
An Osteoconductive Antibiotic Bone Eluting Putty with a Custom Polymer Matrix

 ,
, 

Abstract
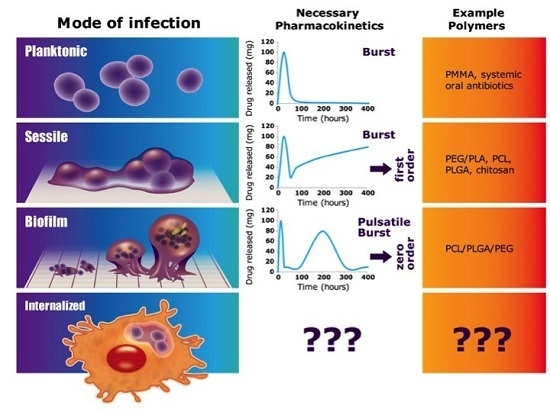
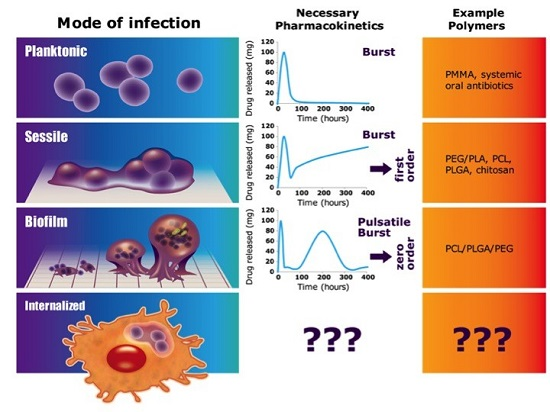
:
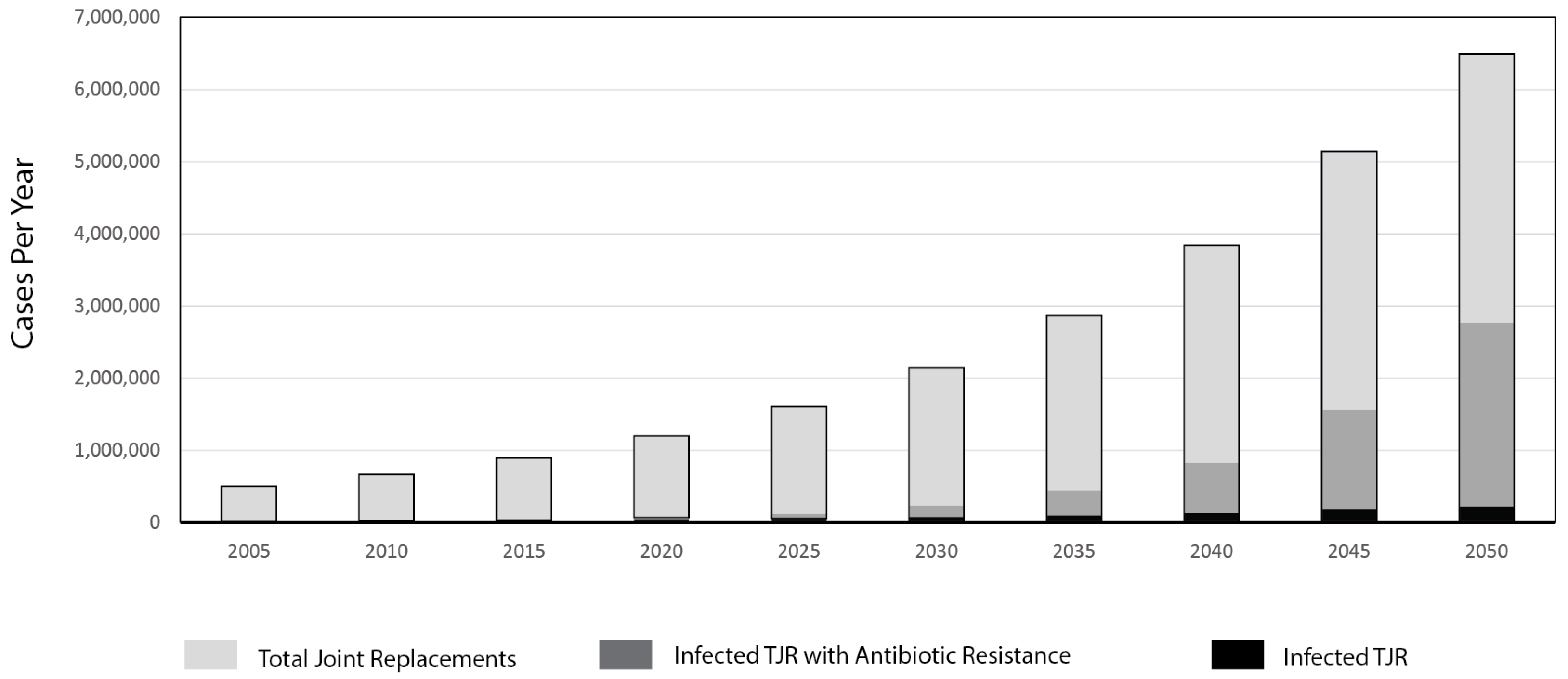
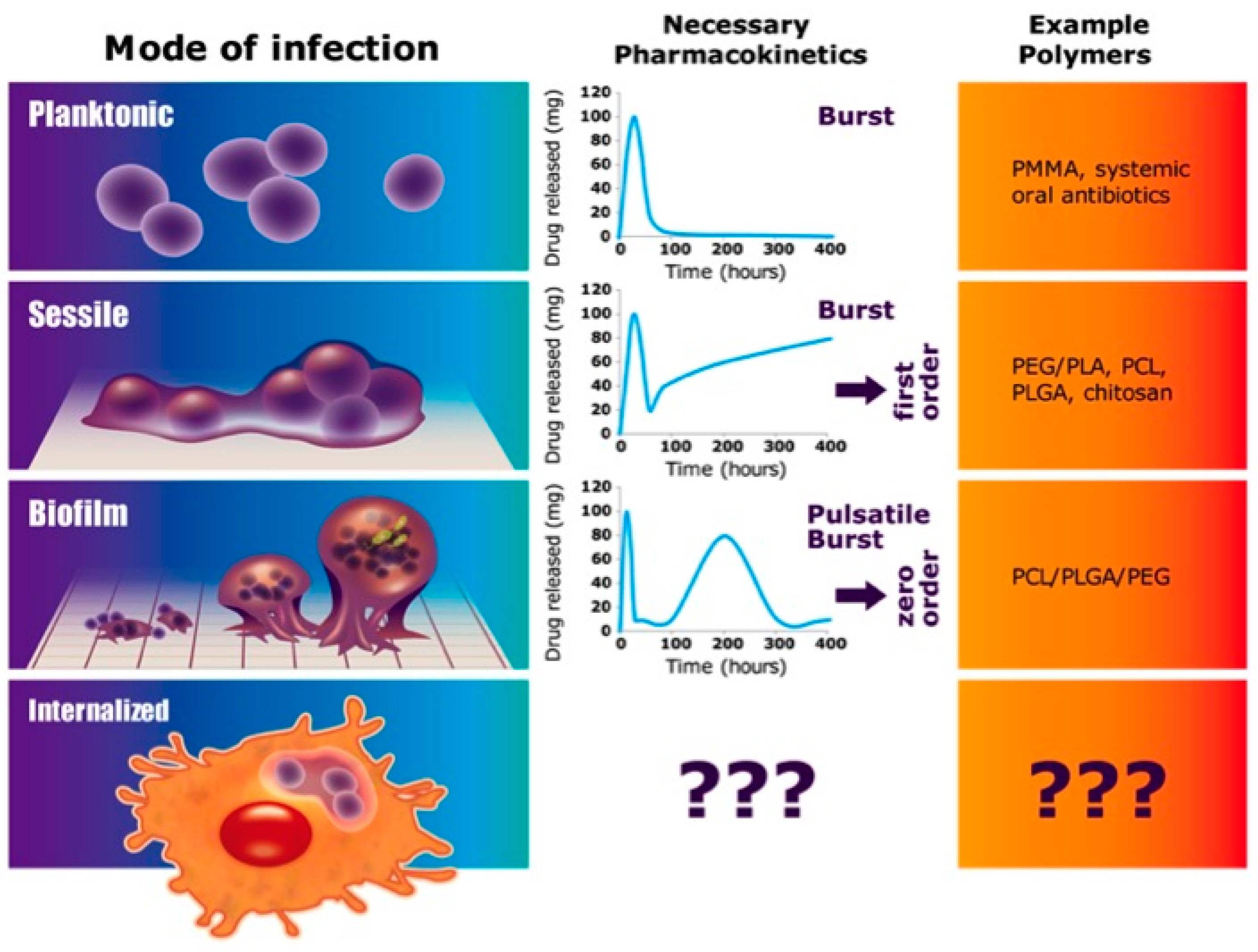
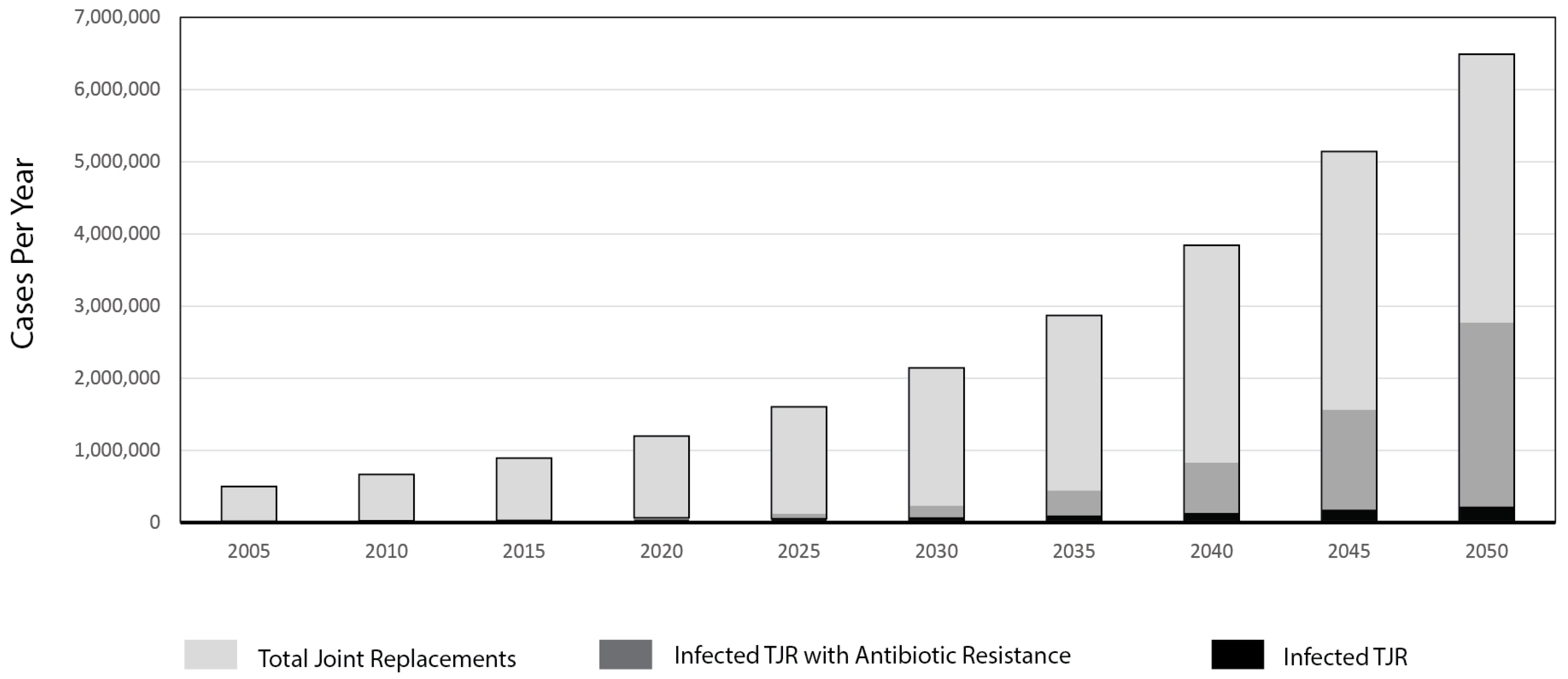
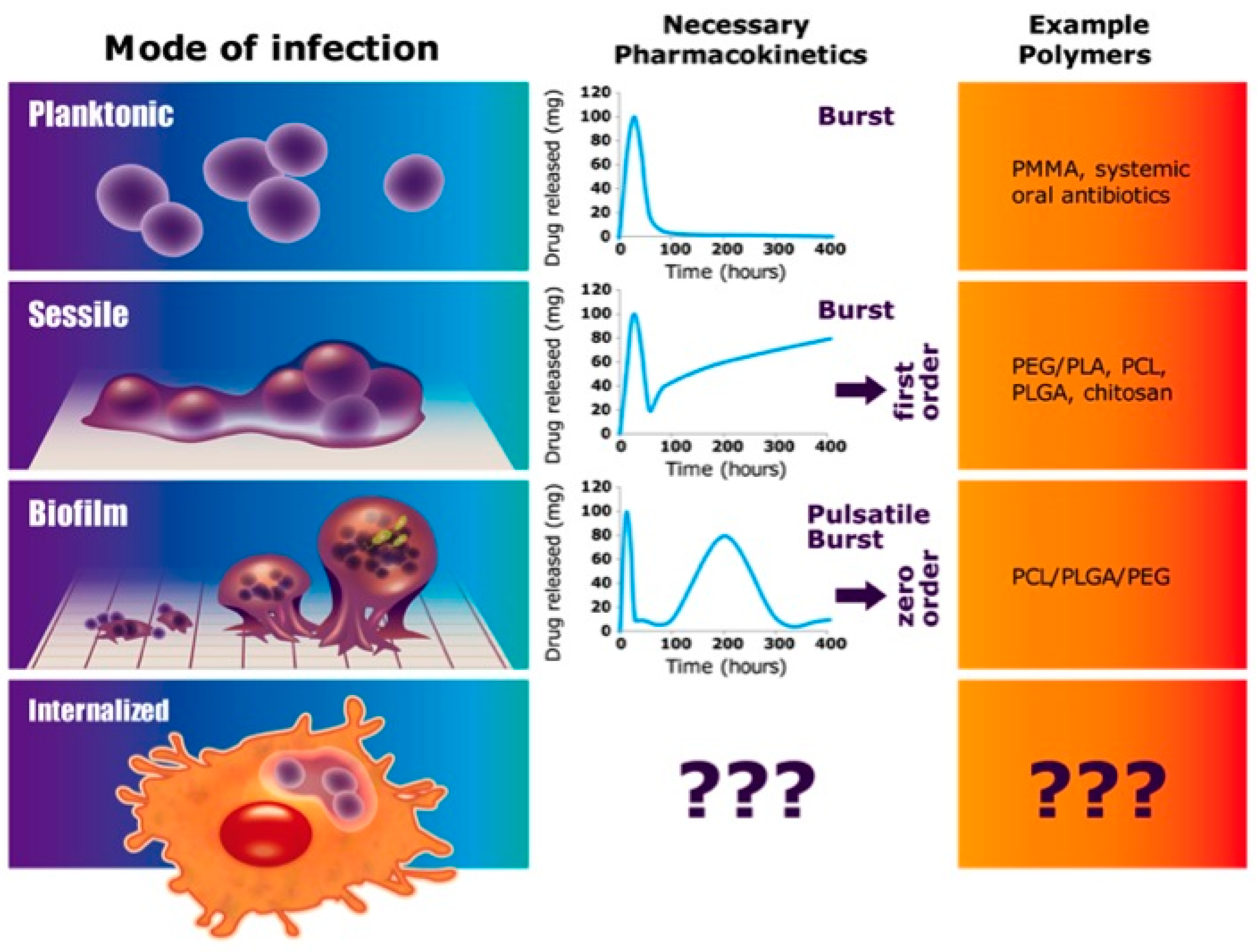
1. Introduction
2. Materials and Methods
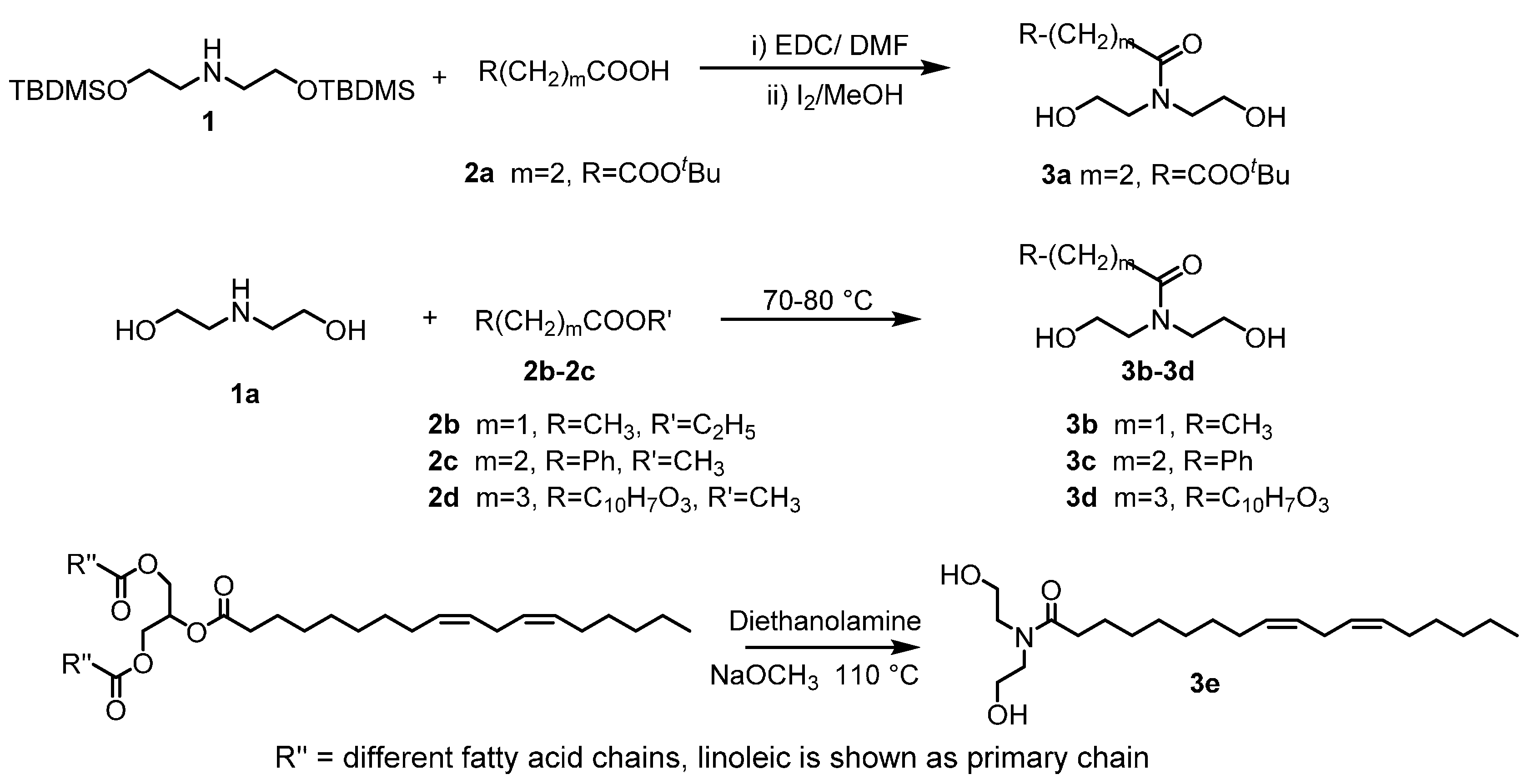
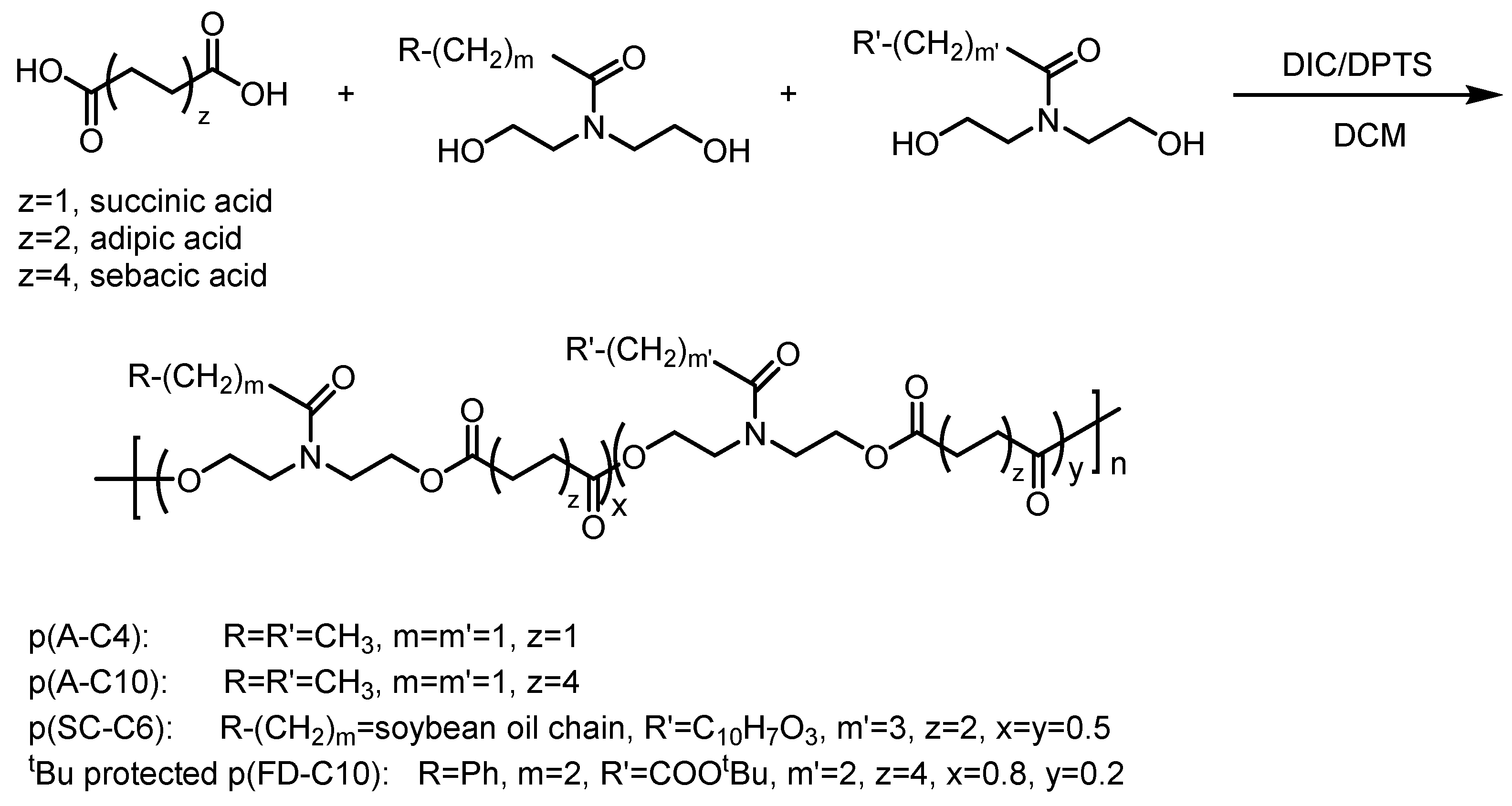
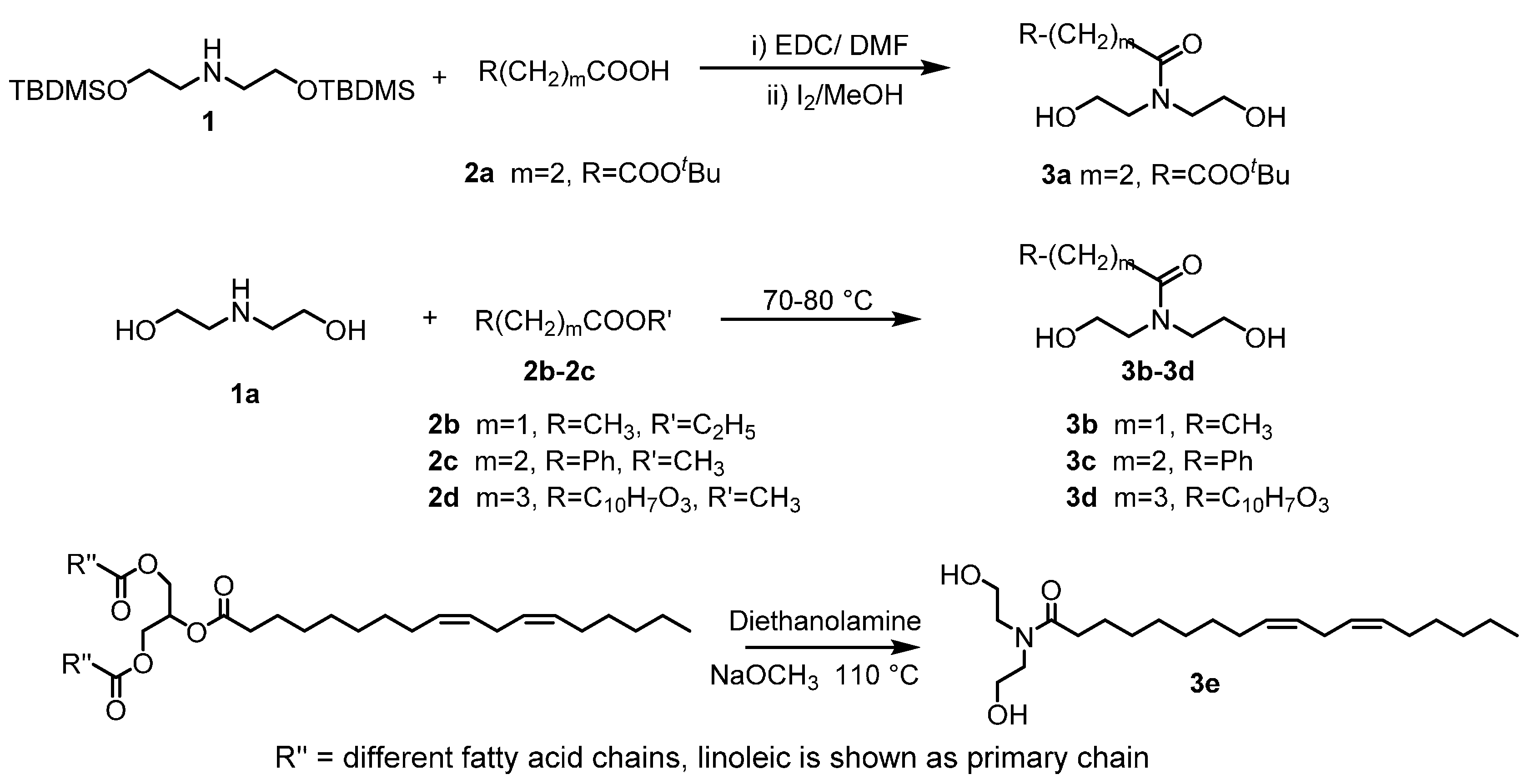
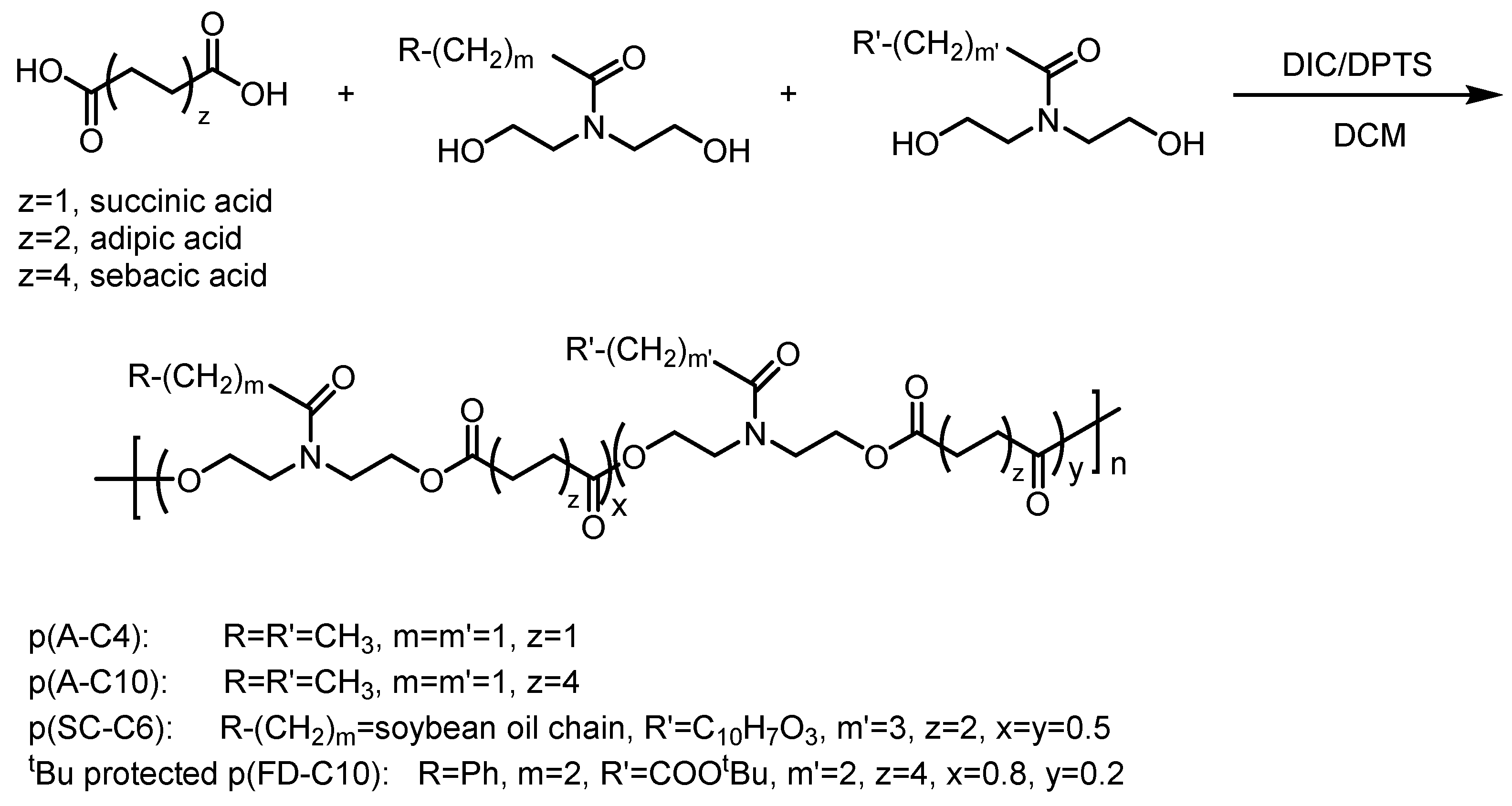
2.1. Custom Polymer Synthesis
2.2. Polymer Characterization
2.3. Putty Formulation
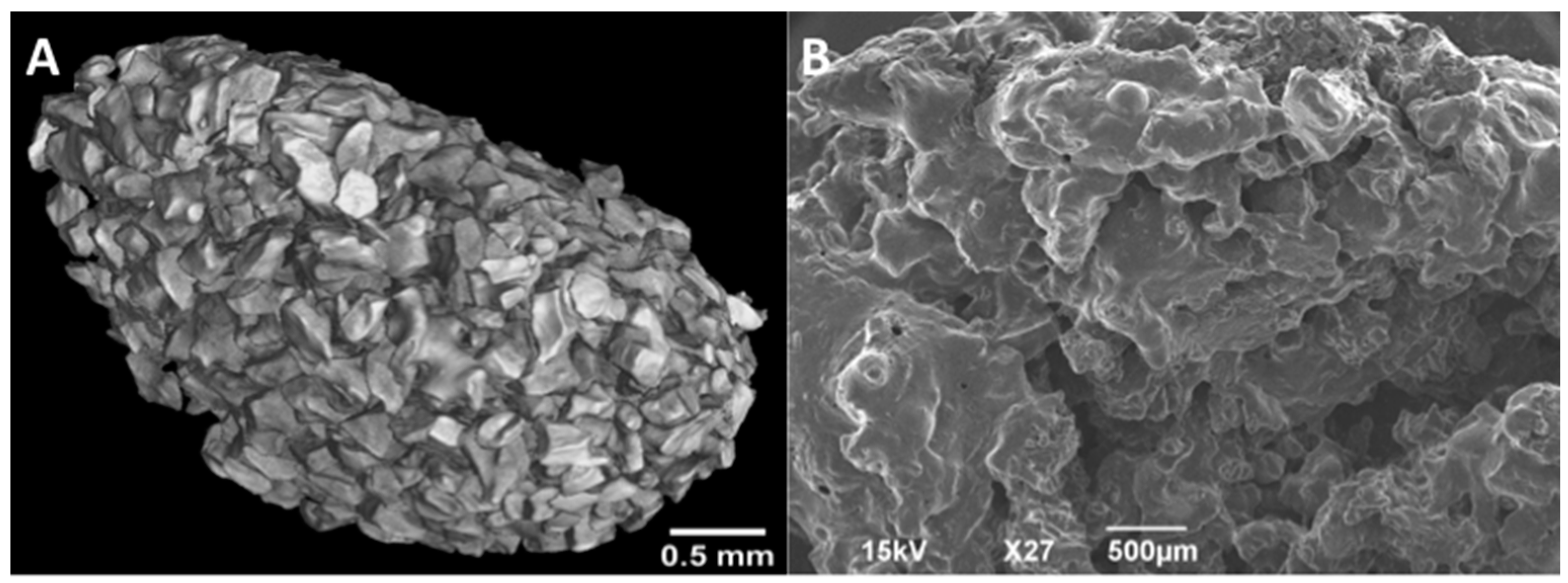
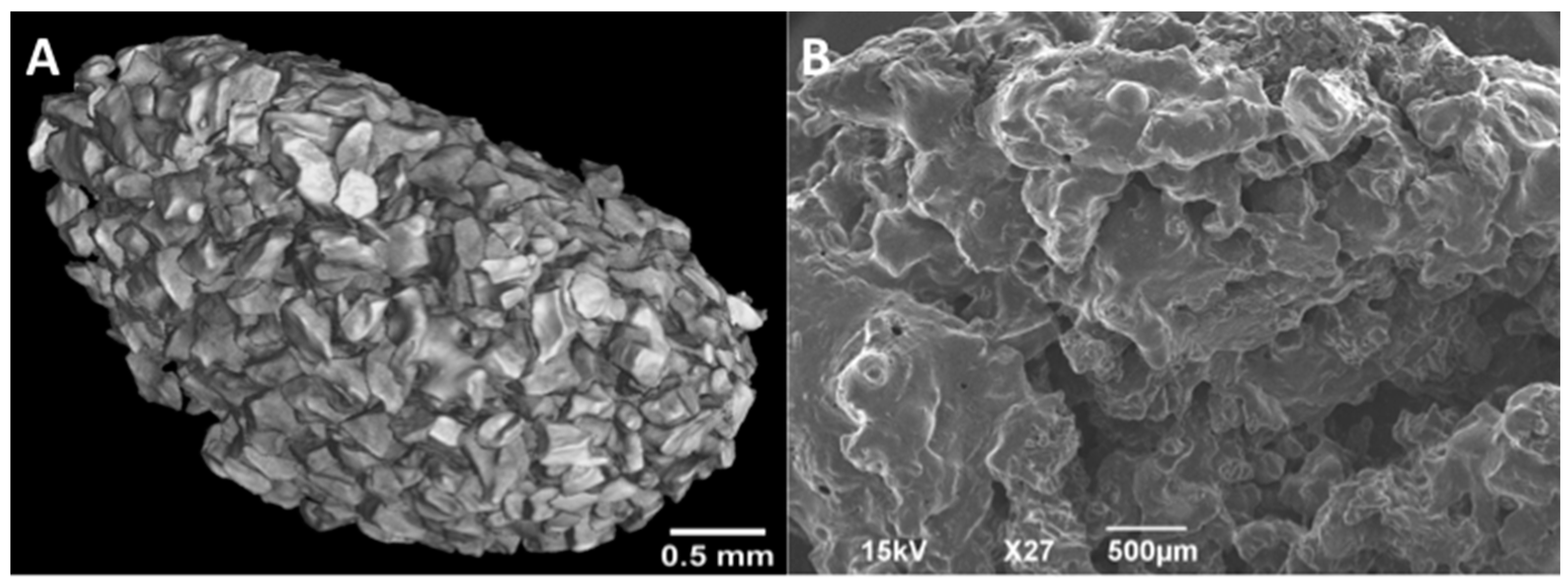
2.4. Imaging
2.4.1. MicroCT
2.4.2. SEM
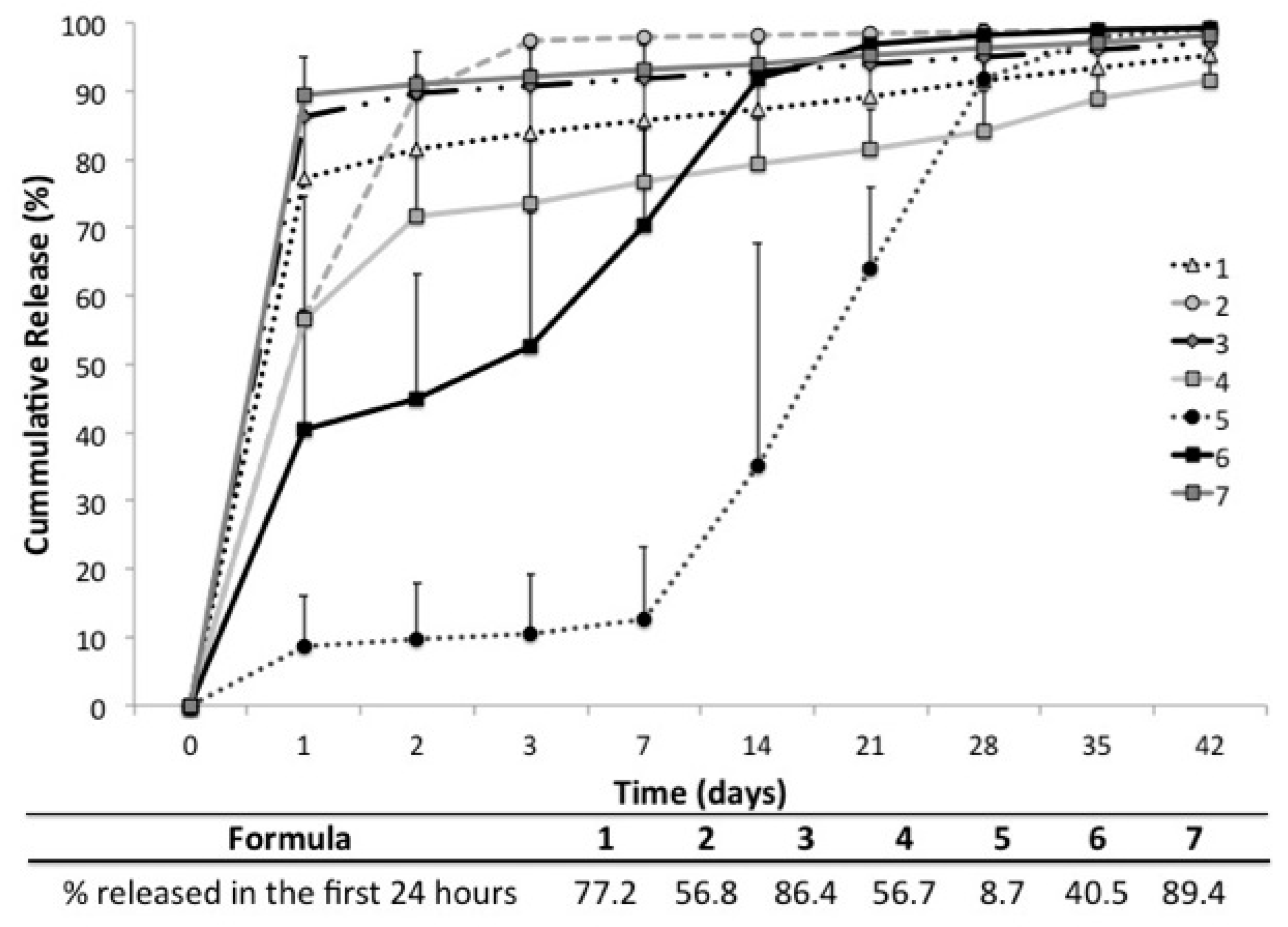
2.5. In Vitro Release
2.6. In Vitro Vancomycin Release Kinetics
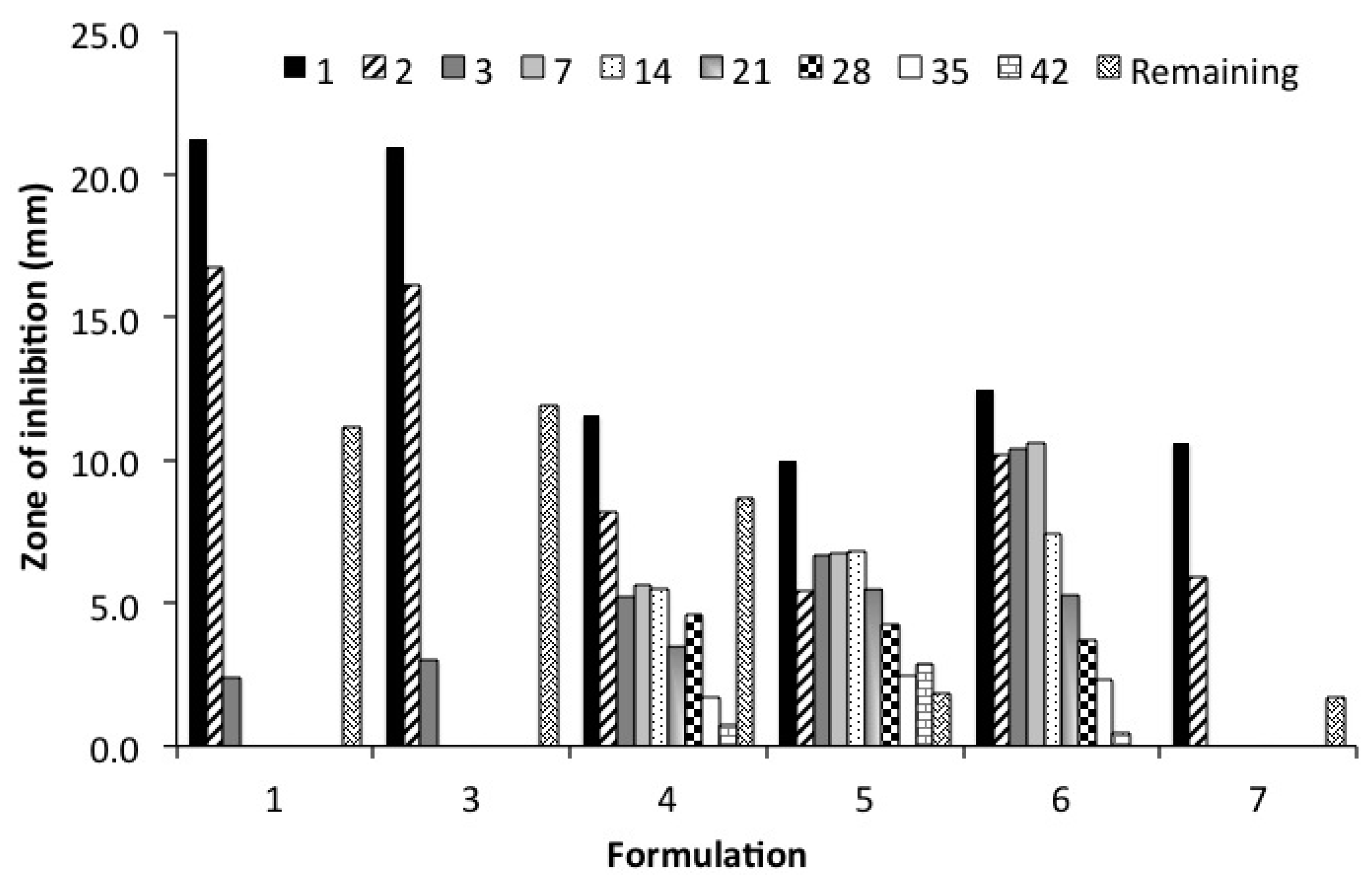
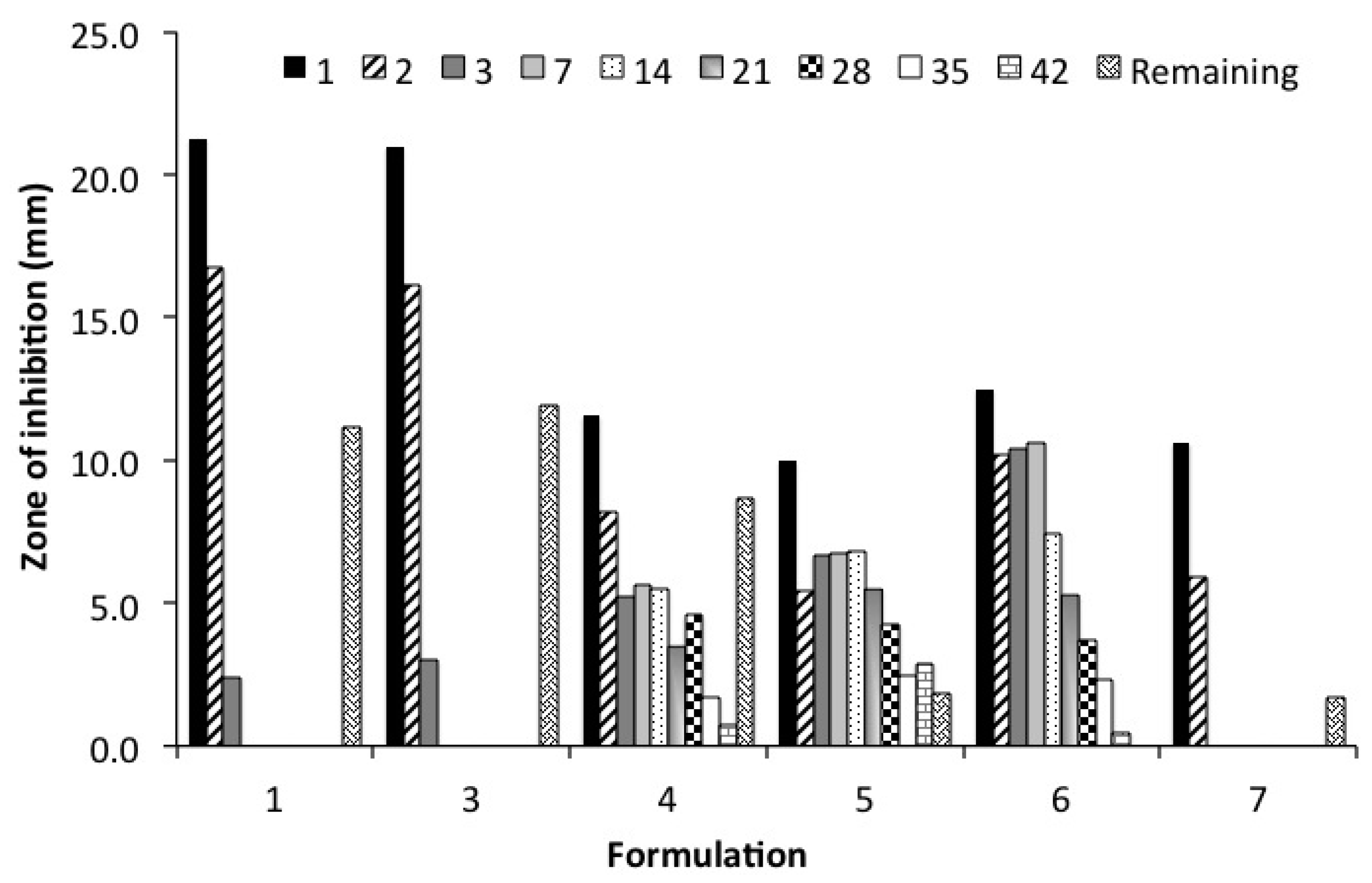
2.7. Kirby Bauer Zone of Inhibition Assay (ZOI)
2.8. Mechanical Characterization
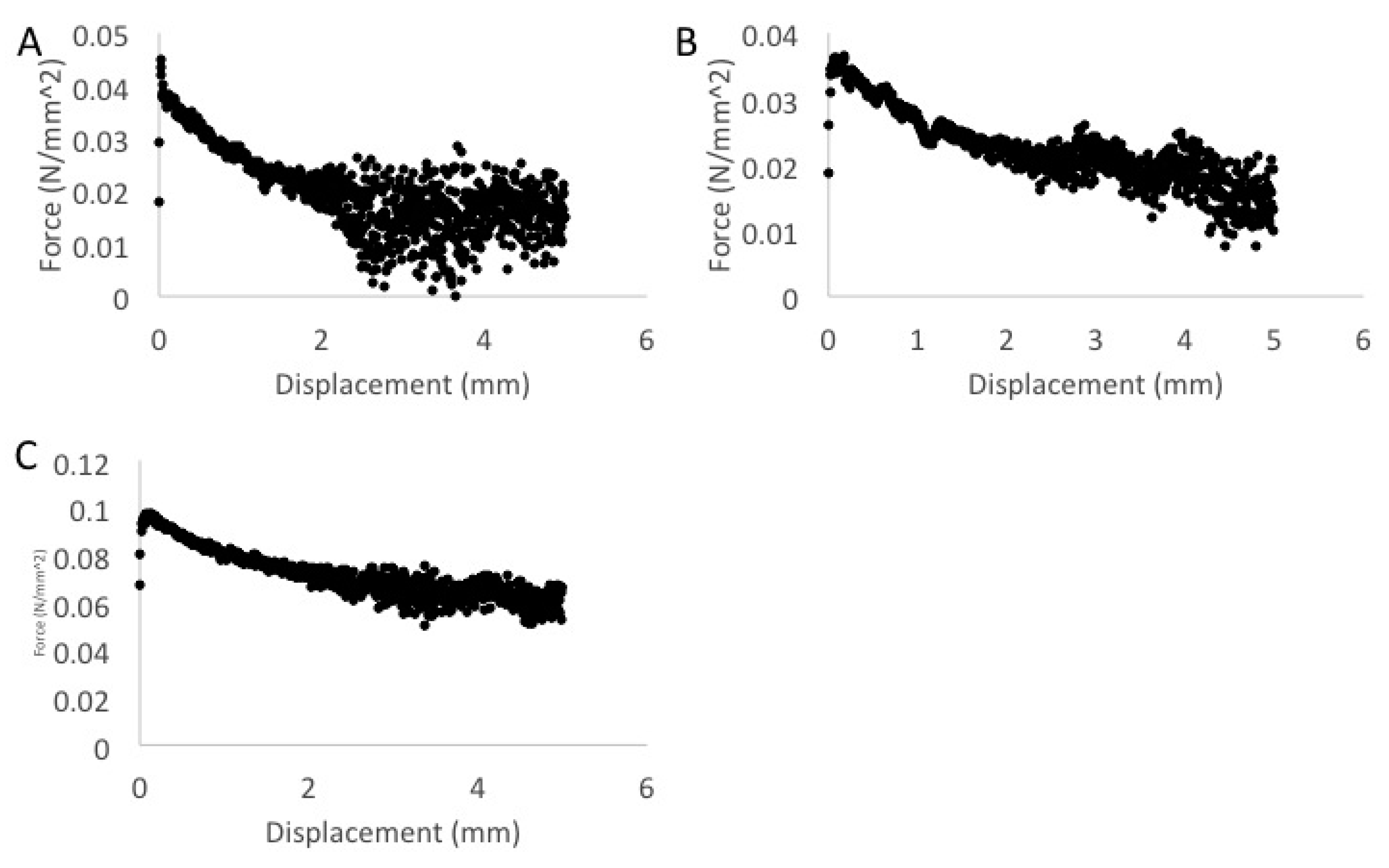
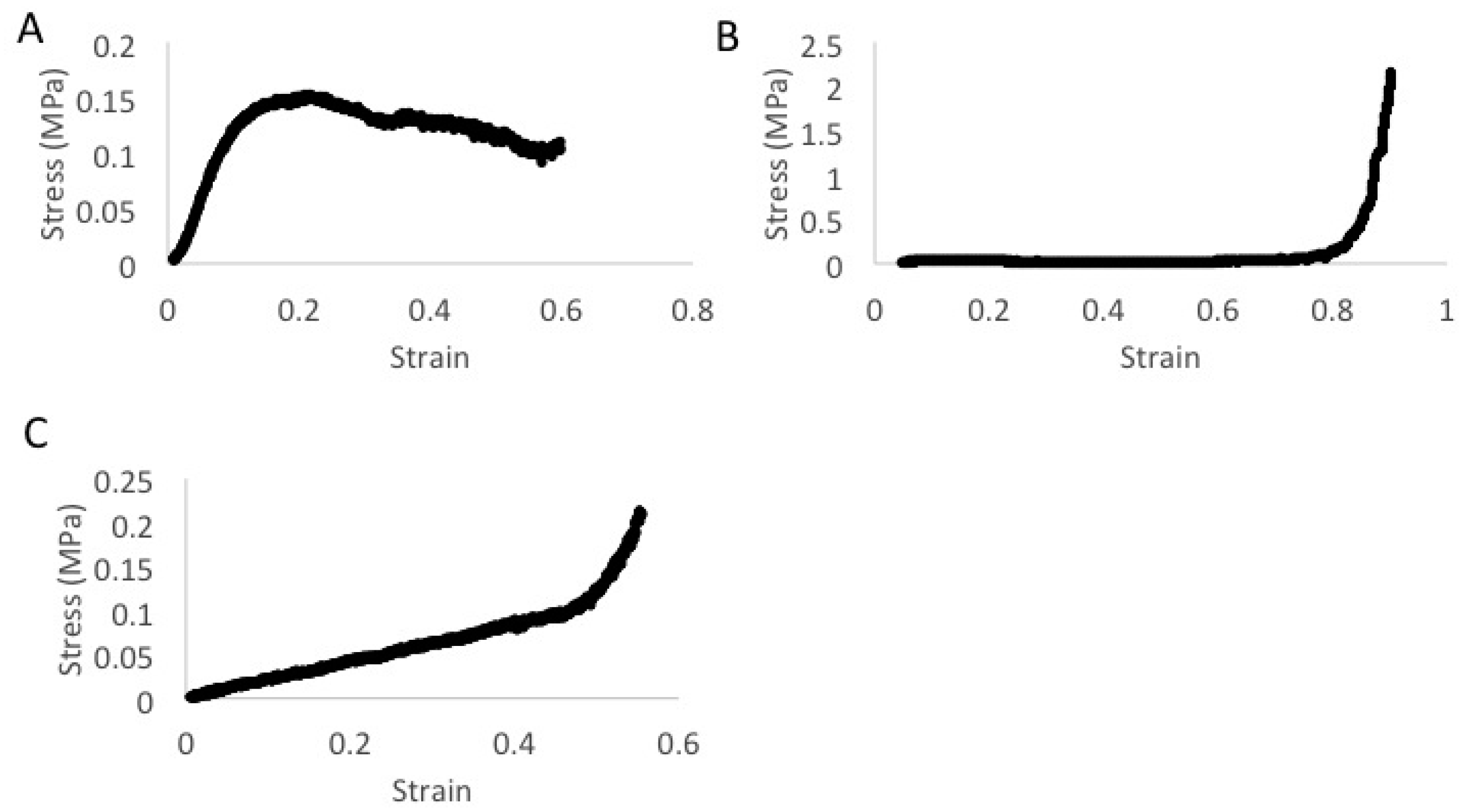
2.8.1. Compression Testing
2.8.2. Tensile Lap Testing
2.9. Statistical Analysis
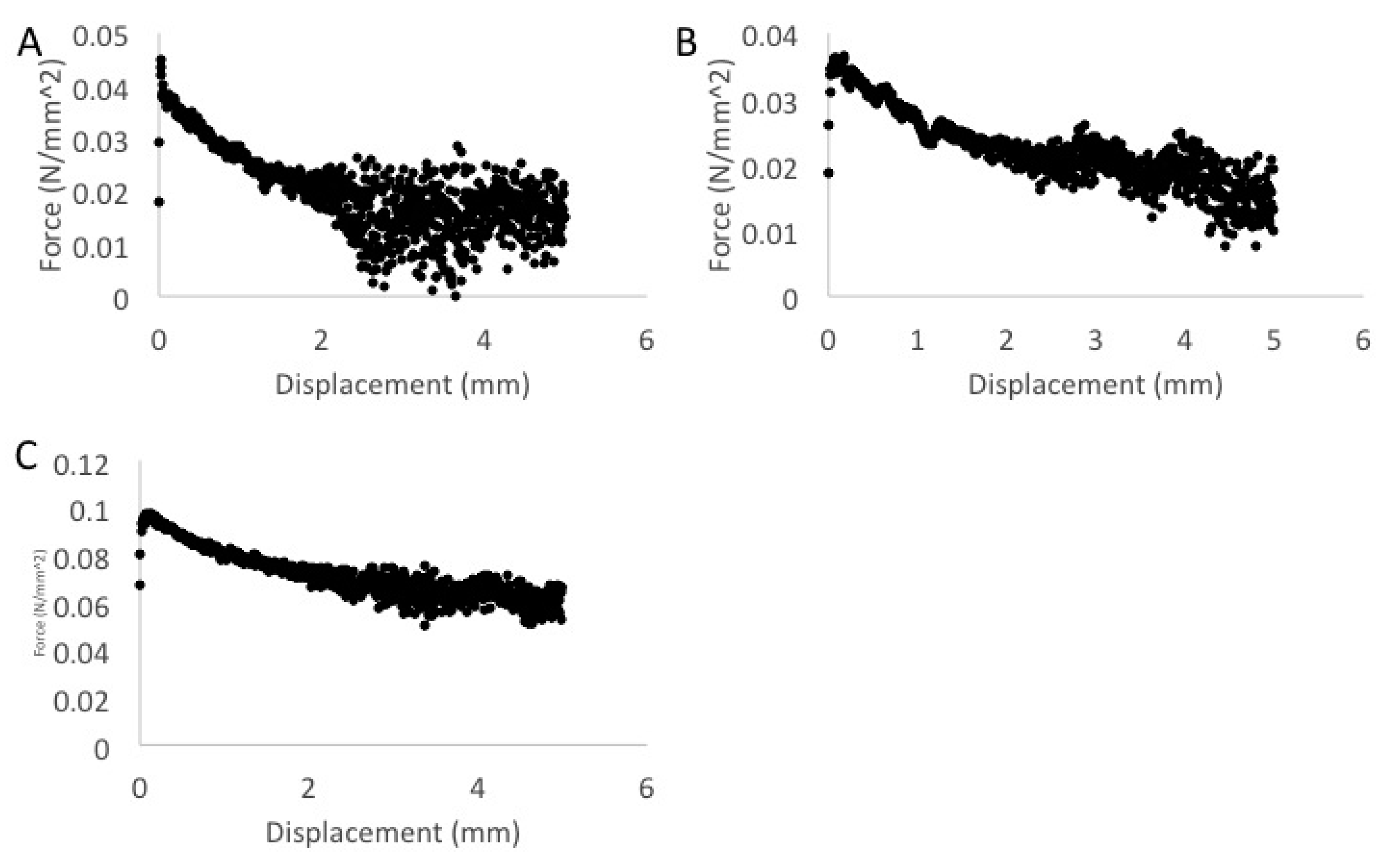
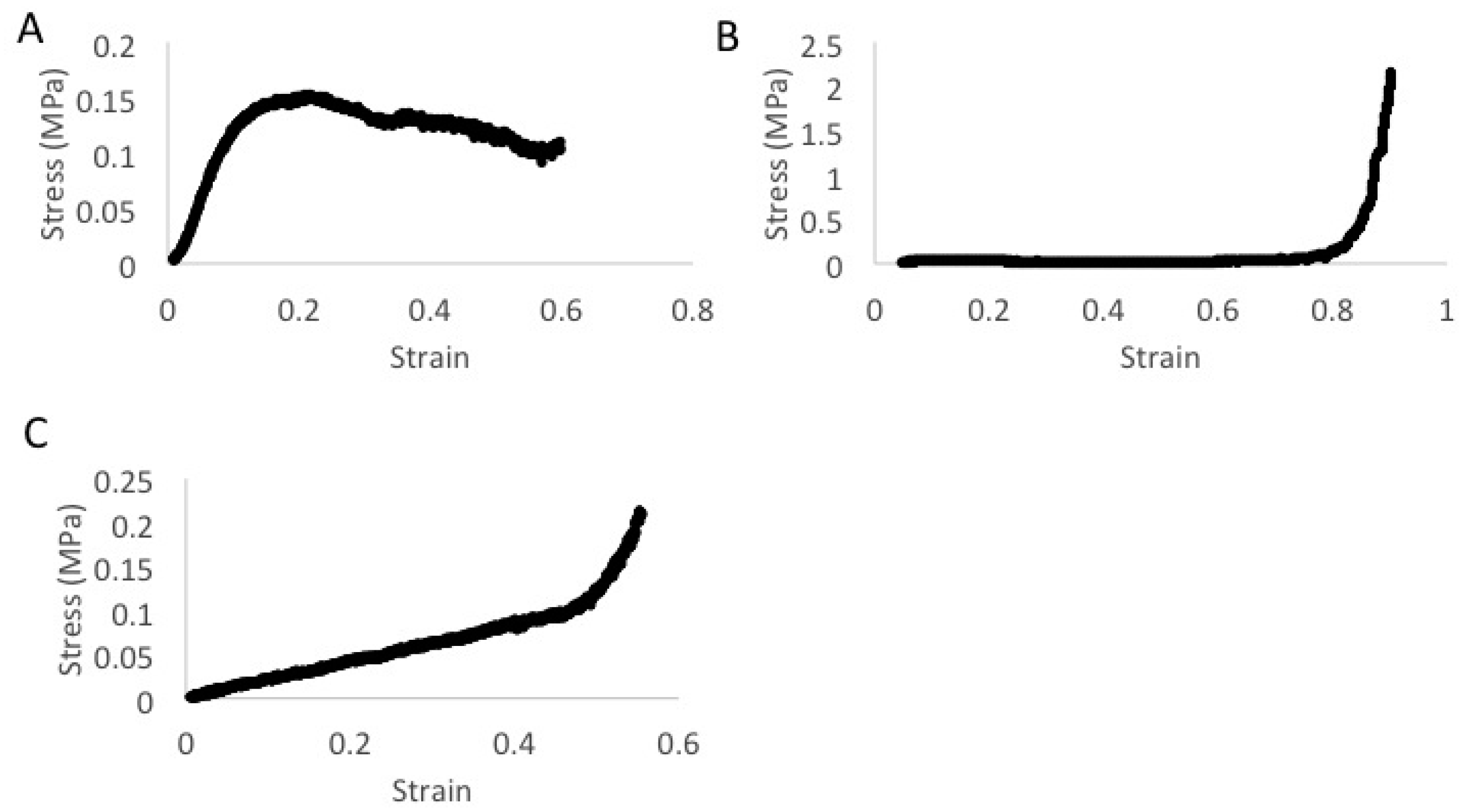
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PCL | Polycaprolactone |
| PLGA | Poly(d,l-lactide-co-glycolide) |
| NMP | N-Methyl-2-pyrrolidone |
| PEG | Polyethyleneglycol |
| BVF | Bone graft void filler |
| ABVF | Antibiotic-eluting bone void filler |
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Zero-order (R2) | First-order (R2) | Korsmeyer-peppas (R2) | Higuchi (R2) | Hixon-crowell (R2) |
|---|---|---|---|---|---|
| 1-A | 0.14 | 0.45 | 0.92 | 0.29 | 0.26 |
| 1-B | 0.34 | 0.66 | 0.74 | 0.54 | 0.54 |
| 1-C | 0.14 | 0.41 | 0.94 | 0.29 | 0.26 |
| 1-D | 0.16 | 0.51 | 0.98 | 0.32 | 0.33 |
| 1-E | 0.14 | 0.45 | 0.93 | 0.29 | 0.26 |
| 1-F | 0.14 | 0.45 | 0.96 | 0.29 | 0.26 |
| 2 | 0.26 | 0.52 | 0.51 | 0.46 | 0.41 |
| 3-A | 0.14 | 0.45 | 0.90 | 0.29 | 0.27 |
| 3-B | 0.15 | 0.39 | 0.93 | 0.30 | 0.26 |
| 3-C | 0.14 | 0.40 | 0.97 | 0.29 | 0.24 |
| 3-D | 0.14 | 0.42 | 0.96 | 0.29 | 0.25 |
| 3-E | 0.14 | 0.47 | 0.63 | 0.29 | 0.27 |
| 3-F | 0.13 | 0.46 | 0.94 | 0.28 | 0.24 |
| 4-A | 0.49 | 0.78 | 0.76 | 0.69 | 0.69 |
| 4-B | 0.27 | 0.71 | 0.83 | 0.43 | 0.53 |
| 4-C | 0.63 | 0.83 | 0.71 | 0.81 | 0.76 |
| 4-D | 0.17 | 0.57 | 0.82 | 0.33 | 0.37 |
| 5-A | 0.89 | 0.84 | 0.77 | 0.80 | 0.86 |
| 5-B | 0.92 | 0.94 | 0.93 | 0.96 | 0.98 |
| 6-A | 0.81 | 0.98 | 0.95 | 0.92 | 0.92 |
| 6-B | 0.32 | 0.74 | 0.84 | 0.53 | 0.59 |
| 6-C | 0.67 | 0.99 | 0.96 | 0.87 | 0.91 |
| 7-A | 0.15 | 0.47 | 0.63 | 0.29 | 0.27 |
| 7-B | 0.13 | 0.46 | 0.94 | 0.28 | 0.24 |
References
- Surgeons, A. Joint Revision Surgery—When Do I Need It. 2007. Available online: http://orthoinfo.aaos.org/topic.cfm?topic=A00510 (accessed on 21 May 2013).
- Trampuz, A.; Widmer, A.F. Infections associated with orthopedic implants. Curr. Opin. Infect. Dis. 2006, 19, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Campoccia, D.; Montanaro, L.; Speziale, P.; Arciola, C.R. Antibiotic-loaded biomaterials and the risks for the spread of antibiotic resistance following their prophylactic and therapeutic clinical use. Biomaterials 2010, 31, 6363–6377. [Google Scholar] [CrossRef] [PubMed]
- Ketonis, C.; Barr, S.; Adams, C.S.; Hickok, N.J.; Parvizi, J. Bacterial colonization of bone allografts: Establishment and effects of antibiotics. Clin. Orthop. 2010, 468, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.G.D.; Bostrom, M.; Brause, B.; Harney, C.; Salvati, E.A. Effective bactericidal activity of tobramycin and vancomycin eluted from acrylic bone cement. Acta Orthop. 2001, 72, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Conterno, L.O.; da Silva Filho, C.R. Antibiotics for treating chronic osteomyelitis in adults. Cochrane Database Syst. Rev. 2009, 3. [Google Scholar] [CrossRef]
- Landersdorfer, C.B.; Bulitta, J.B.; Kinzig, M.; Holzgrabe, U.; Sörgel, F. Penetration of Antibacterials into Bone. Clin. Pharmacokinet. 2009, 48, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N.; Reilly, D.T.; Karchmer, A.W.; Goldie, S.J. Clinical effectiveness and cost-effectiveness of 2 management strategies for infected total hip arthroplasty in the elderly. Clin. Infect. Dis. 2001, 32, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.M.; Lau, E.; Schmier, J.; Ong, K.L.; Zhao, K.; Parvizi, J. Infection burden for hip and knee arthroplasty in the United States. J. Arthroplast. 2008, 23, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Brooks, A.E. Therapeutic strategies to combat antibiotic resistance. Adv. Drug Deliv. Rev. 2014, 78, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Peel, T.N.; Buising, K.L.; Choong, P.F. Diagnosis and management of prosthetic joint infection. Curr. Opin. Infect. Dis. 2012, 25, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.; Masters, S.; Berendt, A.R.; McLardy-Smith, P.; Byren, I.; Atkins, B.L. Guiding empirical antibiotic therapy in orthopaedics: The microbiology of prosthetic joint infection managed by debridement, irrigation and prosthesis retention. J. Infect. 2007, 55, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, M.P.; Seigerman, D.A. The clinical use of allografts, demineralized bone matrices, synthetic bone graft substitutes and osteoinductive growth factors: A survey study. HSS J. 2005, 1, 9–18. [Google Scholar] [CrossRef] [PubMed]
- O’May, G.A.; Brady, R.A.; Prabhakara, R.; Leid, J.G.; Calhoun, J.H.; Shirtliff, M.E. Biofilm Infections; Bjarnsholt, T., Jensen, P.Ø., Moser, C., Høiby, N., Eds.; Springer: New York, NY, USA, 2011; pp. 111–137. [Google Scholar]
- Miclau, T.; Dahners, L.E.; Lindsey, R.W. In vitro pharmacokinetics of antibiotic release from locally implantable materials. J. Orthop. Res. 1993, 11, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Bozic, K.J.; Kurtz, S.M.; Lau, E.; Ong, K.; Chiu, V.; Vail, T.P.; Rubash, H.E.; Berry, D.J. The Epidemiology of Revision Total Knee Arthroplasty in the United States. Clin. Orthop. Relat. Res. 2009, 468, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Richards, R.G. Staphylococci and implant surfaces: A review. Injury 2006, 37, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, R.M. Case Management: Revision Knee and Hip Replacement Surgery. Available online: http://praxis.iuhealth.org/article/revision-knee-and-hip-replacement-surgery (accessed on 28 June 2016).
- Hetrick, E.M.; Schoenfisch, M.H. Reducing implant-related infections: Active release strategies. Chem. Soc. Rev. 2006, 35, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, P.V.; Dinopoulos, H.; Tsiridis, E. Bone substitutes: An update. Injury 2005, 36, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Brooks, A.E.; Grainger, D.W. Antimicrobial Medical Devices in Preclinical Development and Clinical Use. In Biomaterials Associated Infection; Moriarty, T.F., Zaat, S.A.J., Busscher, H.J., Eds.; Springer: New York, NY, USA, 2013; pp. 307–354. [Google Scholar]
- Sevy, J.O.; Slawson, M.H.; Grainger, D.W.; Brooks, A.E. Assay method for polymer-controlled antibiotic release from allograft bone to target orthopaedic infections-biomed 2010. Biomed. Sci. Instrum. 2010, 46, 136. [Google Scholar] [PubMed]
- Brooks, A.E.; Brooks, B.D.; Davidoff, S.N.; Hogrebe, P.C.; Fisher, M.A.; Grainger, D.W. Polymer-controlled release of tobramycin from bone graft void filler. Drug Deliv. Transl. Res. 2013, 3, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Sinclair, K.D.; Davidoff, S.N.; Lawson, S.; Williams, A.G.; Coats, B.; Grainger, D.W.; Brooks, A.E. Molded polymer-coated composite bone void filler improves tobramycin controlled release kinetics. J. Biomed. Mater. Res. B Appl. Biomater. 2013, 102, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Sinclair, K.D.; Grainger, D.W.; Brooks, A.E. A resorbable antibiotic-eluting polymer composite bone void filler for perioperative infection prevention in a rabbit radial defect model. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Jones, Z.; Brooks, A.E.; Ferrell, Z.; Grainger, D.W.; Sinclair, K.D. A resorbable antibiotic eluting bone void filler for periprosthetic joint infection prevention. J. Biomed. Mater. Res. B Appl. Biomater. 2015. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Dhanaraju, M.D.; Sathyamoorthy, N.; Sundar, V.D.; Suresh, C. Preparation of poly(epsilon-caprolactone) microspheres containing etoposide by solvent evaporation method. J. Pharm. Sci. 2010, 5, 114–122. [Google Scholar]
- Swanson, J.P.; Monteleone, L.R.; Haso, F.; Costanzo, P.J.; Liu, T.; Joy, A. A Library of thermoresponsive, coacervate-forming biodegradable polyesters. Macromolecules 2015, 48, 3834–3842. [Google Scholar] [CrossRef]
- Gokhale, S.; Xu, Y.; Joy, A. A library of multifunctional polyesters with “peptide-like” pendant functional groups. Biomacromolecules 2013, 14, 2489–2493. [Google Scholar] [CrossRef] [PubMed]
- Curley, J.; Ortiz, D.; Brooks, B.D.; Brooks, A.E. Evaluation of polycaprolactone encapsulated vancomycin for controlled drug delivery from a bone void filling putty. Biomed. Sci. Instrum. 2016, 52. in press. [Google Scholar]
- Hasan, M.R.; Nodland, J.; Brooks, A.E. A microfluidic platform for continuous monitoring of drug release kinetics. Biomed. Sci. Instrum. 2016, 52. in press. [Google Scholar]
- Budiasih, S.; Jiyauddin, K.; Logavinod, N.; Kaleemullah, M.; Jawad, A.; Samer, A.D.; Fadli, A.; Eddy, Y. Optimization of polymer concentration for designing of oral matrix controlled release dosage form. UK J. Pharm. Biosci. 2014, 2, 54. [Google Scholar] [CrossRef]
- Maria Ann Woodruff, D.W.H. The return of a forgotten polymer-polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Andriano, K.P.; Chandrashekar, B.; McEnery, K.; Dunn, R.L.; Moyer, K.; Balliu, C.M.; Holland, K.M.; Garrett, S.; Huffer, W.E. Preliminary in vivo studies on the osteogenic potential of bone morphogenetic proteins delivered from an absorbable puttylike polymer matrix. J. Biomed. Mater. Res. 2000, 53, 36–43. [Google Scholar] [CrossRef]
- Anseth, K.S.; Wang, C.M.; Bowman, C.N. Reaction behaviour and kinetic constants for photopolymerizations of multi(meth)acrylate monomers. Polymer 1994, 35, 3243–3250. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic acid) controlled release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Modeling of drug release from Swellable polymers. Eur. J. Pharm. Biopharm. 2000, 49, 47–58. [Google Scholar] [CrossRef]
- Kosmidis, K.; Argyrakis, P.; Macheras, P. A reappraisal of drug release laws using monte carlo simulations: The prevalence of the weibull function. Pharm. Res. 2003, 20, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Göpferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New technologies in computer-aided drug design: Toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef] [PubMed]
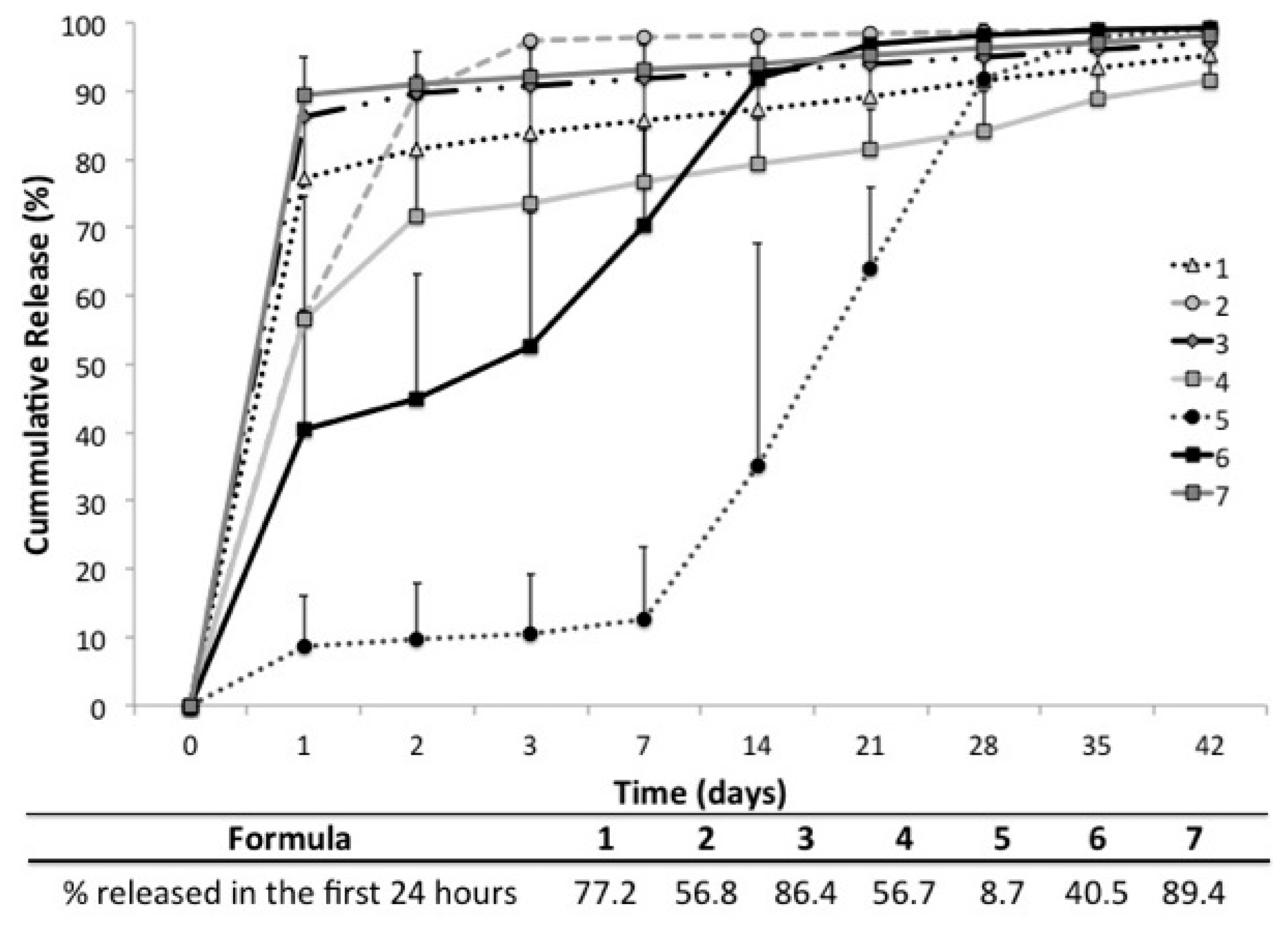
| Formulation (number of replicates) | Geometry | Polymer(s) | N-Methyl-2-pyrrolidone (NMP) | CaCl2 | Vancomycin (10% w/v) |
|---|---|---|---|---|---|
| 1 (n = 6) | Disk | PLGA/PCL/PEG | X | X | RPI |
| 2 (n = 1) | Sphere | PLGA/PCL/PEG | X | X | RPI |
| 3 (n = 6) | Disk | PLGA/PCL/PEG | X | X | Hospira |
| 4 (n = 4) | Disk | p(FD-C10) | X | Hospira | |
| 5 (n = 2) | Disk | p(SC-C6) | X | Hospira | |
| 6 (n = 3) | Disk | p(A-C4) | X | Hospira | |
| 7 (n = 2) | Disk | p(A-C10) | X | Hospira |
| Formulation | Zero-order (R2) | First-order (R2) | Korsmeyer-peppas (R2) | Higuchi (R2) | Hixon-crowell (R2) |
|---|---|---|---|---|---|
| 1 | 0.15 | 0.45 | 0.93 | 0.30 | 0.28 |
| 2 | 0.26 | 0.61 | 0.51 | 0.46 | 0.45 |
| 3 | 0.14 | 0.42 | 0.88 | 0.29 | 0.25 |
| 4 | 0.28 | 0.68 | 0.92 | 0.45 | 0.52 |
| 5 | 0.92 | 0.88 | 0.86 | 0.84 | 0.91 |
| 6 | 0.70 | 0.97 | 0.96 | 0.88 | 0.90 |
| 7 | 0.16 | 0.59 | 0.96 | 0.32 | 0.33 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curley, J.; Hasan, M.R.; Larson, J.; Brooks, B.D.; Liu, Q.; Jain, T.; Joy, A.; Brooks, A.E. An Osteoconductive Antibiotic Bone Eluting Putty with a Custom Polymer Matrix. Polymers 2016, 8, 247. https://doi.org/10.3390/polym8070247
Curley J, Hasan MR, Larson J, Brooks BD, Liu Q, Jain T, Joy A, Brooks AE. An Osteoconductive Antibiotic Bone Eluting Putty with a Custom Polymer Matrix. Polymers. 2016; 8(7):247. https://doi.org/10.3390/polym8070247
Chicago/Turabian StyleCurley, John, Mohammad Raquibul Hasan, Jacob Larson, Benjamin D. Brooks, Qianhui Liu, Tanmay Jain, Abraham Joy, and Amanda E. Brooks. 2016. "An Osteoconductive Antibiotic Bone Eluting Putty with a Custom Polymer Matrix" Polymers 8, no. 7: 247. https://doi.org/10.3390/polym8070247
APA StyleCurley, J., Hasan, M. R., Larson, J., Brooks, B. D., Liu, Q., Jain, T., Joy, A., & Brooks, A. E. (2016). An Osteoconductive Antibiotic Bone Eluting Putty with a Custom Polymer Matrix. Polymers, 8(7), 247. https://doi.org/10.3390/polym8070247