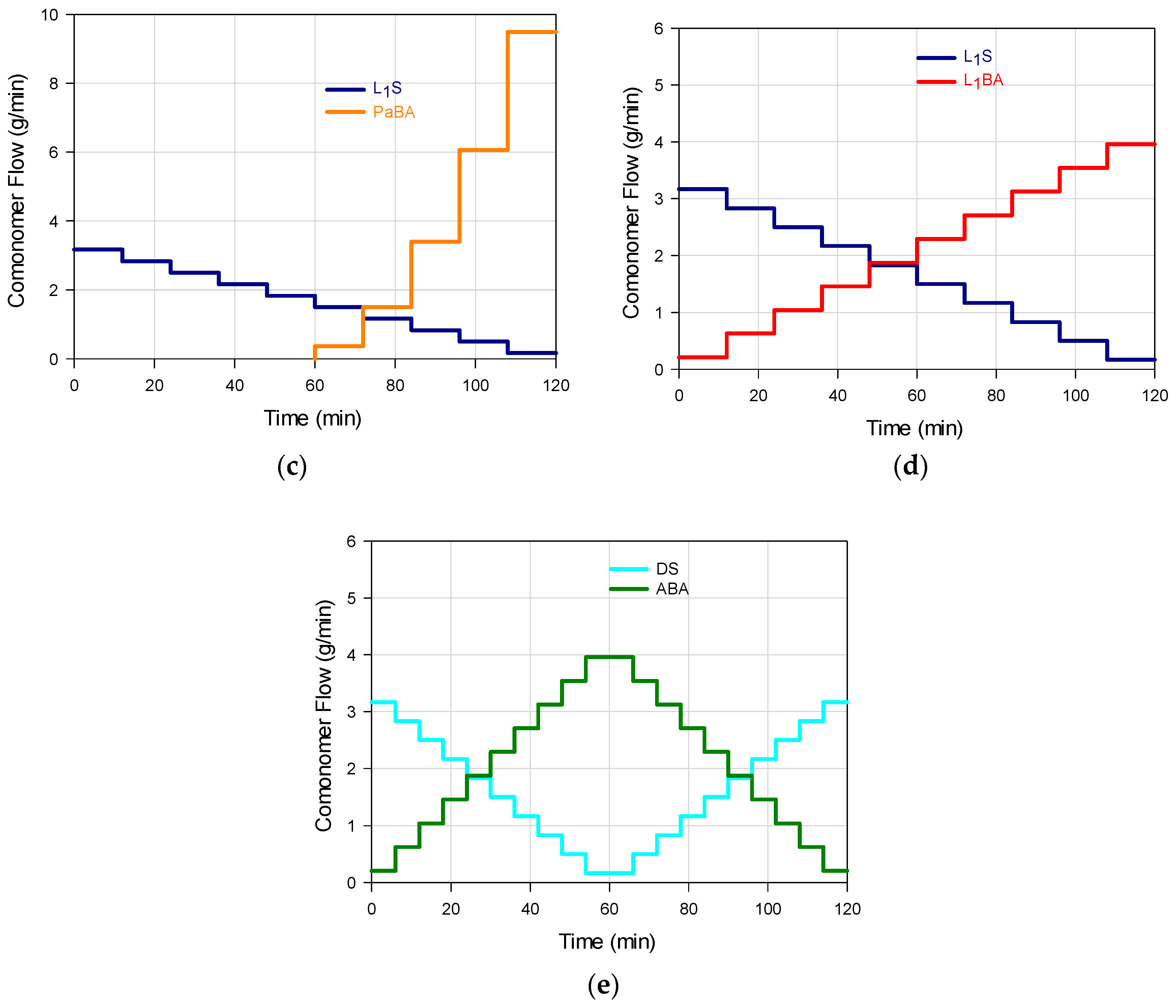
3 were added as a batch to the reaction system. In every stage, the amount added of each salt was the one corresponding to 2% of the total mass of the comonomers to be added in that stage. The feeding flow was modified at the start of each stage, and pumped to the reactor (Masterflex L/S 7528-10) at constant flow rate during the stage. Five feeding profile combinations for the S and BA comonomers were implemented (M
1–M
5), by means of linear (L
1 or L
2), parabolic (Pa), linear ascendant-linear descendant (A), and linear descendant-linear ascendant (D) trajectories. Such individual flow profiles are shown in
Figure 1a–e with the following schemes: L
2S/L
1BA (M
1); PaS/L
1BA (M
2); L
1S/PaBA (M
3); L
1S/L
1BA (M
4); DS/ABA (M
5); in all cases the feeding time was predetermined as 2 h.
For the characterization of each FCC material, seven samples were extracted at predetermined reaction times, to be measured by
1H-NMR (Varian Gemini 2000, Agilent Technologies, Palo Alto, CA, USA) their respective global styrene percentage (
values) and, by gravimetry their respective global conversion values (
values). For the calculations to build up the WCD, the conversion interval (from 0% up to the final experimental conversion value) was discretized in subintervals of 0.1% in width. It was considered that all the chains produced inside of each conversion subinterval
i (characterized by its respective
value) have the same
value, which was estimated with a third order polynomial obtained by fitting the seven above mentioned experimental values; for this, the least squares method was used. The total mass of monomer
j (S or BA) polymerized up to subinterval
i (
) can be calculated with the following equations:
or,
,
being the total amount of monomer (S plus BA) included in the reaction (450 g). Thus, the mass of monomer
j polymerized in the subinterval
i can be estimated as follows:
. Therefore, the mass percentage of S in the chains formed in the subinterval
i can be obtained with the following equation:
. The respective masses of polymer produced with
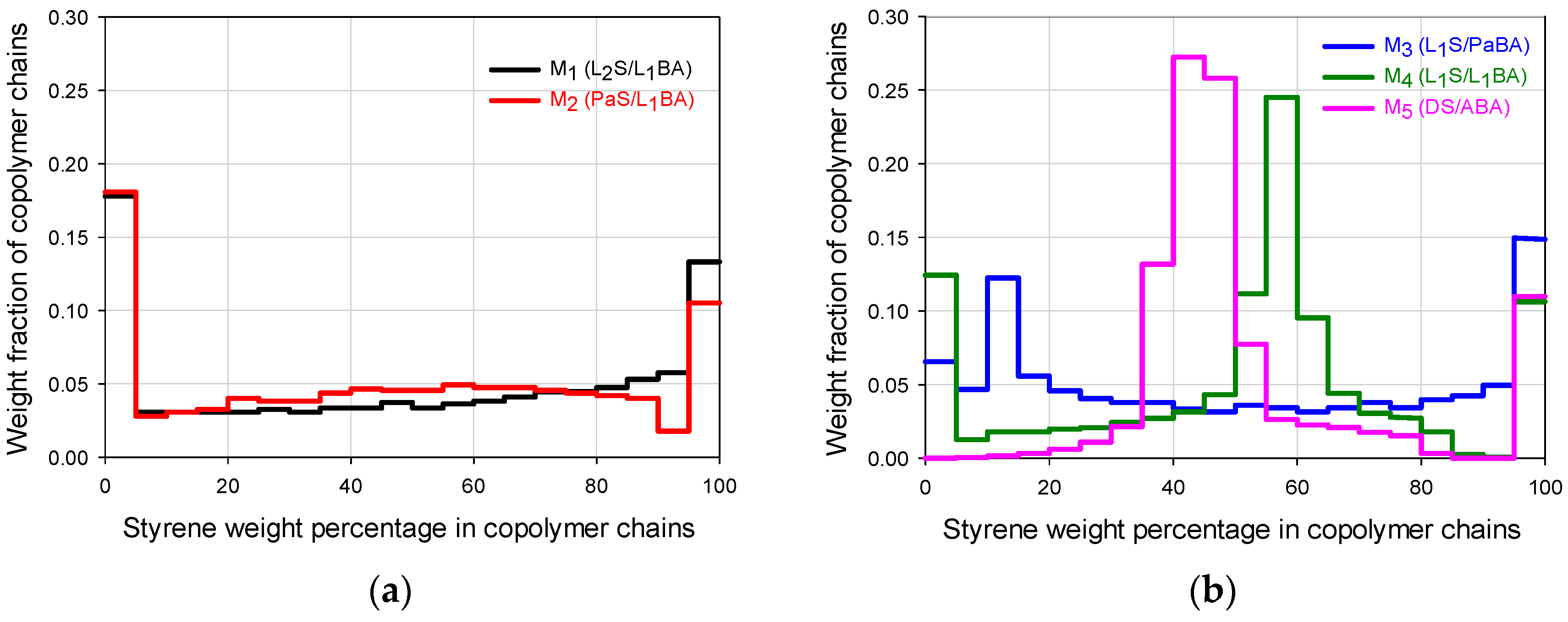
values within the pre-established composition intervals (e.g., 0%–5% of S, 5%–10% of S and so on) were suitably summed up and, with such values, the histograms showing the WCD profiles for the copolymer chains contained in each FCC material at the end of the reaction (
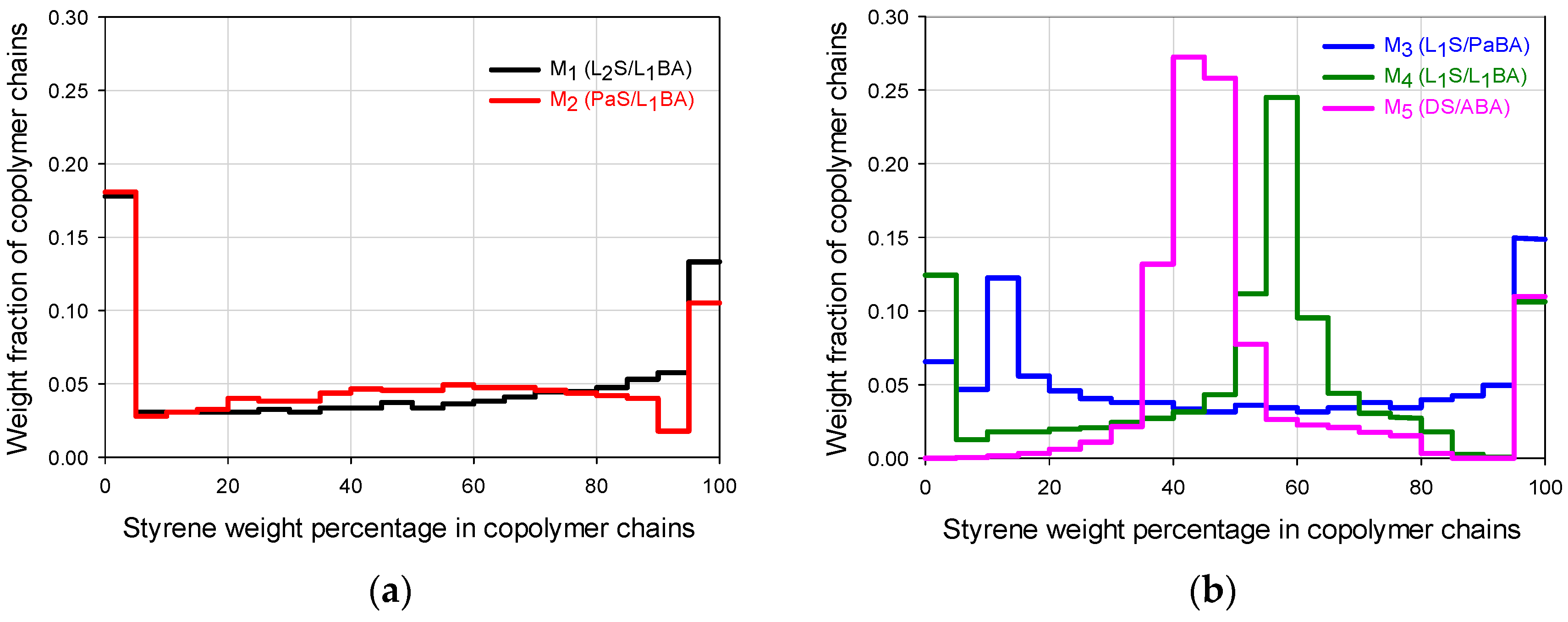
Figure 2a,b) were constructed [
19,
20].
All latexes were dried by evaporation at room temperature; the collected high molecular weight polymers (such as the ones reported before [
18,
19,
20]), were used to prepare samples by compression molding (Schwabenthan polystat 200T) to carry out all mechanical tests. Stress-strain measurements were performed at 25 °C following the procedure of the American Society for Testing Materials (ASTM D638, samples Type IV, crosshead speed: 0.0083 cm/s; Universal testing machine SFM10).
A dynamic mechanical analysis (DMA) was carried out to measure storage and loss moduli as a function of temperature (ASTM D-5023, three point bending clamp, 1 Hz, heating rate: 1 °C/min TA Instruments Q800, Waters Corporation, New Castle, DE, USA).
3. Results and Discussion
The chosen feeding profiles (
Figure 1a–e) allow the diversification of composition histograms that represent the WCDs of the FCCs presented in
Figure 2a,b. The feed trajectories allow the formation of chains that are rich in styrene or butyl acrylate at different moments depending on the flow profile and the monomer reactivities; the continuous change in monomer composition allows a biased change in the instantaneous copolymer composition. The composition profiles shown in
Figure 2a,b define the mechanical properties that are presented in
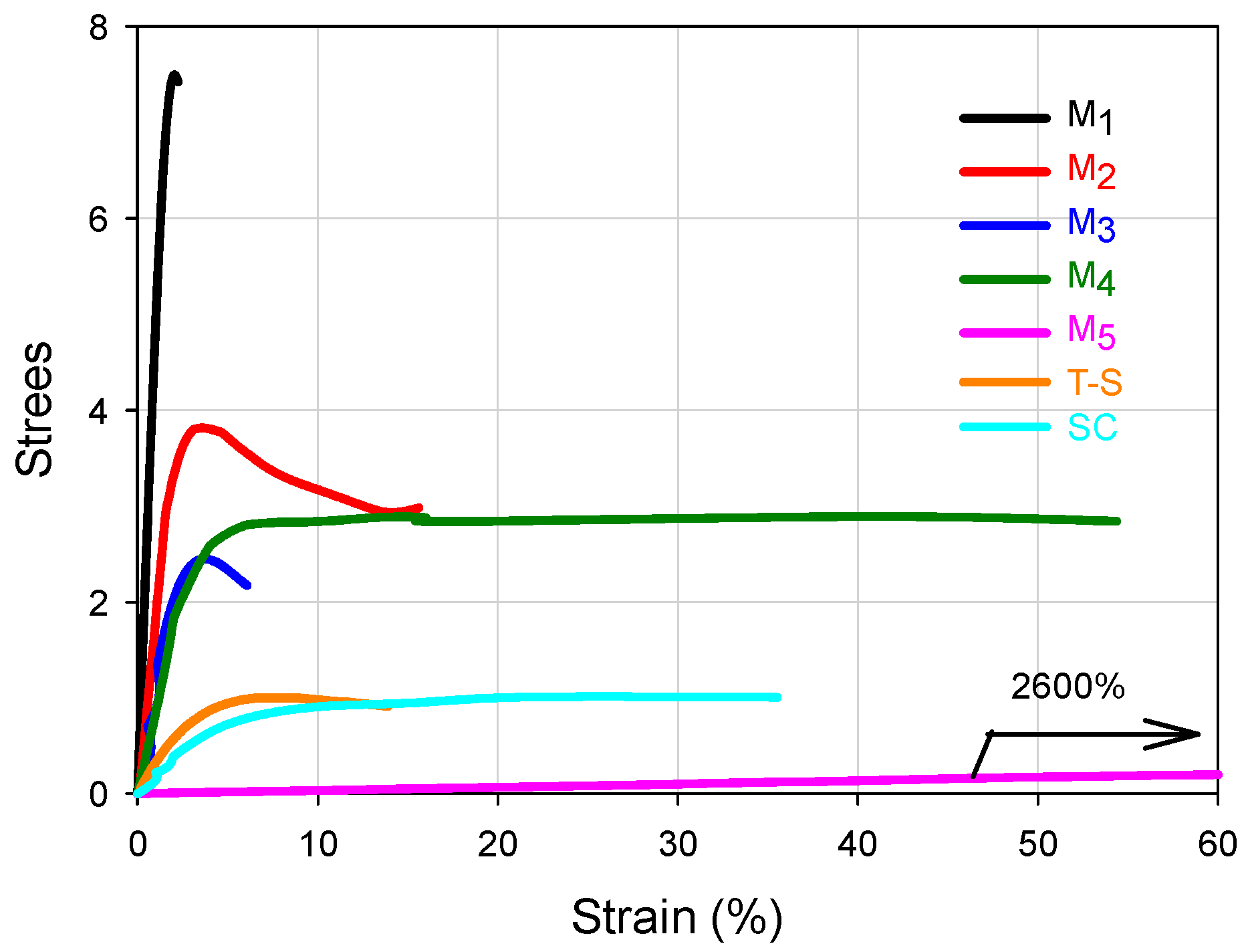
Figure 3,
Figure 4 and
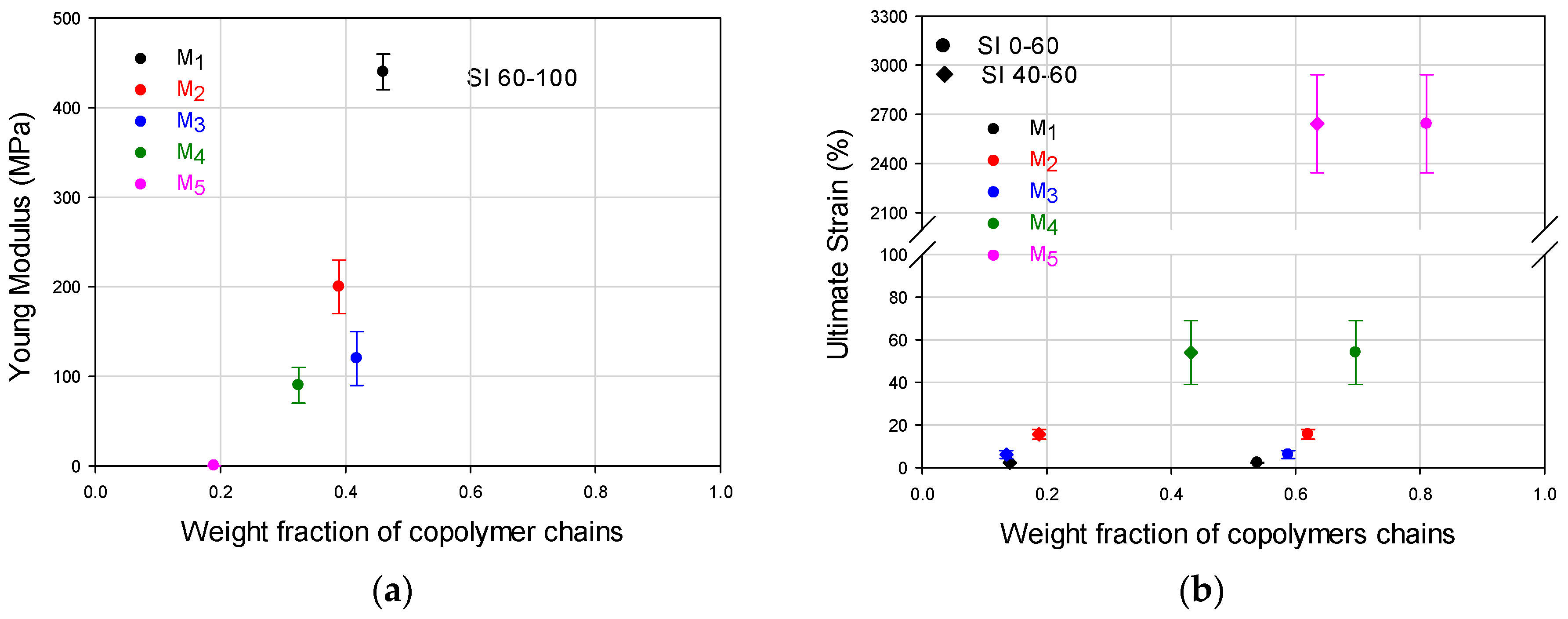
Figure 5. Even though the copolymer composition determines the mechanical performance that may vary from highly elastic (almost pure poly(butyl acrylate) (PBA), to highly rigid behavior (almost pure PS), the deformation capacity and moduli are affected by both components depending on the chains buildup along the composition spectrum. The correlation between the WCD of a copolymer and its correspondent mechanical properties can be disclosed, quantifying in
Figure 2a,b the weight fraction accumulation or wt % of the copolymer chains within specific composition ranges (e.g., 0–60 wt %, which can be designated as styrene composition interval or simply SI 0–60).
For the general FCC behavior, even though the chains that are close to an extreme of the ordinate axis will have significant influence on the property related to the pure homopolymer (i.e., at 0 wt % the behavior stands for PBA and at 100 wt % to PS), the synergic performance also requires the considerable presence of chains that contain both components (as a fundamental difference to T-S materials). In that sense, looking at the FCC histograms as a whole in
Figure 2a,b, the highest mechanical moduli can be expected for M
1 and M
2, but higher deformations can be expected for M
4 or M
5.
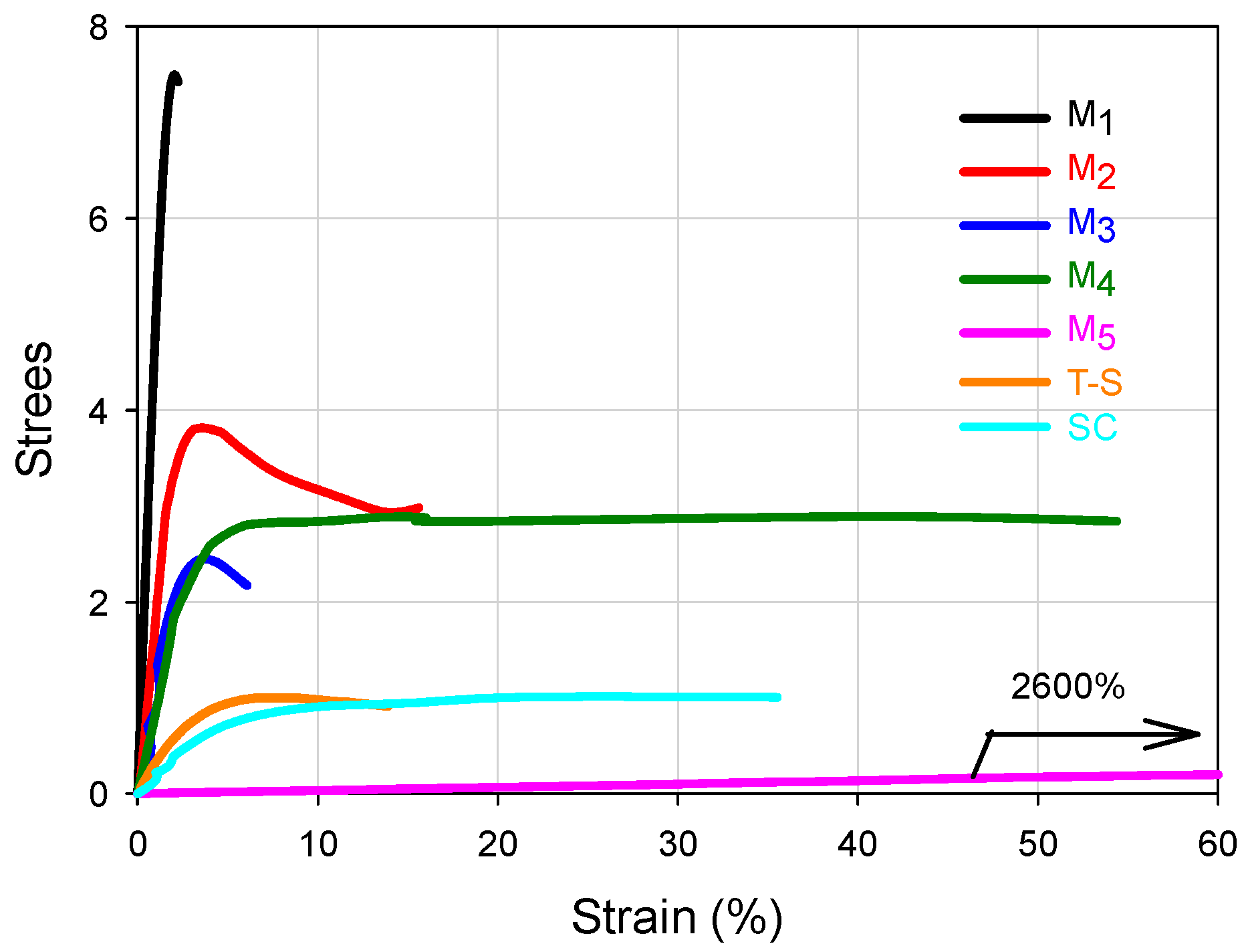
For the FCCs presented in
Figure 3, it can be seen that the set of stress-strain curves at 25 °C encompass an extensive range of mechanical performance, forming in fact a wide failure envelope (although it was obtained at the same temperature and cross-head speed) [
23], which only depends on the WCD of such materials, because they possess the same global composition. The Young’s modulus (
E) and deformation capacity ranks for the FCCs can be seen in
Table 1 and their correlations with the WCD can be established by observing
Table 2. In principle, the higher the modulus, the lower the deformation; that is, basically the FCCs occupy opposite position values in E and deformation ranks. However, looking at the curves of
Figure 3, it can be inferred that the design of the WCDs may be used to obtain a good combination of
E and deformation, searching for considerable rigidity and toughness.
In
Figure 3, M
1 and M
5 clearly show the highest and lowest modulus respectively whose values can be seen in
Table 1. In
Table 2, it can be noticed that they present significant differences in favor of M
1 for the wt % of copolymer chains that are rich in S (SI 70 or 80–100), and even since SI 60–100, because the wt % content of copolymer chains for that range are 46.1 for M
1 versus 18.8 for M
5. In contrast, for the composition ranges below 60 wt % S, the big differences are in favor of M
5 especially for the SI 40–50 region (53.3 wt % for M
5 versus 7.1 wt % of chains for M
1), or the accumulated for SI 0–60 (53.9 for M
1 versus 81.2 for M
5), denoting the high capacity of M
5 for deformation. Such extreme stress-strain behaviors may be used as a basis to look at the general trends for the FCCs.
Comparing the differences for the M
2–M
4 materials regarding Young modulus, in
Figure 3 the curves almost overlap at the origin and that proximity is reflected in
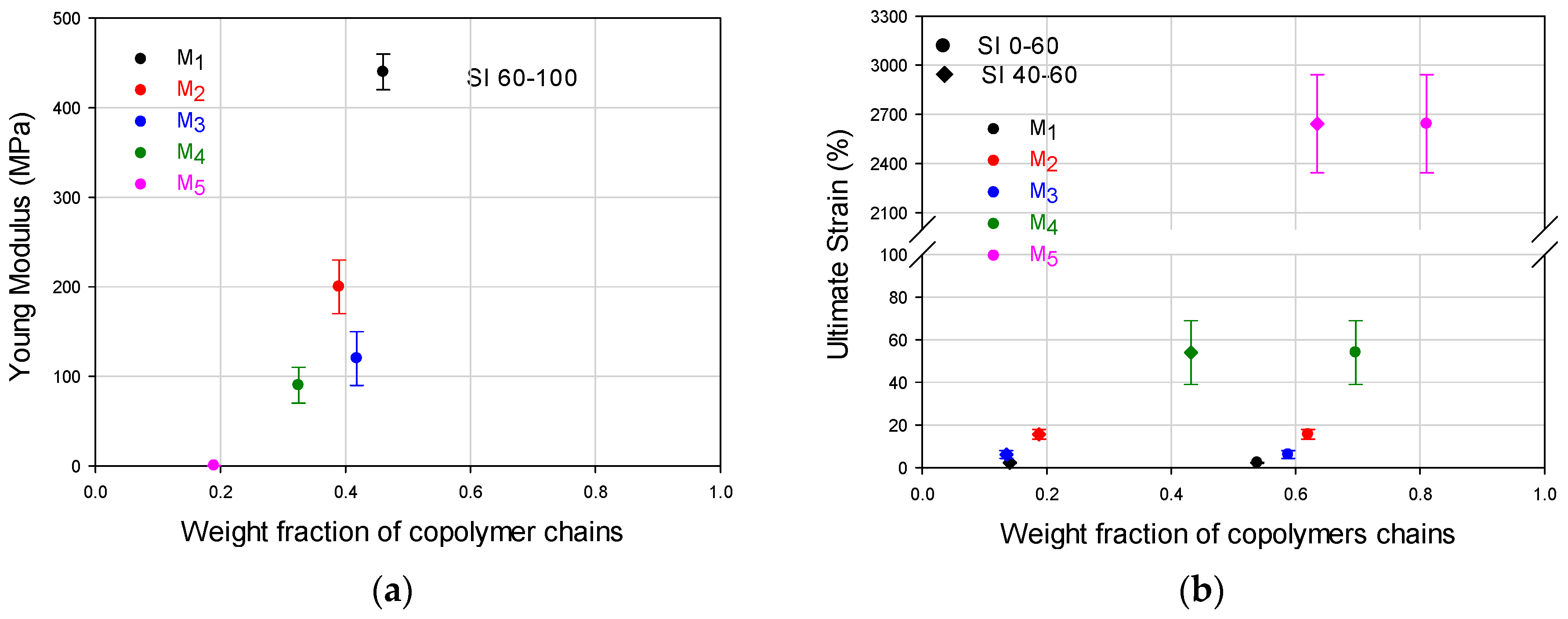
Table 1 considering the standard deviation of the experimental values. Considering the general mechanical behavior the contribution of the chains accumulated in SI 60–100 for the modulus and SI 0–60 for the deformation capacity, those mechanical properties for M
1–M
5 materials are plotted in
Figure 4a,b, to show their trends with weight fraction of copolymer chains. In addition, since M
4 and M
5 show high weight fraction values in SI 40–60, their deformation values are also plotted for that interval (
Figure 4b). In
Figure 4a, the positive slope shows how the modulus increases as the weight fraction increases in the SI 60–100 interval. It can also be seen that the modulus difference is not very significant for M
2, M
3 and M
4; however, looking at the ultimate strain in
Figure 4b and
Table 1, M
2 and M
3 show values close to each other, while M
4 is considerable bigger than both of them (M
4 shows 3.4 and 8.8 times the modulus value of M
2 and M
3 respectively). This behavior clearly shows the synergic performance of M
4, which in fact can also be perceived in
Figure 3 looking at M
2–M
4 curves, and comparing the toughness in
Table 1, where M
4 shows 3.1 and 12.8 times the value of M
2 and M
3 respectively. The positive slope for the deformation trend of the FCCs in
Figure 4b, that can be observed for both composition intervals (SI 0–60 and SI 40–60), denotes the importance of the presence of copolymer chains with intermediate compositions.
Looking at the reference materials in
Figure 3, the T-S polymer shows a low modulus compared to four FCCs and similar deformation capacity to M
2, while the SC shows almost three times its deformation capacity, although it has a lower modulus than the T-S material. Such performances validate the convenience to synthesize copolymers by the procedure proposed here and confirm that in the T-S polymeric materials, the properties combination of the components is not promoted due to phase separation of the homopolymers within the polymer bulk.
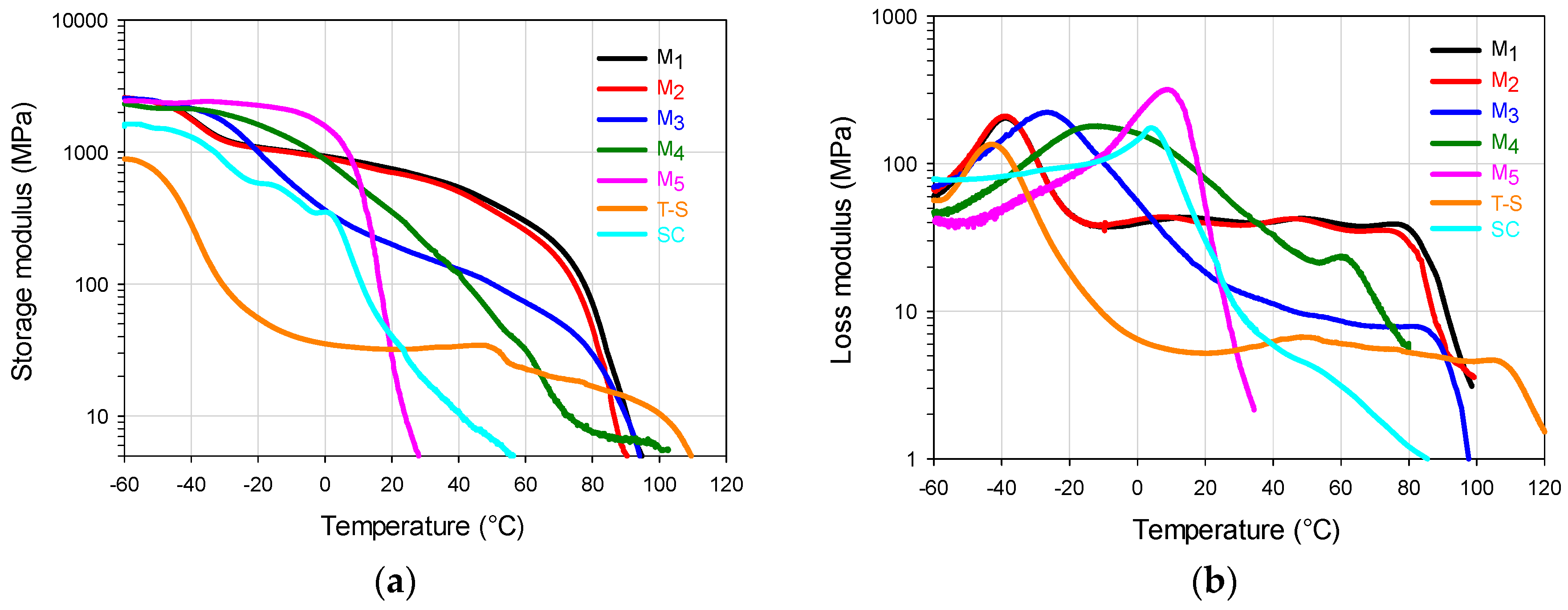
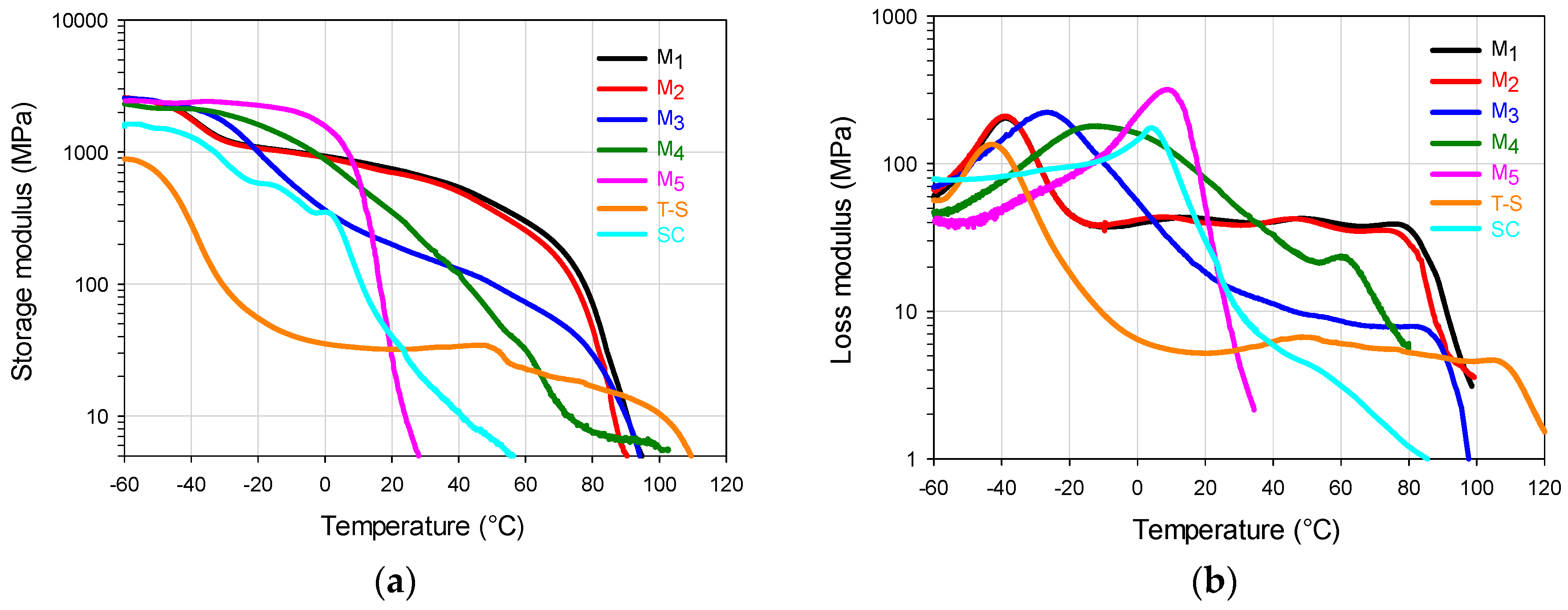
In
Figure 5a, for the storage modulus (
E’) of the two-component polymers it can be seen that the high value in SI 0–60 of the M
5 copolymer promotes the
E’ decay at low temperature, while the high values in SI 60–100 and SI 80–100 of M
1 impart the highest temperature resistance of the FCCs. The M
2 material follows closely the M
1 trajectory up to 40 °C with an
E’ decrease at slightly lower temperature due to its lower value for the SI 60–100, but mainly to its lower value in SI 80–100. The
E’ decay that M
1 and M
2 materials show at PBA
Tg essentially correspond to the high value in SI 0–20, that both show in
Figure 2a and
Table 2. For the SI 0–30, the M
3 copolymer that shows the highest value in
Table 2 presents a steeper
E’ decay that starts slightly above PBA
Tg, but its high value in SI 80–100 allows a thermal resistance before the catastrophic E’ final decay at 80 °C.
The M4 copolymer does not show a phase separation tendency in E’, starting a smooth decay at 0 °C and, around 40 °C it presents a crossover point with the M3 curve, denoting a slightly higher temperature resistance for the latter material, when both approach the terminal zone for E’. The SC shows very poor temperature resistance for the E’ value, while the T-S shows a marked decay at PBA Tg (more than one order of magnitude) followed by a plateau that starts at −20 °C that presents a final decay close to 100 °C. Its low E’ performance as in the static test can be attributed to the lack of interaction between components.
For loss modulus (
E”) in
Figure 5b, the M
5 FCC shows a copolymer type behavior with a peak at approximately 10 °C with an almost vertical decay afterwards, denoting an elastomeric behavior at room temperature. M
1 and M
2 copolymers follow very similar trajectories (as in
E’ behavior) in the whole test temperature range. The peak around −40 °C is assigned to a glass transition response, due to the chains accumulated in the SI 0–20 (
Figure 2a); then,
E” maintains a certain plateau value, until it falls at 80 °C, in accordance with
E’ decay (
Figure 5a). M
3 material, shows a wide peak at −25 °C, because there are a considerable amount of chains in the SI 0–20; then, it shows a smooth decay as it did in the
E’ curve (
Figure 5a). At around 40 °C, it starts a plateau like behavior due to the influence of the SI 60–100, until its final decay at approximately 80 °C.
The M4 material shows a very wide peak from −40 to 40 °C (the widest of all polymeric materials) indicating the presence of copolymer chains with a wide range of compositions around the highest peak value (approximately at −11 °C); that trajectory is highly influenced by the chains composition profile in the SI 0–70 range. The slow decrease of E” that starts at ca. 0 °C continuous until it presents a shoulder formation tendency at 60 °C, which is a signal influenced by the accumulation of chains in the SI 70–100 range (chains rich in S); the final modulus decay occurs at lower temperature than M1–M3 materials due to its lower SI 70–100 value. The high E” value from −55 °C to almost 40 °C, indicates a high energy dissipation capacity (area under the curve) within that temperature range. For the reference materials, the SC presents a copolymer type behavior with a lower but slightly wider peak than the one presented by M5 (accounting for the dissipation capacity since −60 °C), that deviates slightly at 20 °C towards its final decay, denoting the random structure with very low temperature resistance. The T-S material shows a peak at −45 °C showing a temperature peak value close to PBA Tg with a significant E” decay (due to is high PBA content) that is followed by a plateau from 0 to ca. 100 °C (showing there a small peak), denoting the glass transition temperature values that resemble the ones of the pure homopolymers.
In summary, the methodology proposed here to design the properties of a FCC by variations in the WCD of the chains and its correlation with the mechanical properties has been demonstrated, showing also an unusual failure envelope involving diverse mechanostatic performances of copolymers obtained using the same chemical system and global comonomer composition, tested at the same temperature and cross head speed. Additionally, as stated in this work, the correlation between WCD profiles and the mechanical properties above described using FRP reactions (establishing the structure characteristics required for synergic behavior) has an equivalent with controlled radical polymerization (CRP). Recently, for the S/BA system using also a semicontinuous emulsion process with a multistage feeding technique [
18,
19,
20]), Guo et al. tested two types of gradient copolymers obtained by CRP that exhibited yielding and elastic deformation [
24], whose copolymer chains composition varied from one component to the other, according to their former report [
25]. Although they did not present a discussion about their behavior related to the specific structures of those gradient copolymers, the explanation for that behavior is in accordance with the synergic contribution of the components proven here (as it was also demonstrated in chemical blends); the presence of long segments of each one of the components along with transition chain segments of intermediate compositions of monomers A and B in between each molecule, should lead to the optimization of polymer properties. The conformed polymer bulk would be similar to the copolymers above described using FRC. In another report, similar results on damping performance were found for materials obtained by FRC [
20] and CRP [
21] for the S/BA system. The equivalence in properties has to do with the richness in S and BA units. For the FRC case, such richness occurs in different chains, and for the CRP case it occurs within one molecule. Such component richness in high MW chains is important for mechanical properties in both cases, because the contribution of each component is enhanced in that way; it is well known that MW is an important parameter for mechanical properties [
26,
27]. Additionally, the polydispersity index should also be considered if looking for uniform properties in the product [
28], or for easy processing, particularly for high MW polymers [
29]. To improve the synthesis processes, the use of programmed feeding can help to control comonomer addition in FRC [
30] or CRP [
31,
32].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}