1. Introduction
Polyesters are currently among the most competitive biodegradable polymers commercialized as both commodity and speciality products. Applications range from materials for solving “white pollution concerns” caused by non-degradable polymers to materials with good properties to fulfil, for example, the highly restrictive requirements of the biomedical field [
1,
2,
3]. The most common polyesters are prepared by ring-opening polymerization of lactones (e.g., polyglycolide, polylactide, and poly(ɛ-caprolactone)) [
4,
5], but the production of poly(alkylene dicarboxylate)s has attracted a great deal of attention as well, especially for commodity applications [
6].
Poly(butylene succinate) (PBS) is considered as one of the most important biodegradable poly(alkylene dicarboxylate)s commercialized up to now, because of its considerable combination of desired properties (e.g., high biodegradation rate, elastic modulus, and melting point) [
7,
8]. Such features generally worsen when another diol or aliphatic dicarboxylic acid is employed, making it very interesting to incorporate rigid units such as aromatic dicarboxylates to avoid any performance drop. Polyesters derived from aromatic dicarboxylic acids are much less electrophilic and, thus, far less sensitive to hydrolysis than aliphatic polyesters. However, aromatic polyesters may possess higher melting temperature (
Tm’s) and better mechanical properties. Therefore, it is an attractive challenge to incorporate aliphatic units randomly and determine if it is possible to optimize the properties in the direction of a satisfactory rate of biodegradation while retaining good mechanical properties. In the range of 30 mol % to 60 mol % of terephthalic acid, which is of particular interest since such materials provide useful material properties; the degradation rate drops linearly with the content of the aromatic acid [
9,
10]. Furthermore, these units may reduce the final cost of the polymer. Therefore, BASF and Novamont have commercialized aliphatic/aromatic polyesters derived from 1,4-butanediol and either adipic and terephthalic acids (i.e., Ecoflex™ and Origo-Bi™, respectively).
Biodegradation of these polymers by microorganisms or in a compost site is well proved, with the degradation process being promoted by a lipase-like hydrolase enzyme [
11,
12,
13,
14].
The biodegradation rate of aliphatic/aromatic polyesters depends on different interplaying factors such as: (a) chain microstructure (e.g., random or blocky disposition); (b) aromatic content; and (c) the degree of crystallinity [
15,
16,
17,
18,
19,
20,
21]. For example, note that a comonomer content close to 50% and a random microstructure is expected to result in a decrease in the degree of crystallinity and logically an increase in the degradation rate.
The present work has been focused on aliphatic/aromatic polyesters derived from 1,4-butanediol, different aliphatic dicarboxylic acids (i.e., succinic, adipic, and sebacic acids) and a high content of terephthalic acid (i.e., a nominal value of 70 molar % with respect to the total dicarboxylic acid content was selected in order to explore the possibility of maintaining the biodegradable characteristics at such a high content). In this case, one should expect a low degradation rate as a result of such high aromatic content and the relatively high degree of crystallinity. Incorporation of hydrophilic hydroxyapatite (HAp) nanoparticles may increase both biodegradability and biocompatibility, and can be considered as an interesting option that will be evaluated for the proposed copolymers. To this end, copolymers with a 2.5 wt % of HAp will be synthesized as well.
HAp (Ca
10(PO
4)
6(OH)
2) is a bioceramic that forms part of the majority of the inorganic components of hard and connective tissues such as bones, teeth, and tendons. Nowadays, development of Hap-based nanocomposites and biodegradable polymers is attracting increasing interest since the presence of hydroxyl groups in the added nanoparticles may enhance polymer degradation. Furthermore, HAp particles can provide osteoconductivity and osteogravitivity as well as improved mechanical properties [
22].
Hybrid HAp/biodegradable polymer composites can be prepared by different techniques, such as hot press moulding, plasma spraying, solution compounding, and also in situ polymerization, which is the most frequently employed method [
23]. Distribution of nano-HAp particles in the organic matrix and final mechanical properties of the composite mainly depend on the interface compatibility between organic and inorganic phases. A good adhesion is expected when the hybrid is prepared by in situ polymerization, taking the capability of hydroxyl groups of the HAp surface to bind carboxyl groups of the growing poly(alkylene dicarboxylate) into account. Nevertheless, some limitations may also be expected as a consequence of the low concentration and reactivity of hydroxyl groups on the nanoparticle surface, which could lead to a reduced number of grafted molecules.
2. Experimental Section
Materials: Terephthalic acid (TA) was supplied by Shahid Toundgoyan Petrochemical Complex (Mahshar, Iran). Nanohydroxiapatite (n-HAp) was purchased from Beijing DK Nano Technology Co., Ltd., Beijing, China. 1,4-butanediol (BDO), sebacic acid (SeA), adipic acid (AdA) and succinic acid (SuA) were bought from Daejung Chemical & Metal Co., Ltd., Shiheung, Korea. Titanium butoxide (TBT) as polycondensation catalyst was purchased from Merck Co. (Darmstadt, Germany).
African green monkey kidney fibroblast-like (COS-7) and epithelial-like Madin–Darby Canine Kidney (MDCK) cells (ATCC, Manassas, VA, USA) were used in this work. Fetal bovine serum (FBS) was purchased from Gibco (Thermo Fisher Scientific Inc., Alcobendas, Spain). Lipases from pancreas porcine and Pseudomonas cepacia were purchased from Sigma Aldrich (Barcelona, Spain).
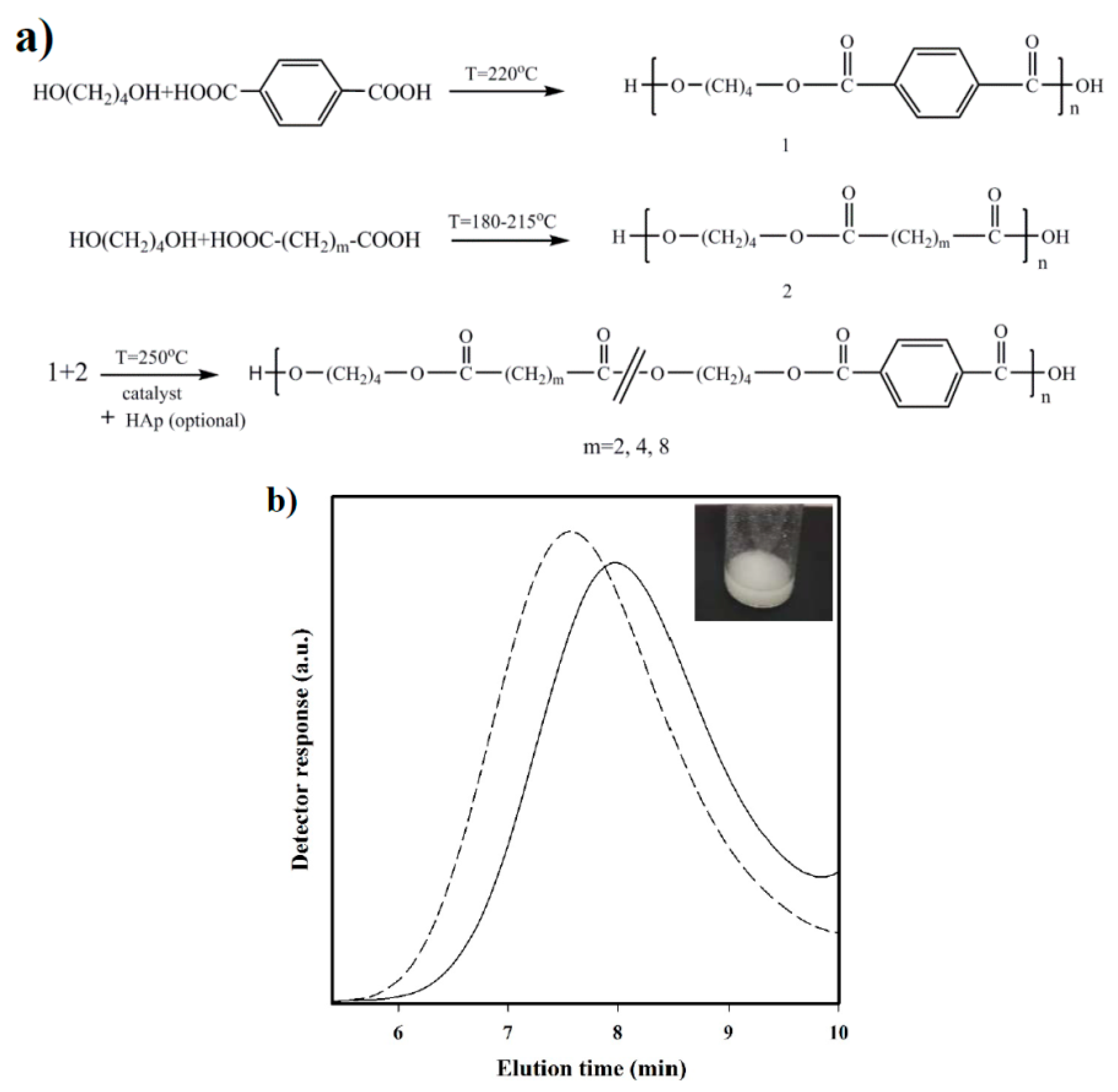
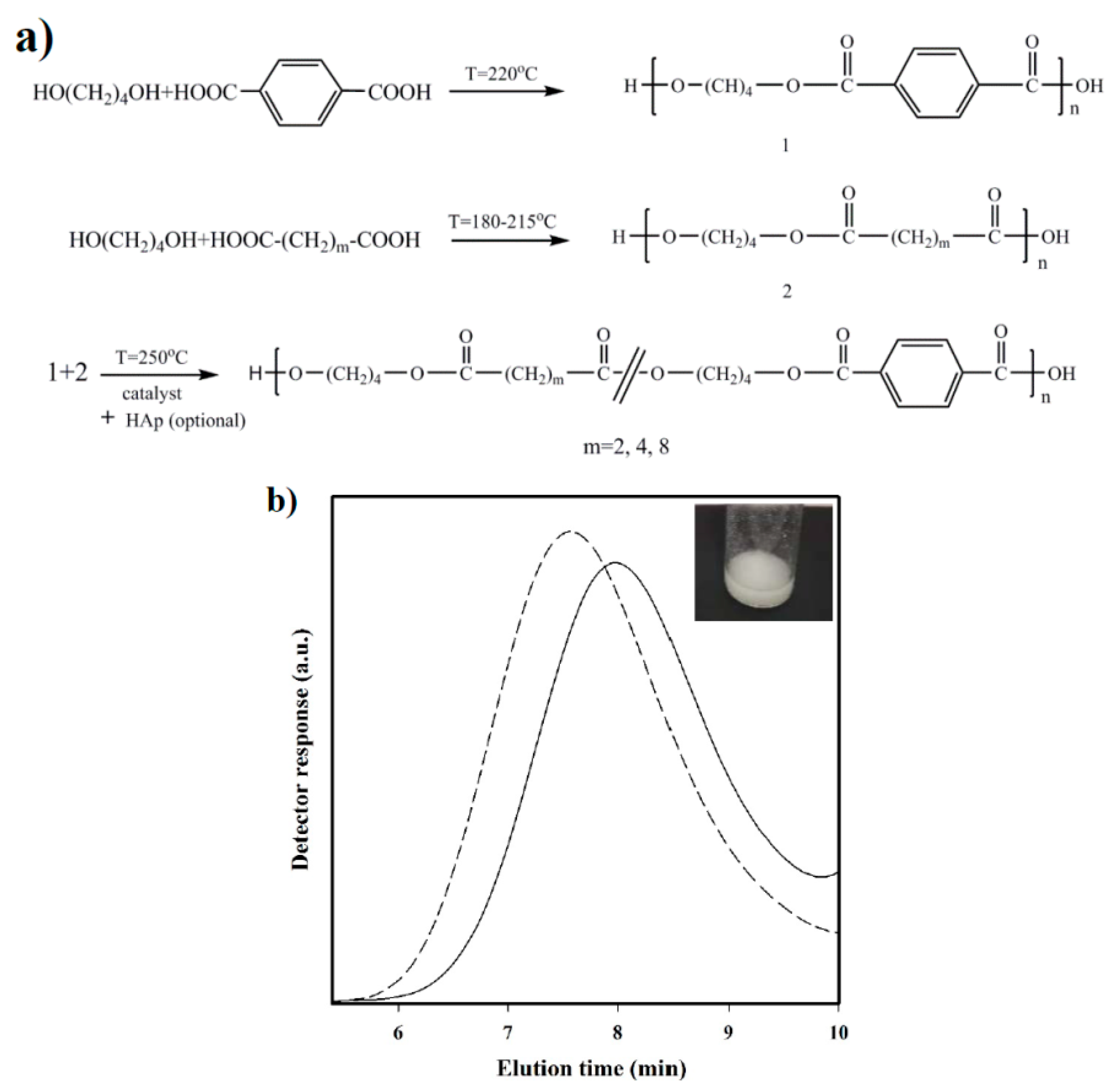
Polymerization: Synthesis of the three selected copolymers named as polybutylene succinate-
co-terephthalate (PBST), poly(butylene adipate-
co-terephthalate) (PBAdT) and poly(butylene sebacate-
co-terephthalate) (PBSeT) was performed in a home-made 1-L high-pressure reactor with effective stirring, following a two-step melt polycondensation procedure.
- (a)
Synthesis of prepolymers from 1,4-butanediol and the selected dicarboxylic unit. Polycondensation reactions were carried out using an excess of 1,4-butanediol (BDO) (i.e., 1.7:1 [OH]:[COOH]). After loading the reactor with 650 g of a mixture of BDO and the appropriate dicarboxylic acid, the reaction mixture was firstly stirred for 30 min at 140 °C under a pressure of 3–3.5 bars. A flow of N2 was provided to keep the required pressure while an electric condenser allowed separating the condensed water and the excess of alcohol. Reaction temperature was then increased to 215, 207, 200, and 220 °C, for sebacic, adipic, succinic, and terephthalic acid derivatives, respectively. The reaction was stopped when no more water could be recovered (approximately after 180 min). In fact, the reaction extent could be evaluated by weighing the water that was recovered at regular time intervals.
- (b)
Vacuum polycondensation and thermal transesterification between aliphatic and aromatic prepolymers. The appropriate mixtures constituted by a 0.3:0.7 molar ratio of prepolymers derived from the aliphatic and aromatic acids and TBT as catalyst (1.4 mmol for 1 mol of dicarboxylic acid) were transferred to the reactor for 10 min at 200 °C and atmospheric pressure. Temperature was subsequently increased up to 250 °C while a vacuum (of 20 mbar) was applied. Once the mixer torque reached the desired value, the reaction was stopped (approximately after 150 min). Copolymers were dissolved in 1,1,1,3,3,3-hexafluroisopropanol (HFIP), precipitated in water, washed several times with water, methanol, and ether, and finally dried in a vacuum desiccator.
The chosen procedure differs from the conventional syntheses based on thermal polycondensation processes from an appropriate ratio of comonomers. The advantage of the proposed synthesis is that copolymers with different composition (i.e., having different comonomer ratios) can be easily obtained if desired from the corresponding homopolymers. Nevertheless, the time and reaction temperature of the second step appear highly important to achieve a random microstructure that should be checked.
Nanocomposites were synthesized based on the same protocol, except for the addition of a 2.5 wt % n-HAp at the beginning of copolymerization step. Nanocomposites will be referred to as PBST-HAp, PBAdT-Hap, and PBSeT-HAp depending on the aliphatic dicarboxylic unit.
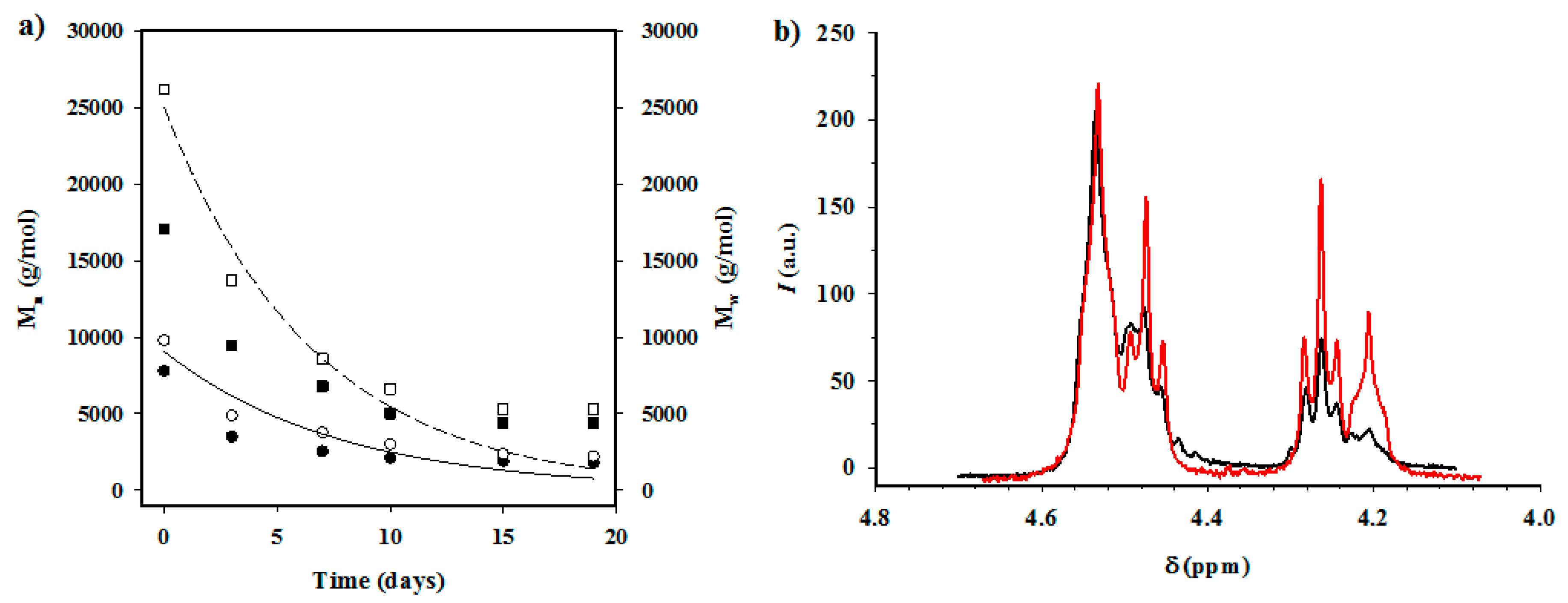
Measurements: Molecular weights were estimated by size exclusion chromatography (GPC) using a liquid chromatograph (Shimadzu, model LC-8A, Tokyo, Japan) equipped with an Empower computer program (Waters, Milford, MA USA). A PL HFIP gel column (Polymer Lab, Böblingen, Germany) and a refractive index detector (Shimadzu RID-10A, Tokyo, Japan) were employed. The polymer was dissolved and eluted in HFIP containing CF3COONa (0.05 M) at a flow rate of 1 mL/min (injected volume 100 μL, sample concentration 2.0 mg/mL). The number and weight average molecular weights were calculated using polymethyl methacrylate standards.
Carboxylic acid end-group content was calculated, 1 g of polymer was dissolved in 20 mL chloroform and titrated using 0.05 N KOH in methanol with red phenol as the indicator. The carboxylic acid content [COOH] in meq/kg was measured using the following equation:
where
v and
v0 are the volume of the titrant (mL) for titration of the chloroform solution with and without the analyte, respectively,
m is the titrant concentration (N), and
w is the weight (g) of the polymer sample.
Infrared absorption spectra were recorded in the 4000–600 cm−1 range with a Fourier Transform FTIR 4100 Jasco spectrometer (Jasco International Co. Ltd., Tokyo, Japan) dotted with a Specac model MKII Golden Gate attenuated total reflection (ATR) cell.
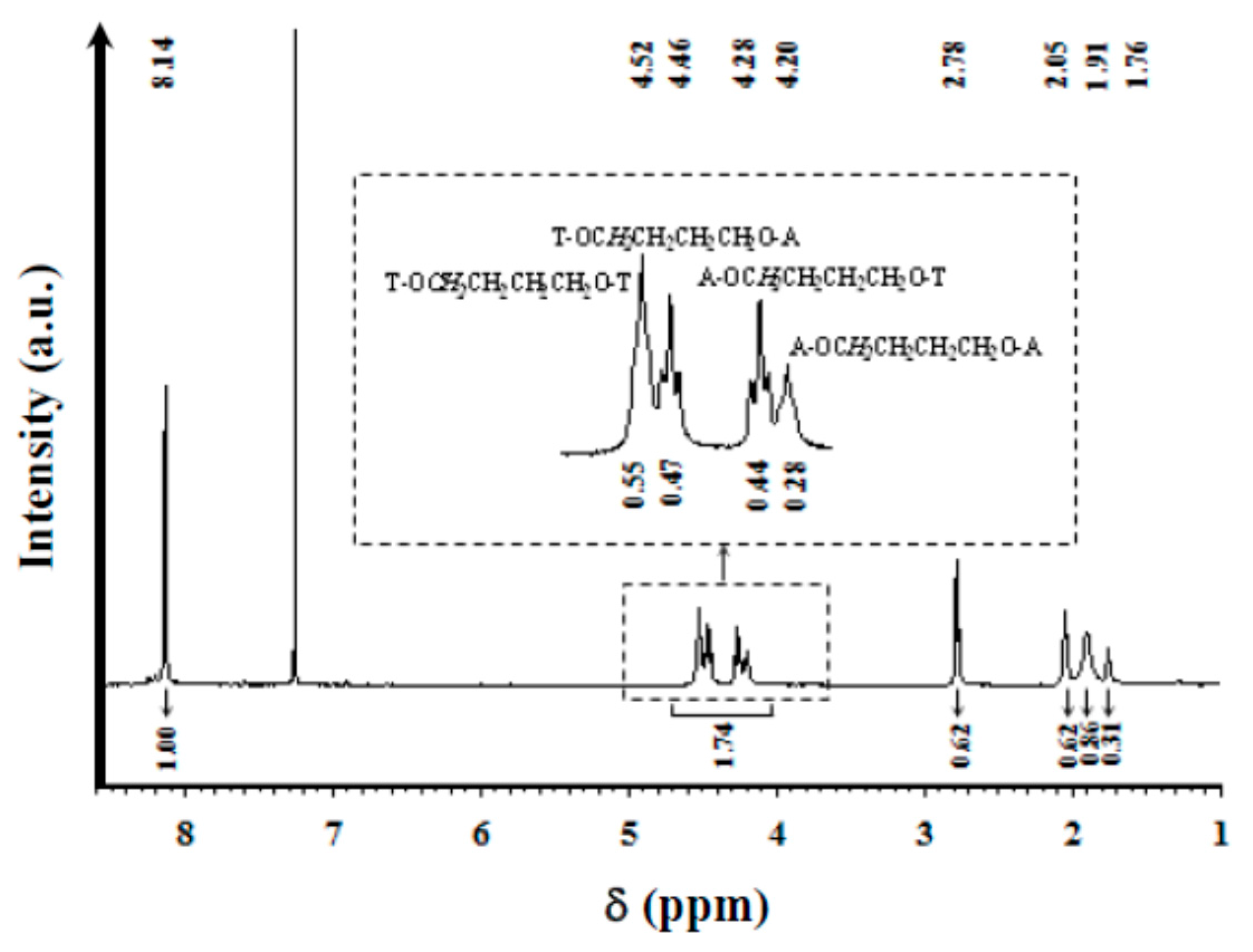
1H NMR spectra were acquired with a Bruker AMX-300 spectrometer (Bruker Co., Bremen, Germany) operating at 300.1 MHz. Chemical shifts were calibrated using tetramethylsilane as an internal standard. A mixture (1:1 v/v) of deuterated chloroform and trifluoroacetic acid (TFA) was used as the solvent.
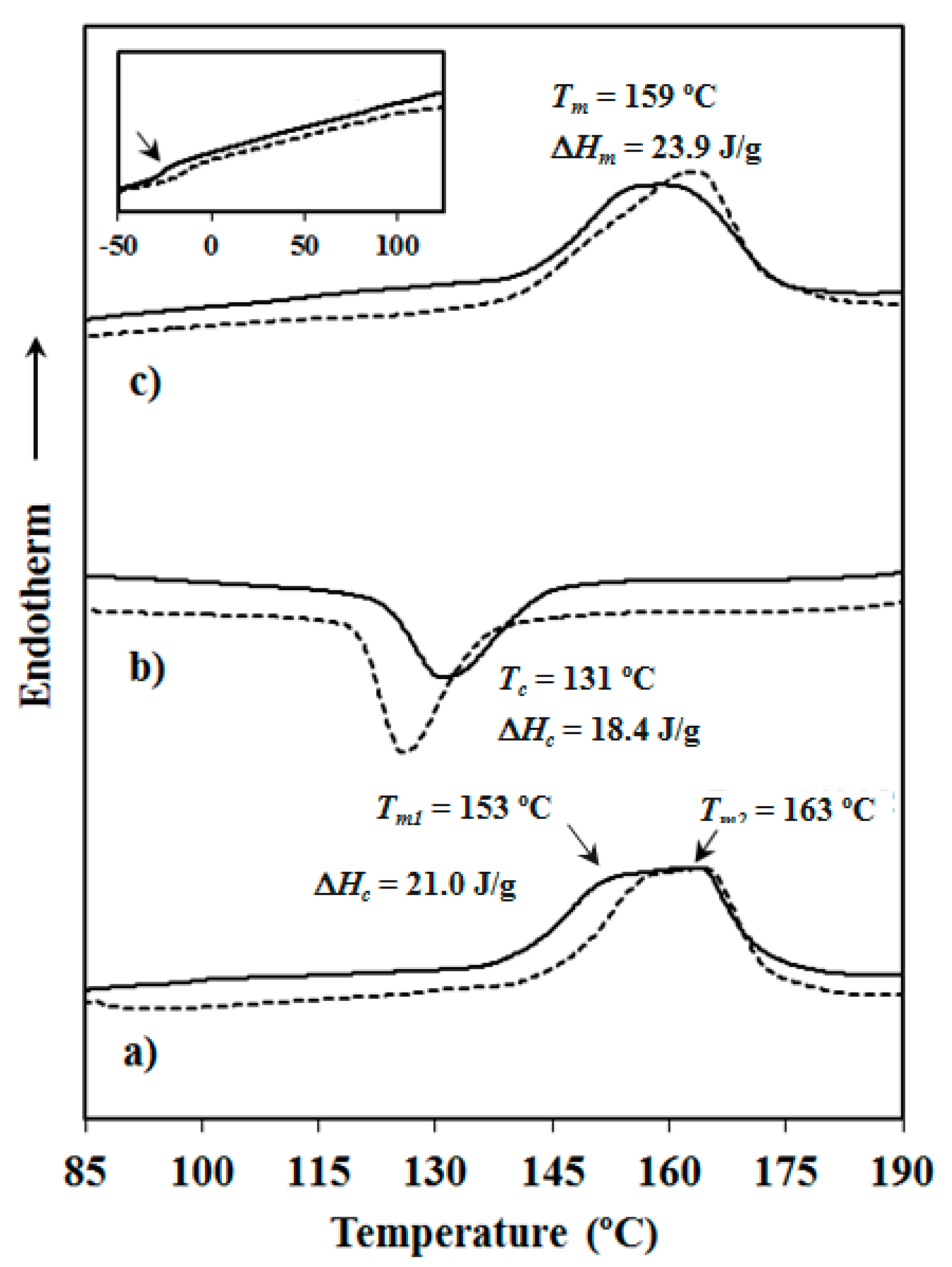
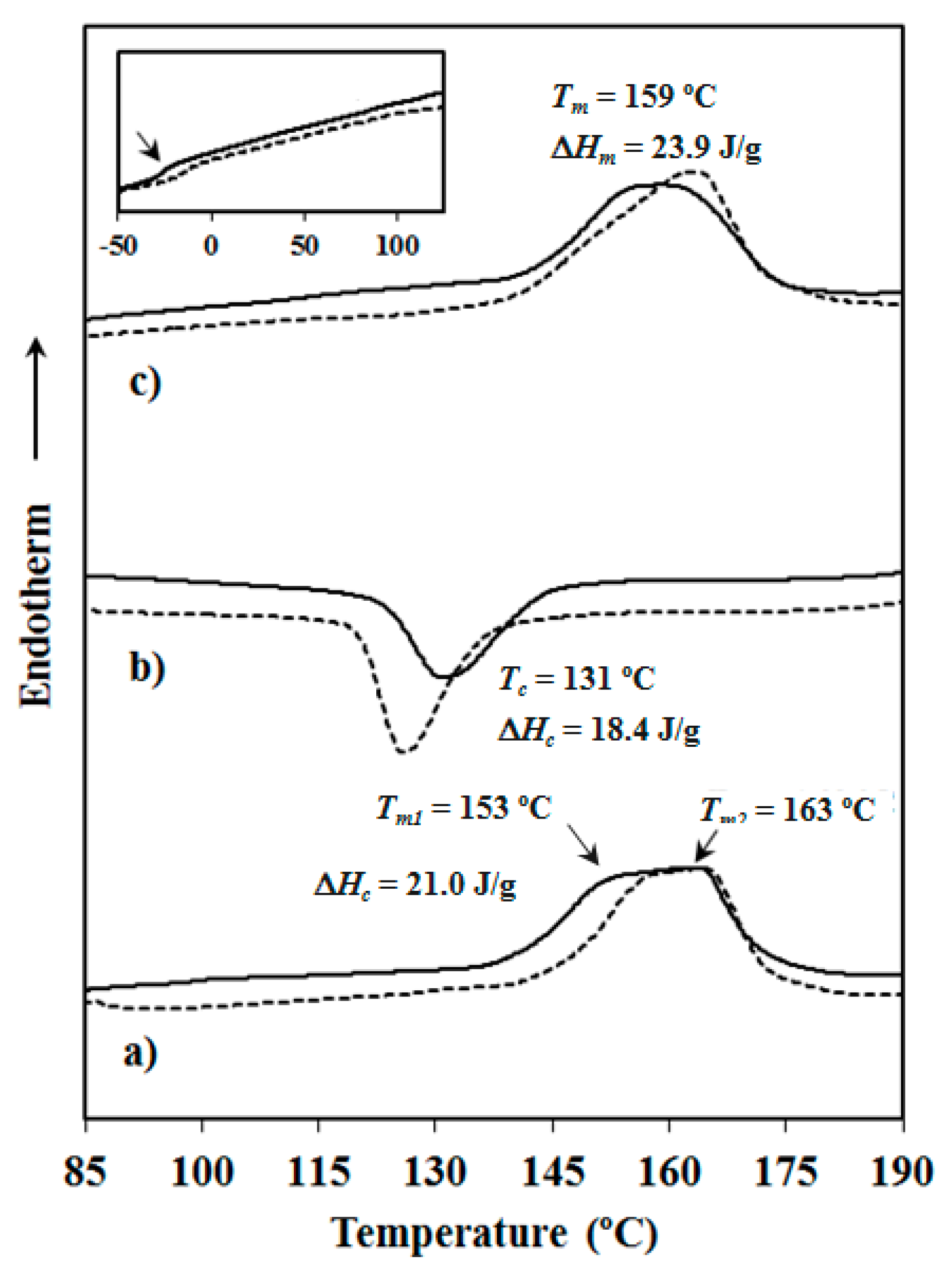
Calorimetric data were obtained by differential scanning calorimetry with a TA Instruments Q100 series (TA Instruments, New Castle, DE, USA) with Tzero technology equipped with a refrigerated cooling system (RCS). Experiments were conducted under a flow of dry nitrogen with a sample weight of approximately 5 mg and calibration was performed with indium. Tzero calibration required two experiments: the first experiment was performed without any sample while in the second case sapphire disks were used. A first heating run (20 °C/min) was performed to determine melting temperature and enthalpy of the as-synthesized sample. To erase the thermal history the sample was then kept in the melt state for three minutes and subsequently cooled to room temperature (10 °C/min) to obtain crystallization data. Finally, a second heating run (20 °C/min) was carried out to characterize the melt crystallized sample.
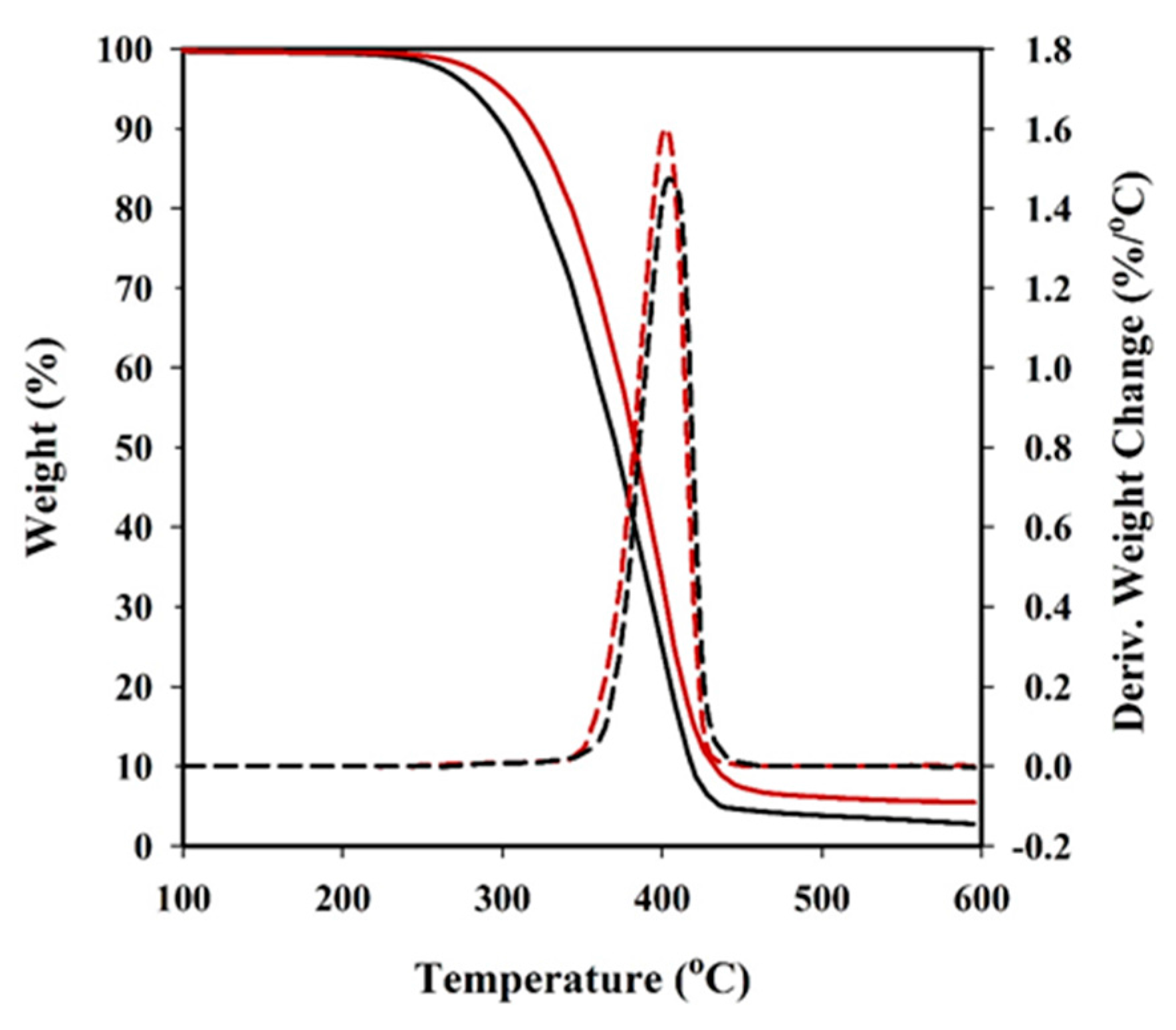
Thermal degradation was studied at a heating rate of 10 °C/min with around 5 mg samples in a Q50 thermogravimetric analyser (TGA) of TA Instruments, under a flow of dry nitrogen. Test temperatures ranged from 50 to 600 °C.
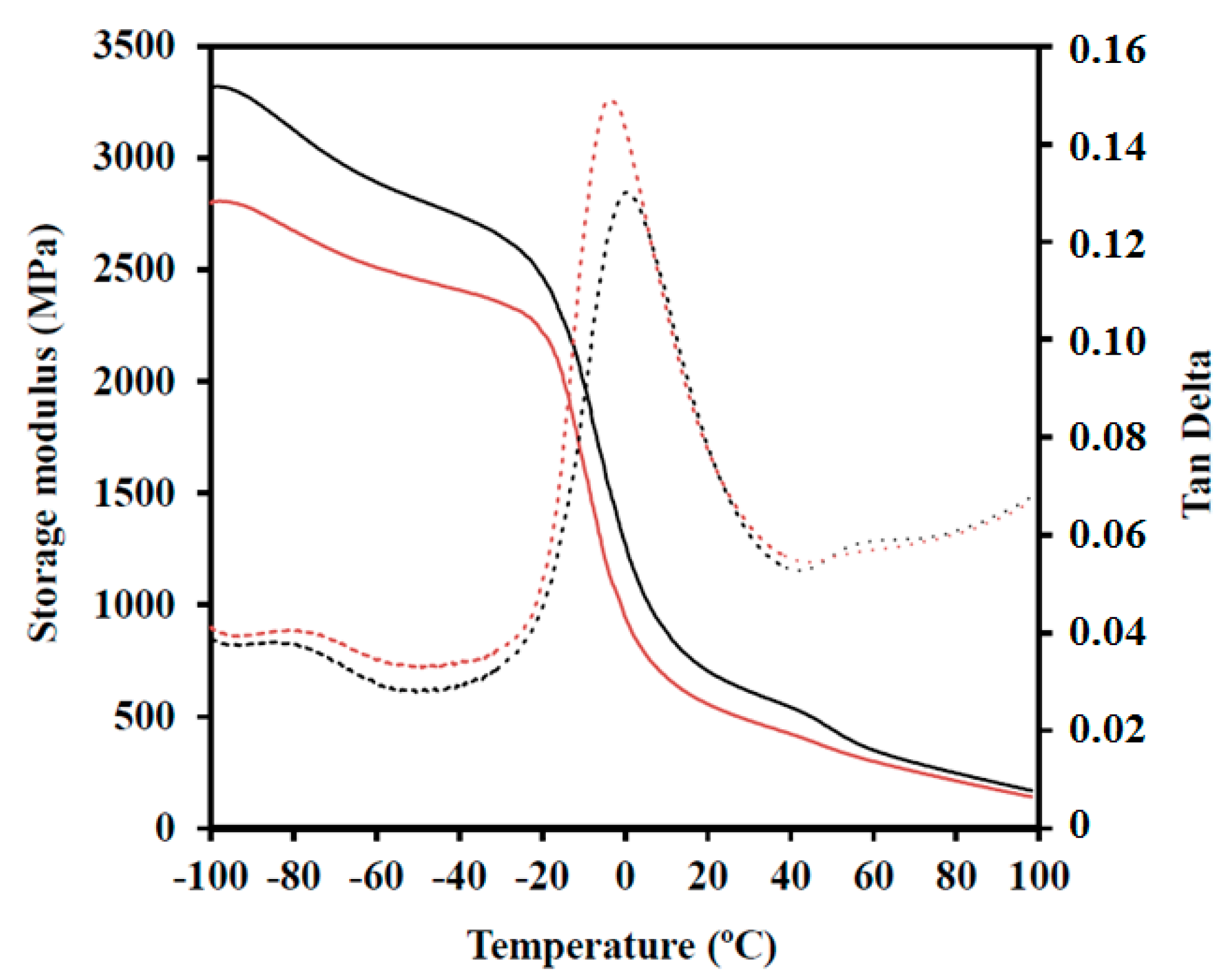
A TA Instruments DMA Q800 (TA Instruments, New Castle, DE, USA) was used to study the dynamic mechanical properties of materials. Prismatic rectangular samples (ca. 10 × 12 × 1.3 mm3) were analysed in single-cantilever mode at 1 Hz and 10 μm strain amplitude at 3 °C/min from −100 to 100 °C.
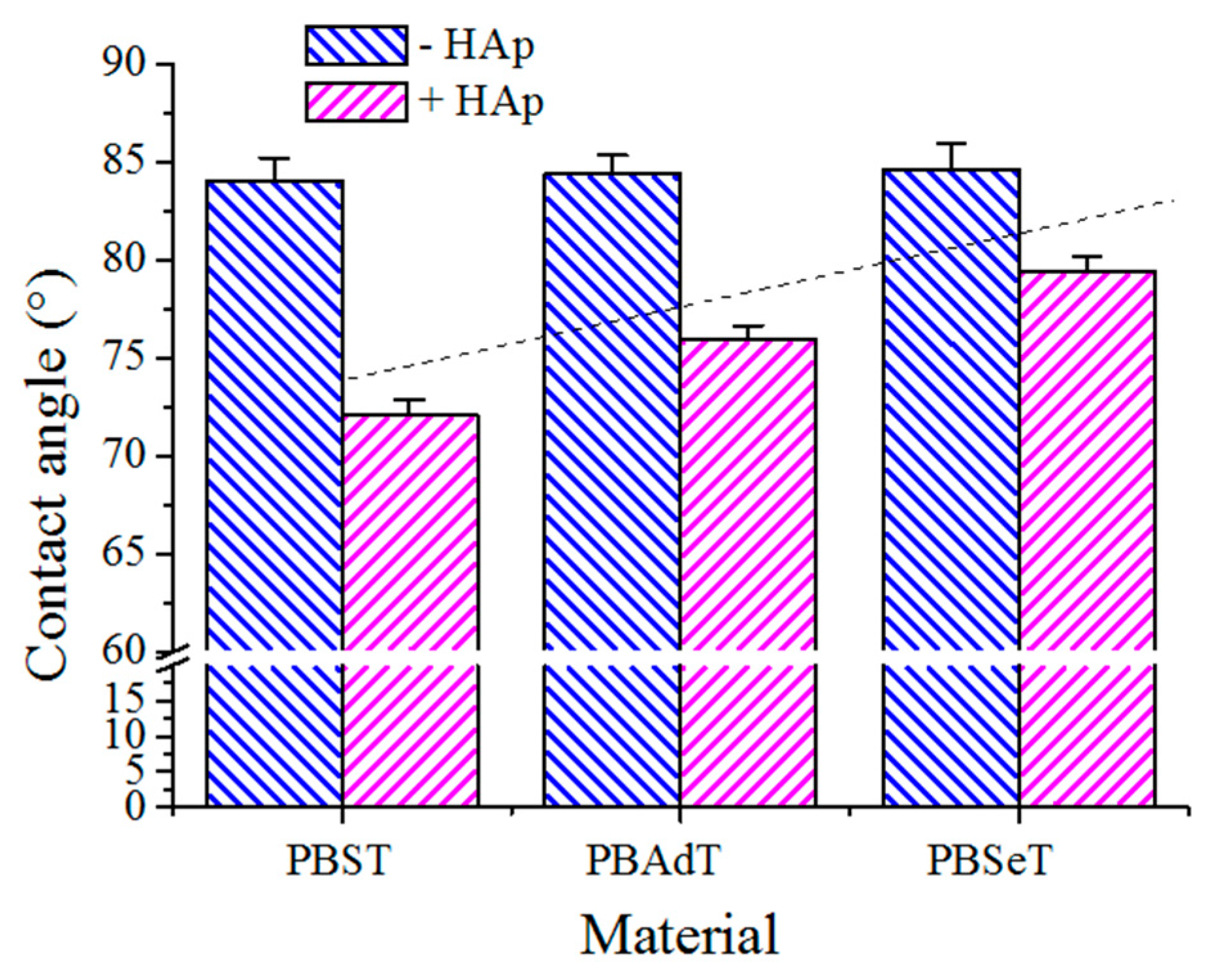
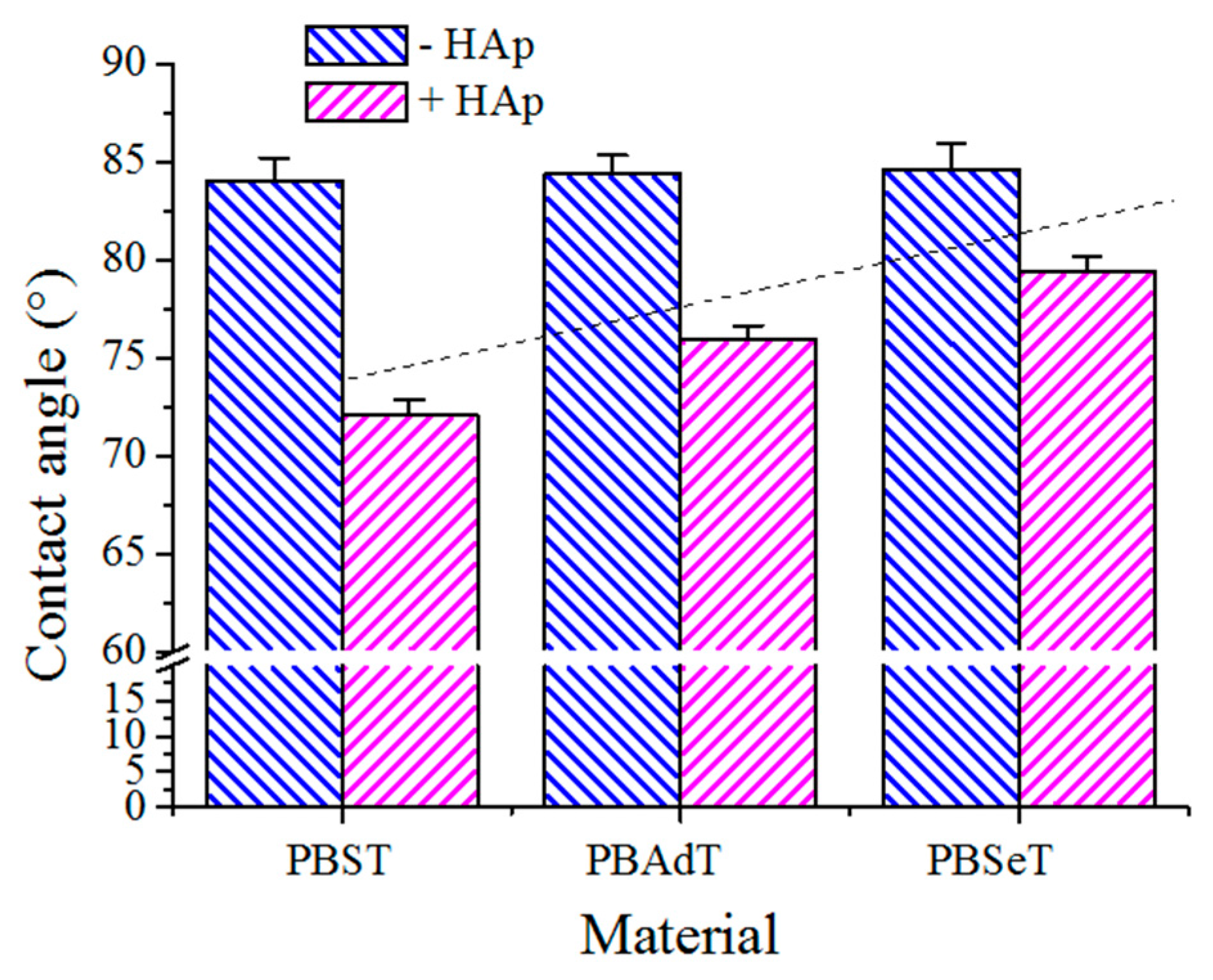
Contact angles (CA) were measured at room temperature with sessile drops using an OCA-15 plus Contact Angle Microscope (DataPhysics Instruments GmbH, Filderstadcity, Germany) and SCA20 software. Contact angle values of the right and left sides of distilled water drops were measured and averaged. Measurements were performed 10 s after the drop (0.5 µL) was deposited on the sample surface. All CA data were obtained by averaging between six measurements on different surface locations.
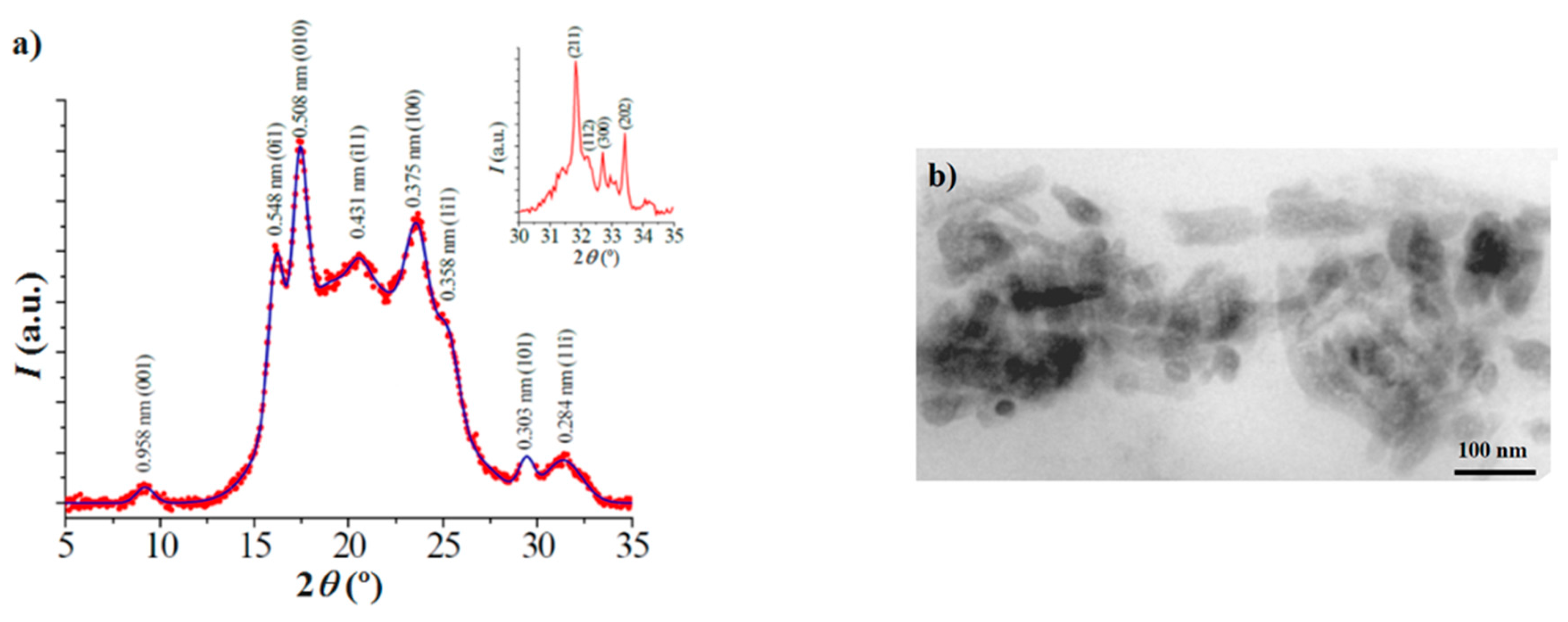
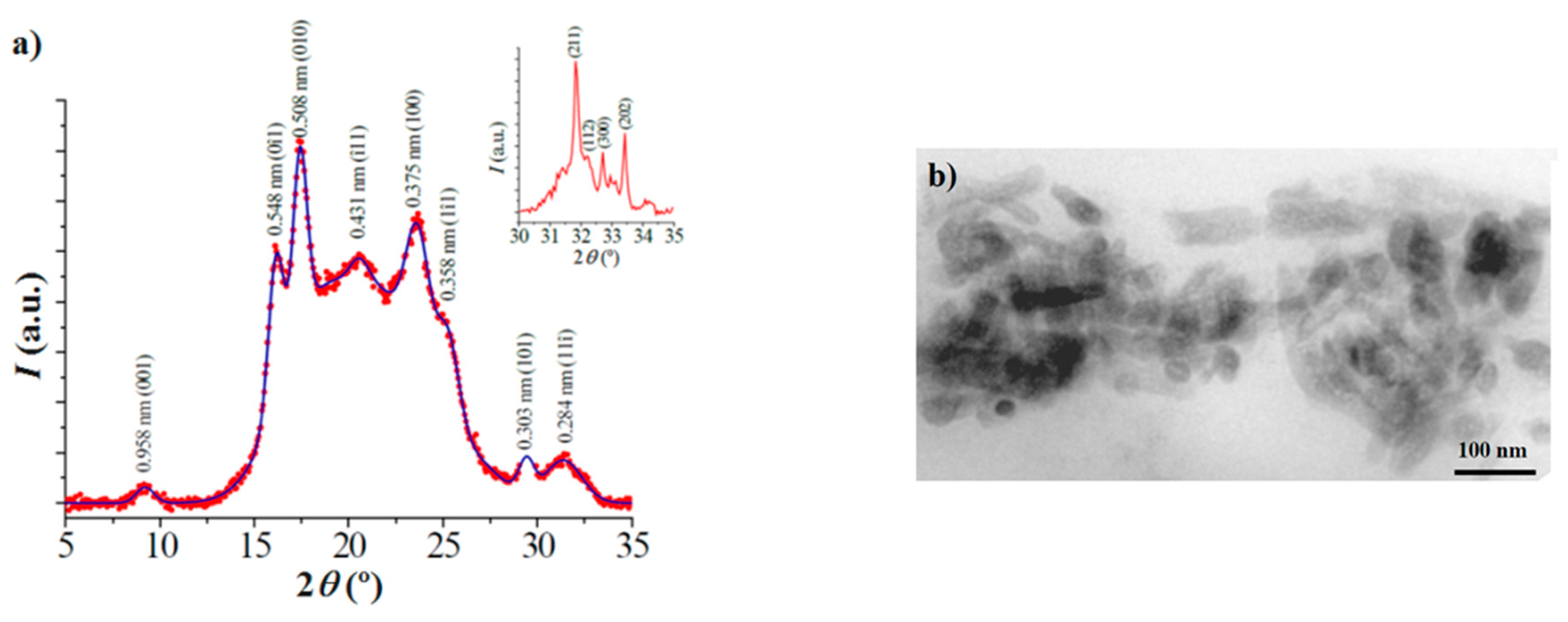
Distribution of HAp nanoparticles in the composites was evaluated by means of a Philips TECNAI 10 electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) at an accelerating voltage of 80 kV. Samples were prepared by embedding the nanocomposite specimens. A low viscosity modified Spurr epoxy resin was employed to embed the specimens before curing and cutting in small sections. In this case, a Sorvall Porter-Blum microtome (Norwalk, CT, USA) equipped with a diamond knife was utilized. The thin sections were collected in a trough filled with water and lifted onto carbon coated copper grids.
X-ray diffraction patterns were acquired using a Bruker D8 Advance model (Bruker, Karlsruhe, Germany) with Cu Kα radiation (λ = 0.1542 nm) and the geometry of Bragg–Brentano, theta–2theta. A one-dimensional Lynx Eye detector (Bruker, Karlsruhe, Germany) was employed.
Degradation studies: Thermally moulded films were used for biocompatibility and degradation studies. Briefly, polymers were heated up to 25 °C above their melting point for 2 min using a hydraulic press equipped with heating plates and a temperature controller (Specac, Orpington, UK). Pressure was progressively increased to 3–4 bars. Polymer films with a thickness of about 150 µm were recovered after cooling the mould to room temperature and subsequently cut to the desired size.
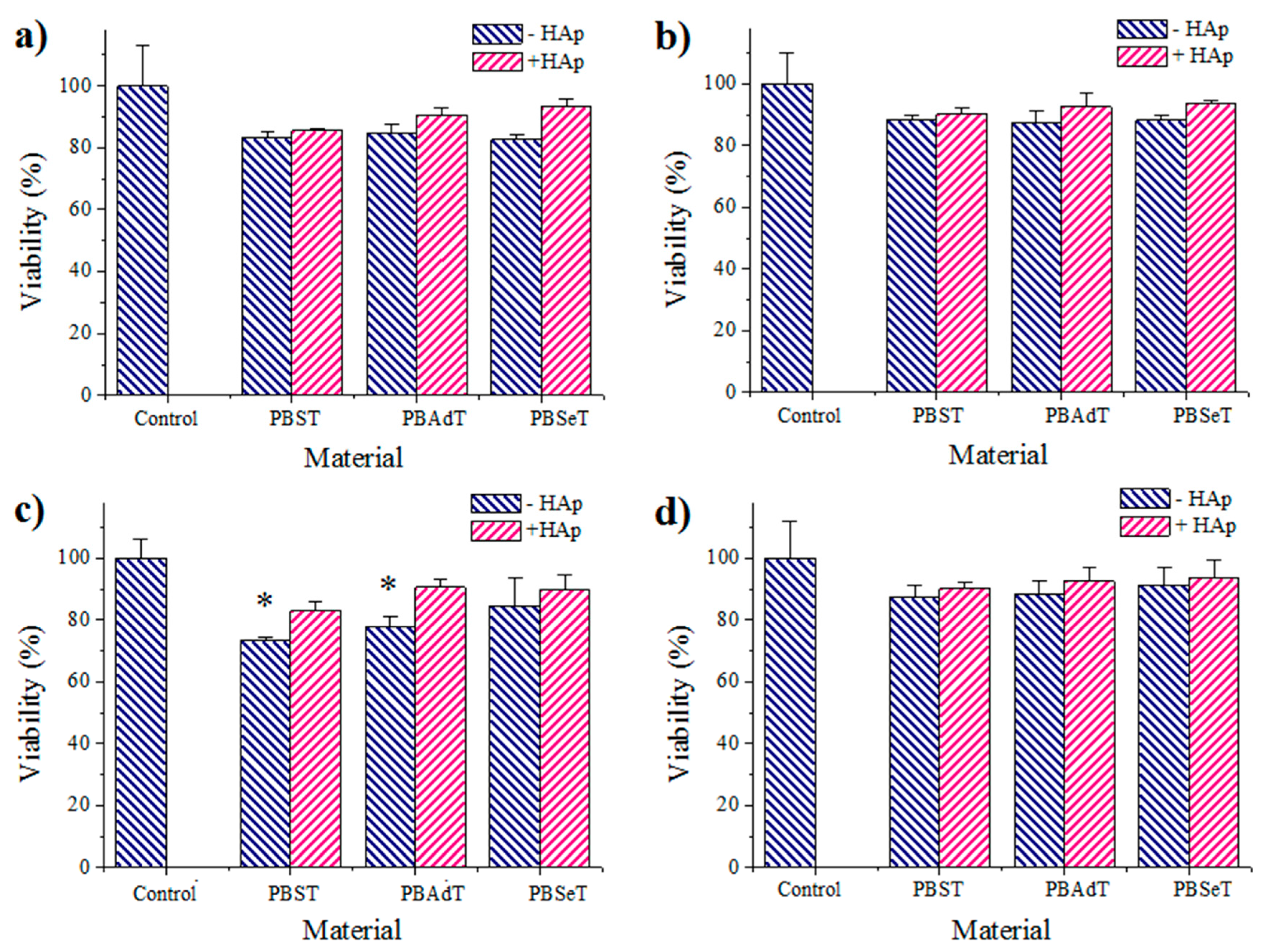
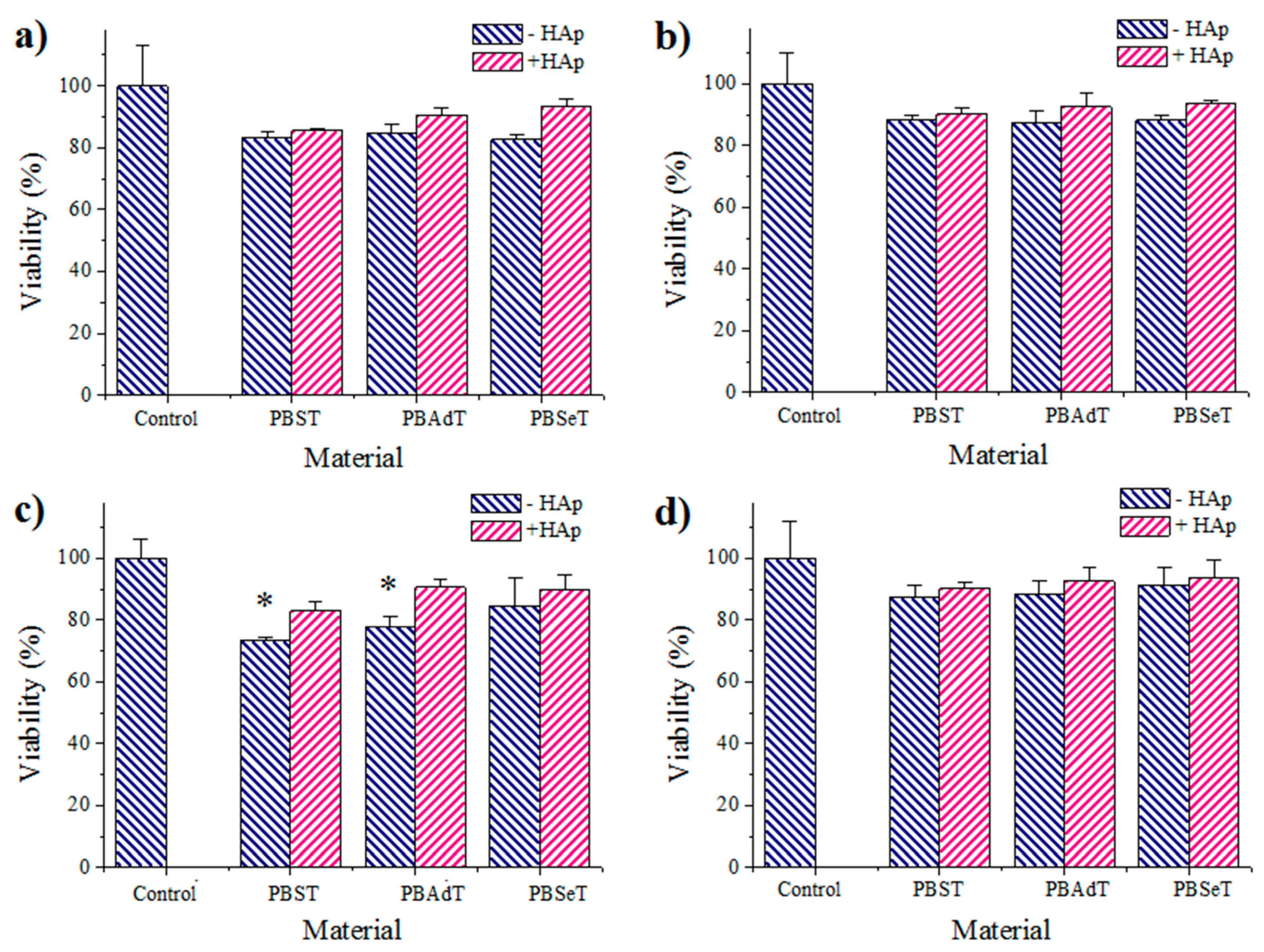
Cell adhesion and proliferation assays: COS-7 and MDCK cells were cultured in Dulbecco’s modified Eagle medium (DMEM) as previously reported [
24]. Square pieces (10 × 10 × 0.15 mm
3) of press molten films were placed and fixed in each well of a multi-well culture plate with a small drop of silicone (Silbione
® MED ADH 4300 RTV, Bluestar Silicones France SAS, Lyon, France). They were then sterilized by UV radiation in a laminar flux cabinet for 15 min. Thereafter, the samples were stabilized for 24 h in 1 mL of medium under culture conditions. For cell adhesion and proliferation assays, aliquots of 50–100 μL containing 5 × 10
4 and 2 × 10
4 cells, respectively, were seeded onto the films in each well containing 1 mL of medium, and incubated for 24 h (adhesion assay) or seven days (proliferation assay). Samples were evaluated by the standard adhesion and proliferation method [
24]. The used procedure is based on a simple modification of the ISO10993-5:2009 standard test that describes the appropriate methodology to assess the in vitro cytotoxicity of medical devices. Finally, the cellular viability on materials was evaluated through the MTT assay [
24]. The study was carried out using five replicates and the results were averaged. Samples with adhered and grown cells on the films were fixed with 2.5%
w/
v formaldehyde at 4 °C overnight. They were subsequently dehydrated and processed for observation via scanning electronic microscopy.
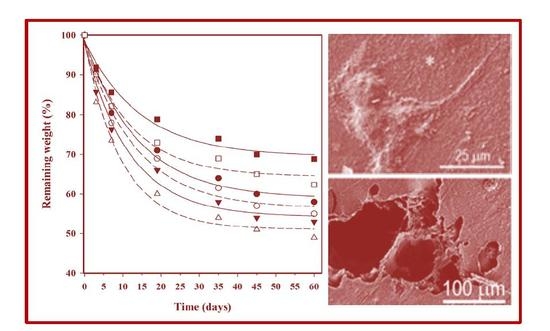
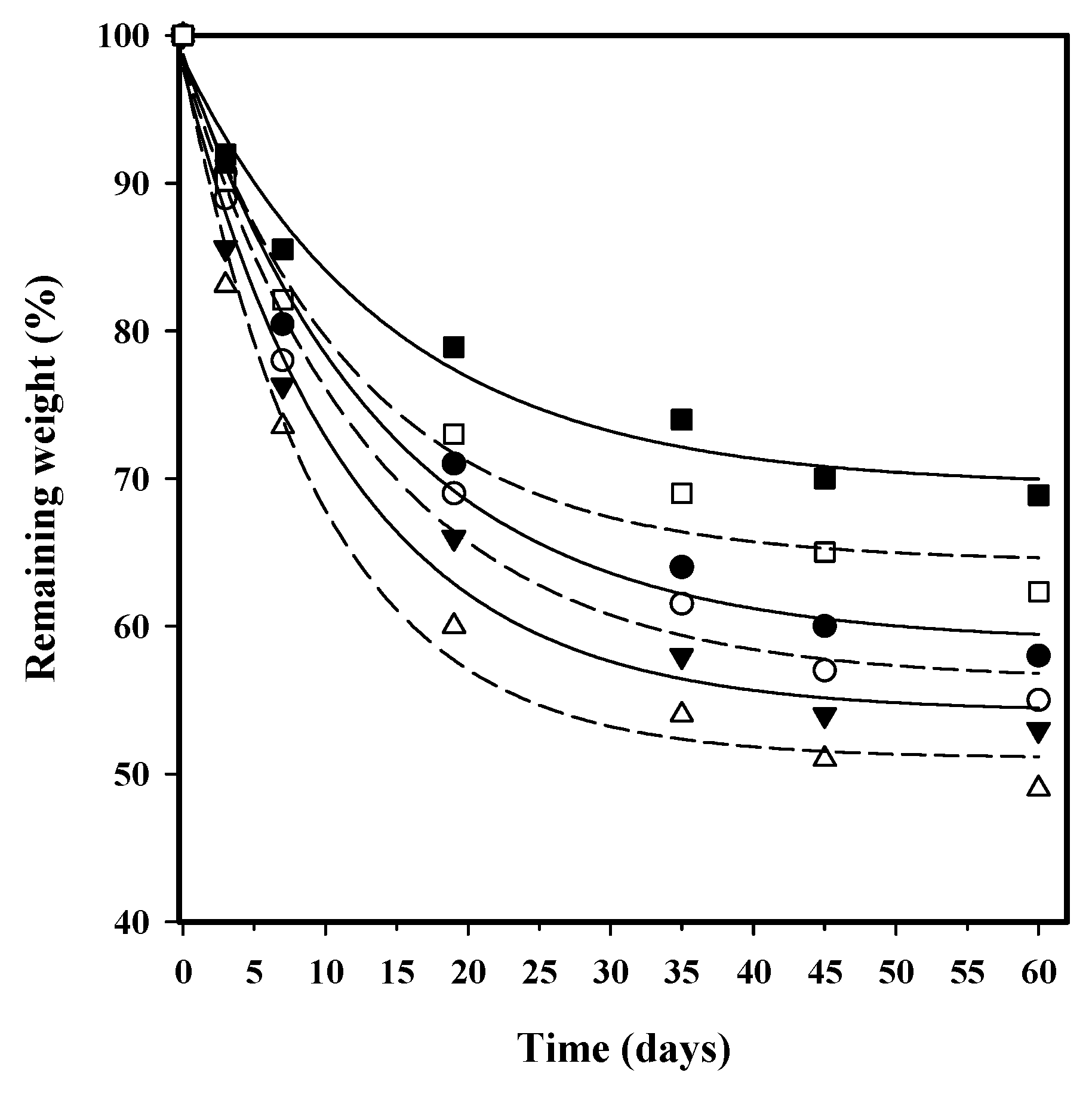
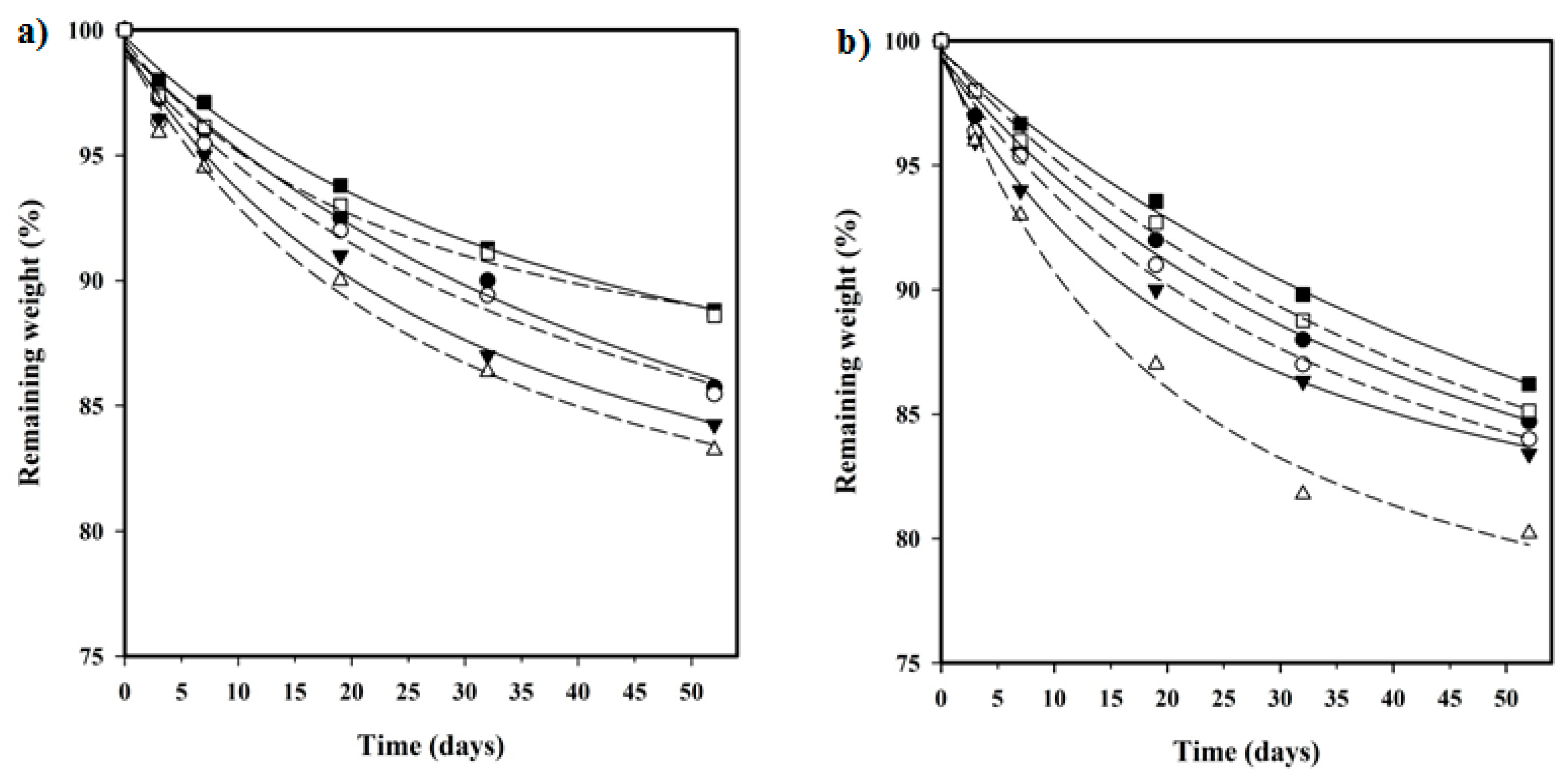
In vitro hydrolytic degradation assays were carried out in a pH 7.4 phosphate buffer (19.268 g of Na2HPO4·12H2O and 1.796 g of KH2PO4 in 1 L of deionized water) at 37 °C and at the accelerated condition provided by raising temperature to 70 °C. Samples (2 × 2 cm2 square pieces) were kept under orbital shaking in bottles filled with 20 mL of the degradation medium and sodium azide (0.03 wt %) to prevent microbial growth for the selected exposure times. The samples were then thoroughly rinsed with distilled water, dried to constant weight under a vacuum, and stored over P4O10 before analysis. Degradation studies were performed in triplicate and the results were averaged.
Enzymatic studies were carried out with lipases from a porcine pancreas (56–60 U/mg) and Pseudomonas cepacia (≥30 U/mg) using three replicates. All samples (1 × 1 cm2 square pieces) were exposed to 2 mL of pH 7.4 phosphate buffer at 37 °C. The buffer contained the enzyme along with sodium azide (0.03% w/v) and calcium chloride (5 mM). Solutions were replaced every 48 h to prevent enzymatic activity loss. Samples were extracted, washed, and dried as indicated before.
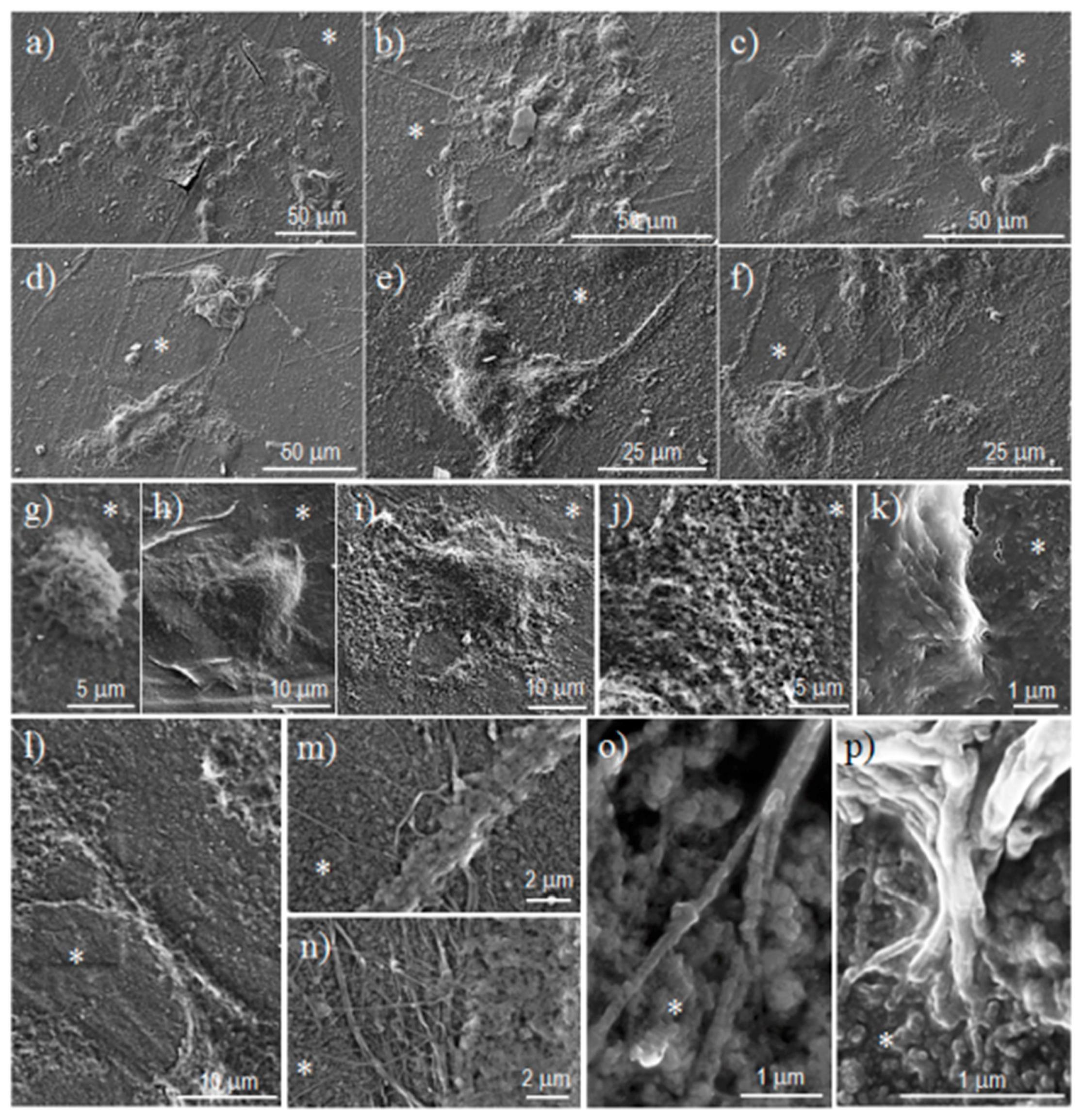
Scanning electron microscopy (SEM) was utilized to examine the morphology of films after different times of exposure to the selected degradation media. Carbon coating was accomplished with a Mitec k950 Sputter Coater (fitted with a film thickness monitor k150x) (Quorum Technologies Ltd., West Sussex, UK). SEM micrographs were obtained with a Zeiss Neon 40 EsB instrument (Carl Zeiss, Oberkochen, Germany).
Weight retention (
Wr) of the specimens was addressed by the percentage ratio of weight after degradation (
Wd) to initial weight before degradation (
W0):


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}