
Amyloid Beta Aggregation in the Presence of Temperature-Sensitive Polymers


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
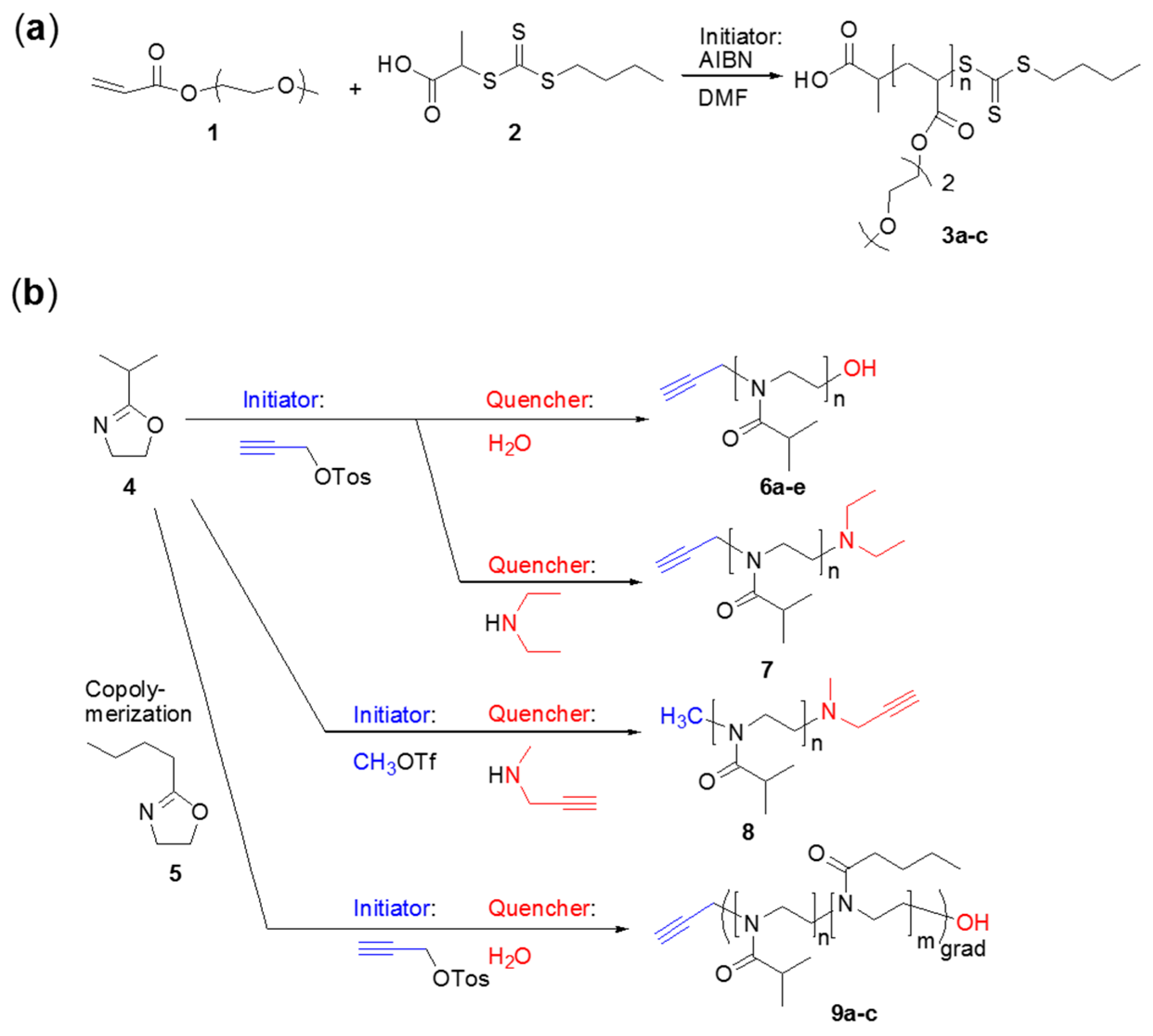
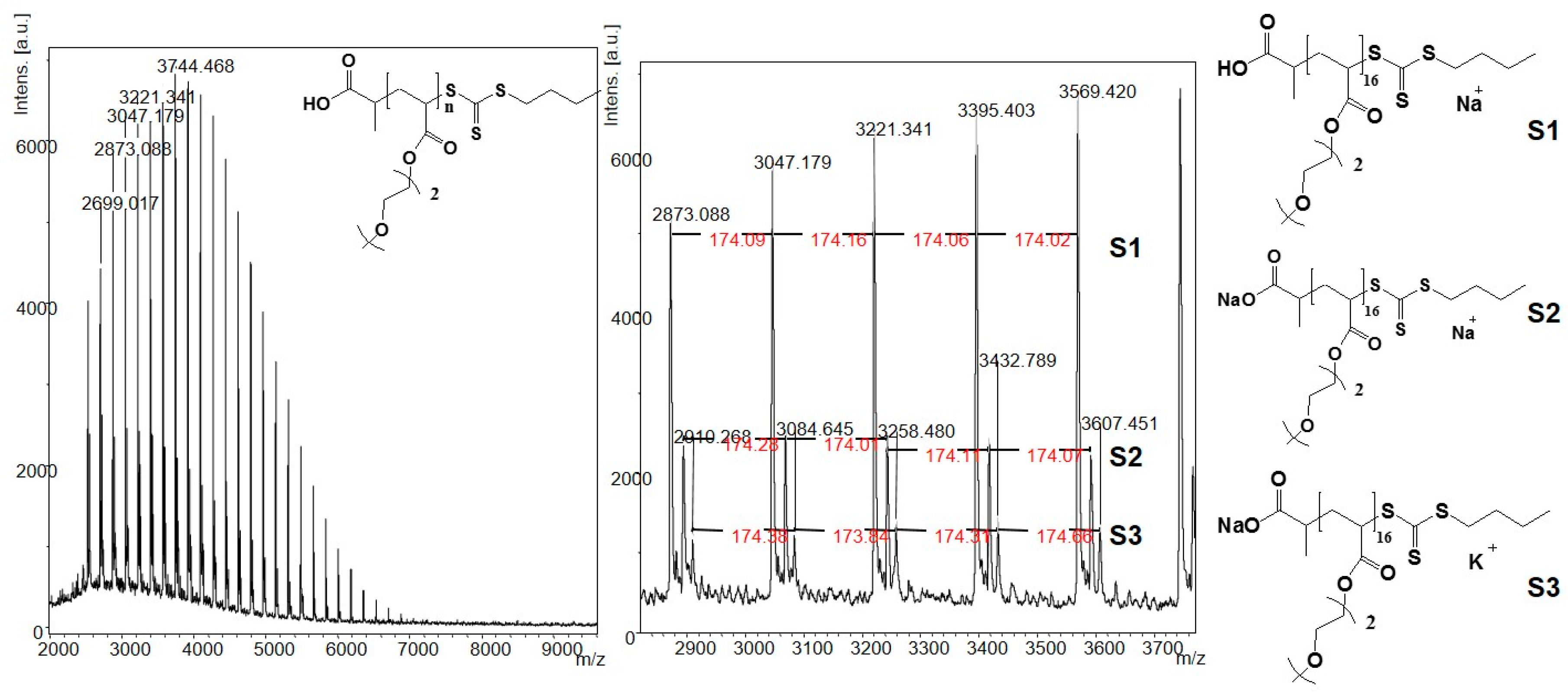
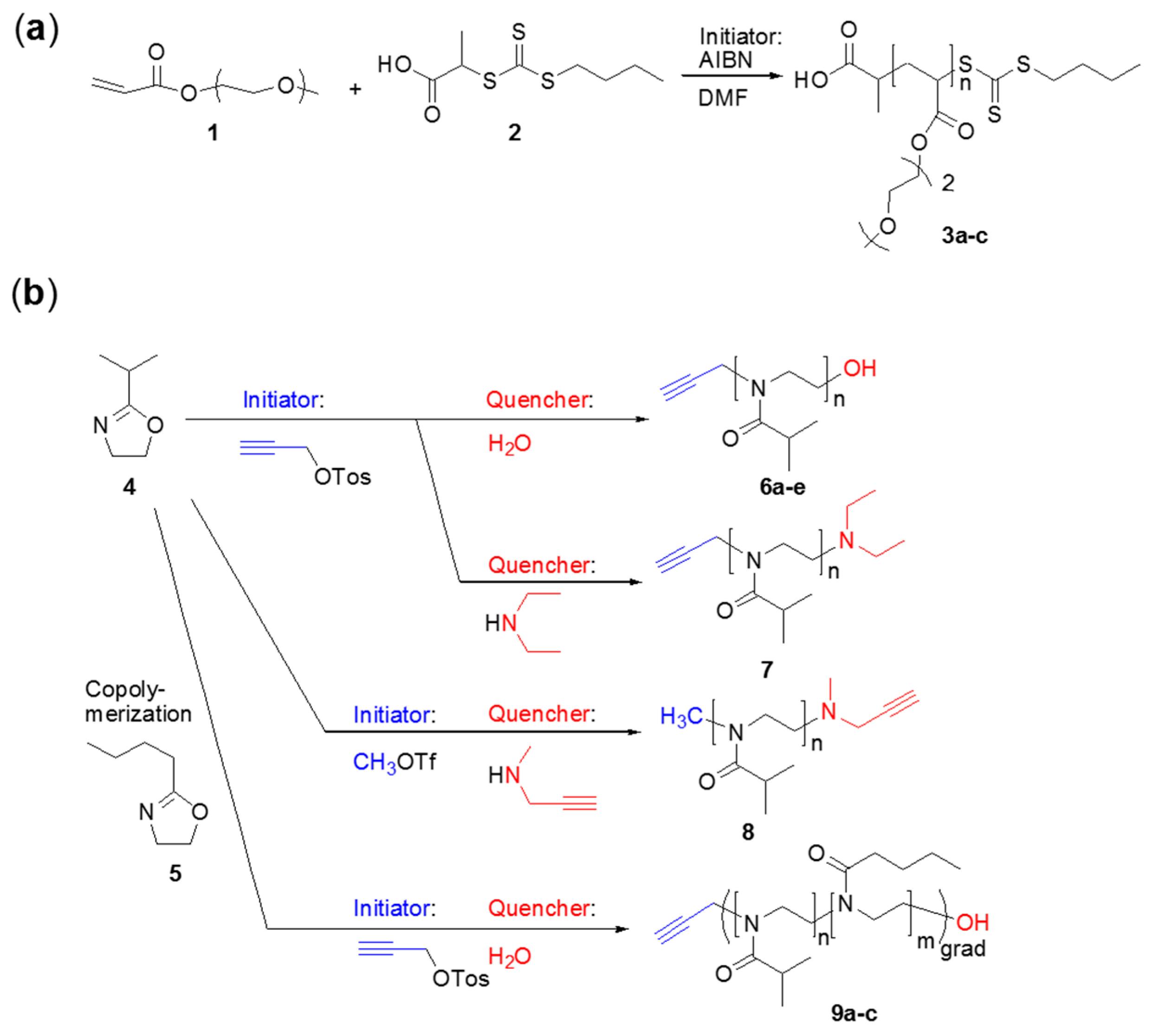
2.3. General Procedure for the Syntheses of the Poly(methoxy di(ethylene glycol)acrylates) 3
2.4. Syntheses of the Poly(oxazolines) 6–9
2.5. General Procedure for the Syntheses of the Poly(oxazolines) 6–8
2.6. General Procedure for the Syntheses of Poly(2-isopropyl-2-oxazoline-grad-2-n-butyl-2-oxazoline) 9
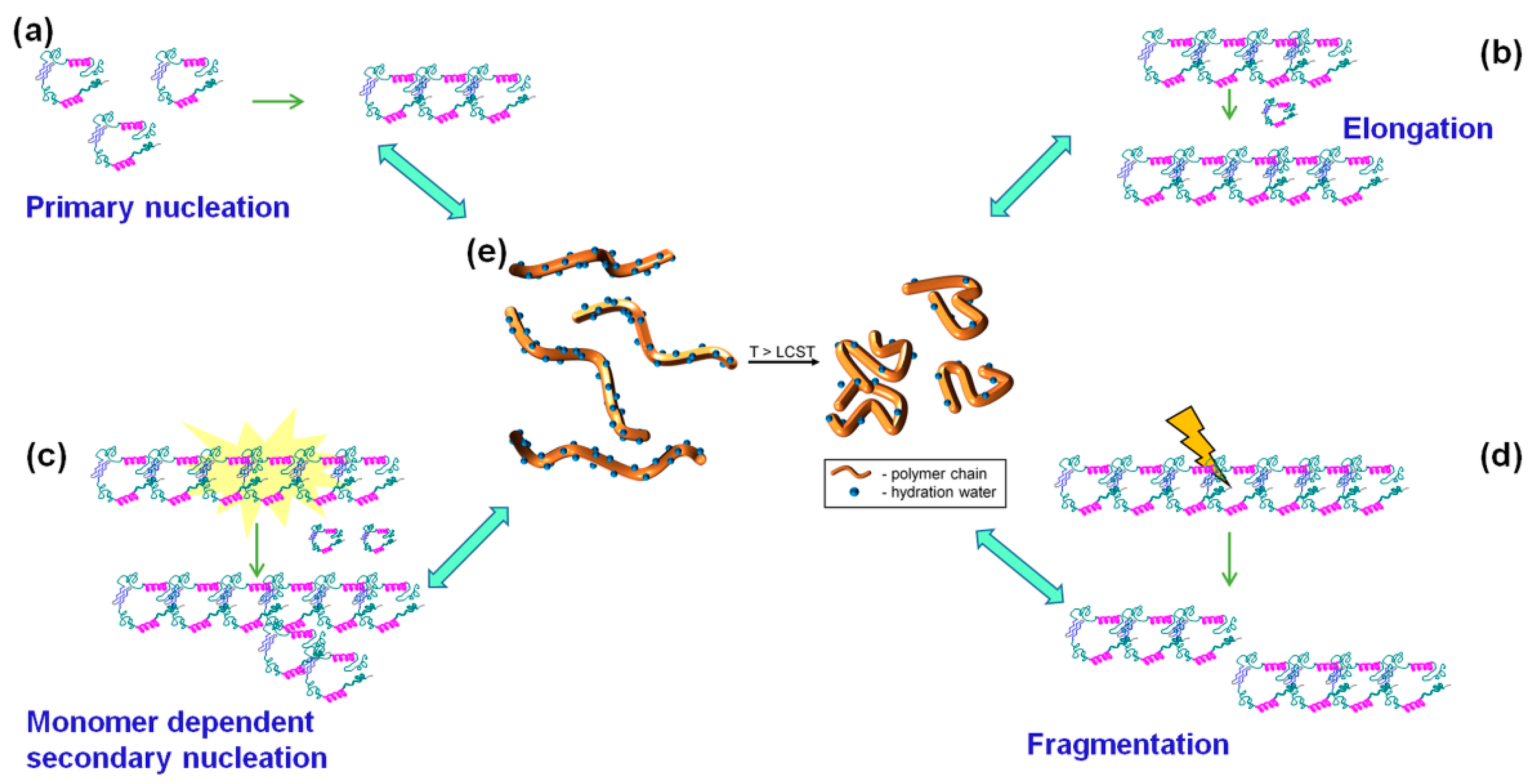
3. Results and Discussion
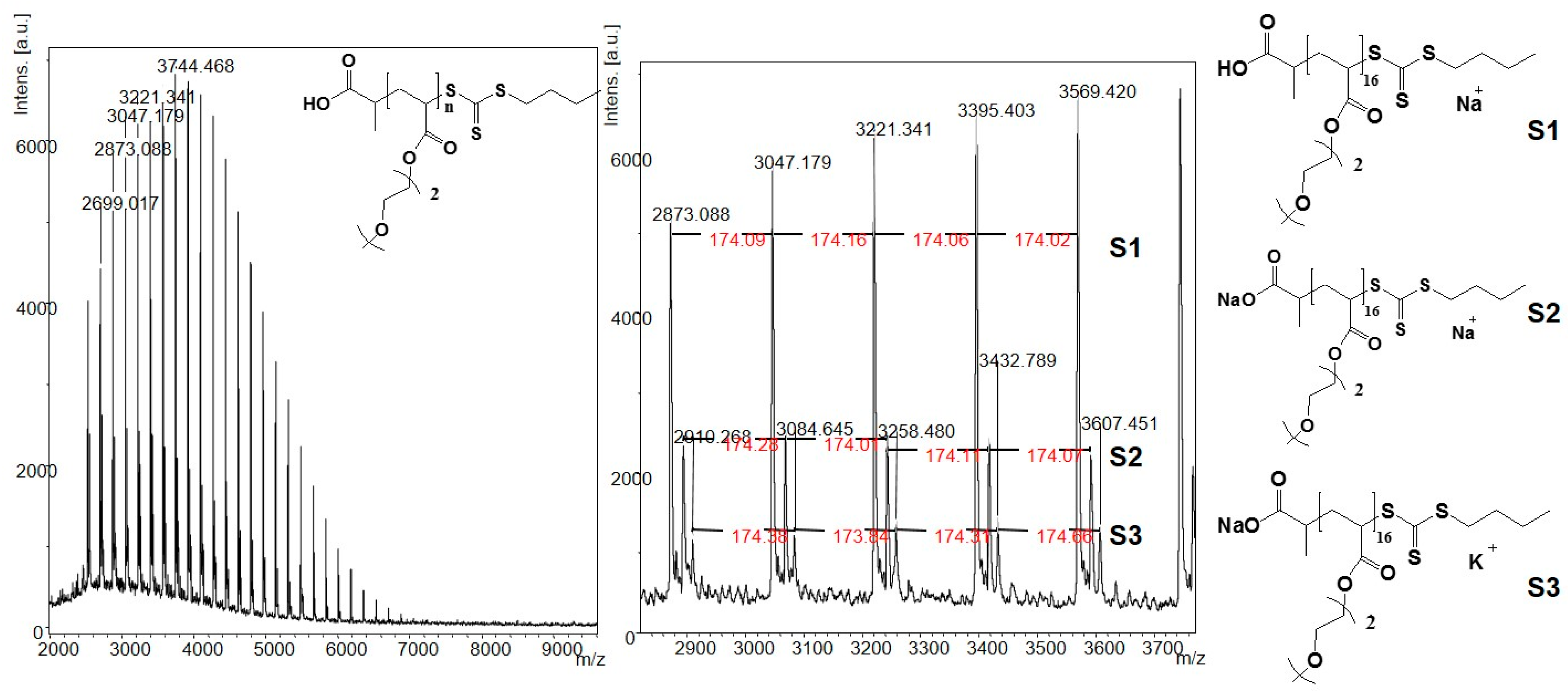
3.1. Syntheses of the Polymers
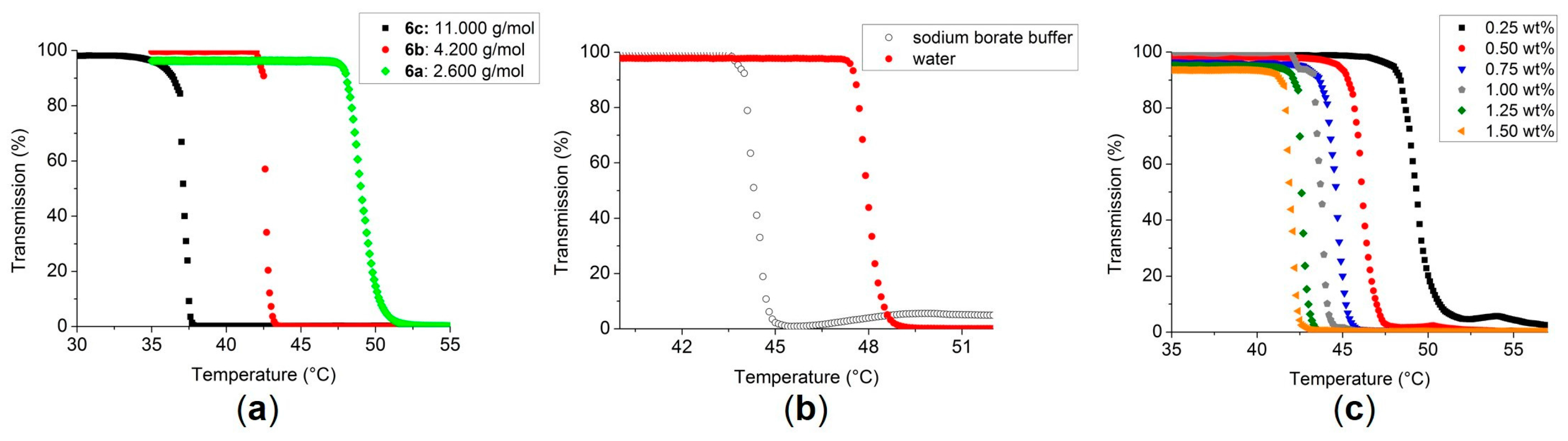
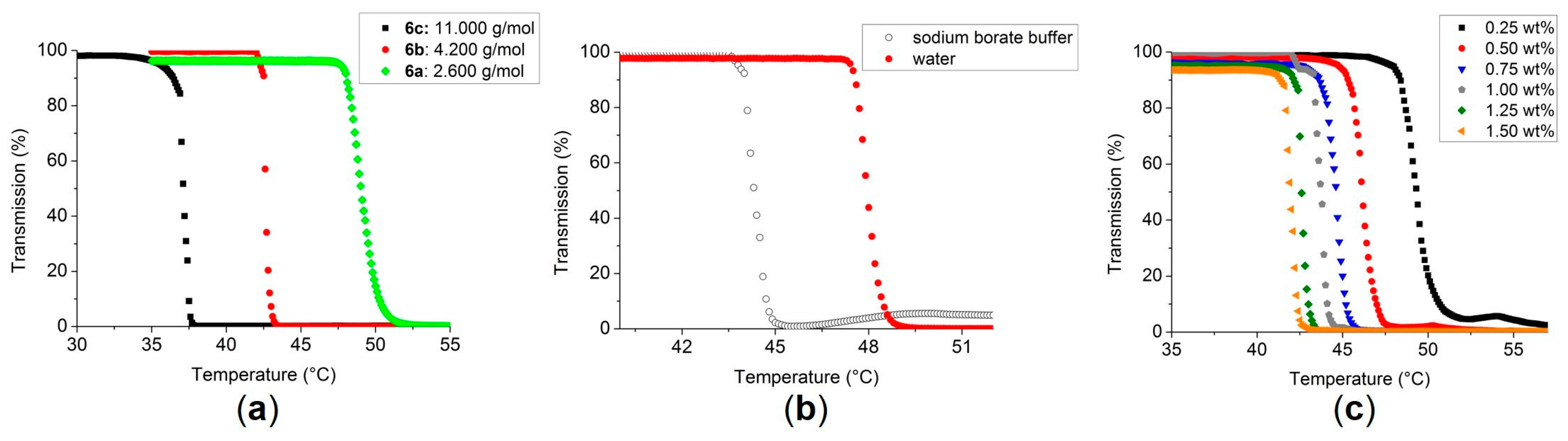
3.2. Turbidimetry of the Polymers 3, 6–9
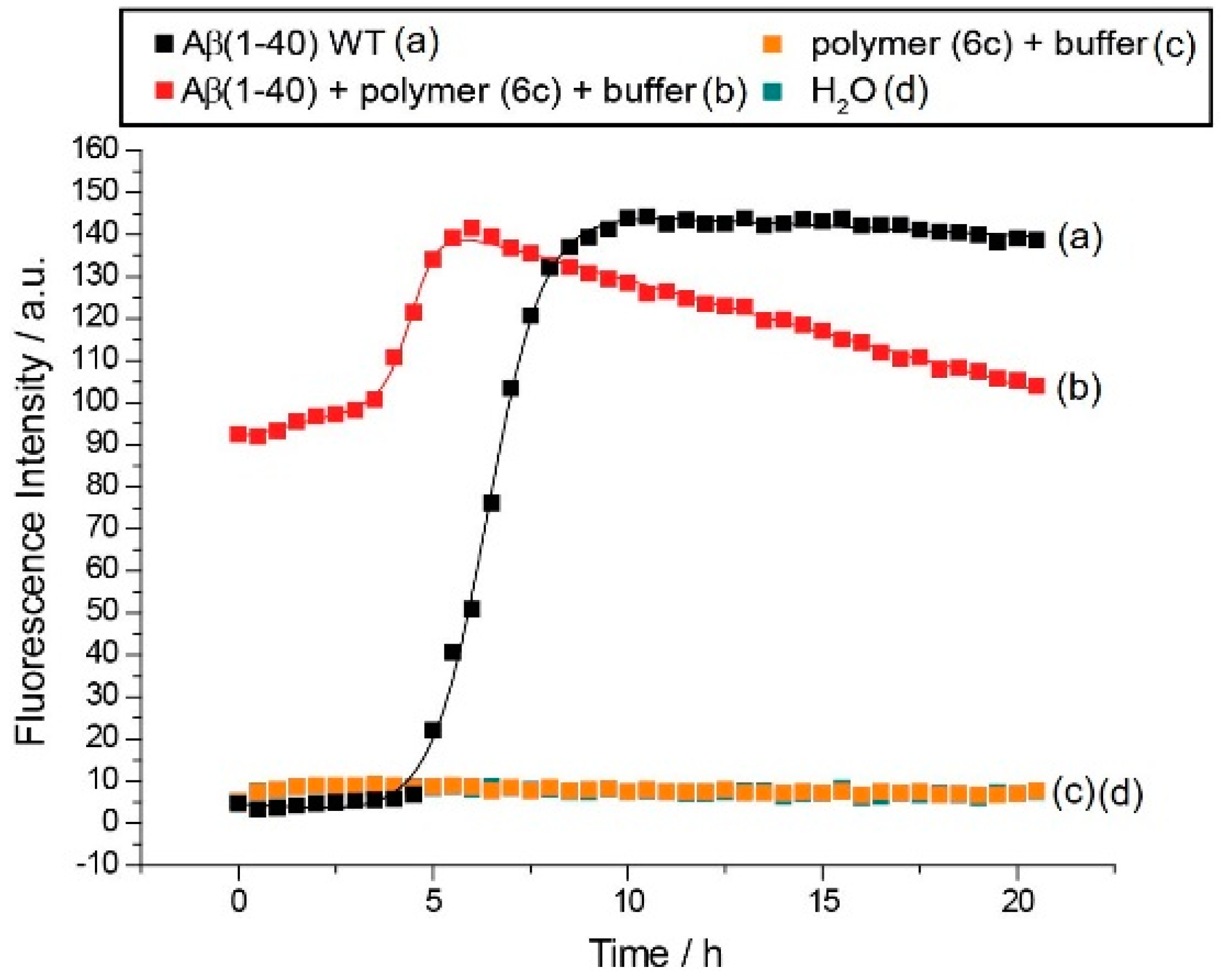
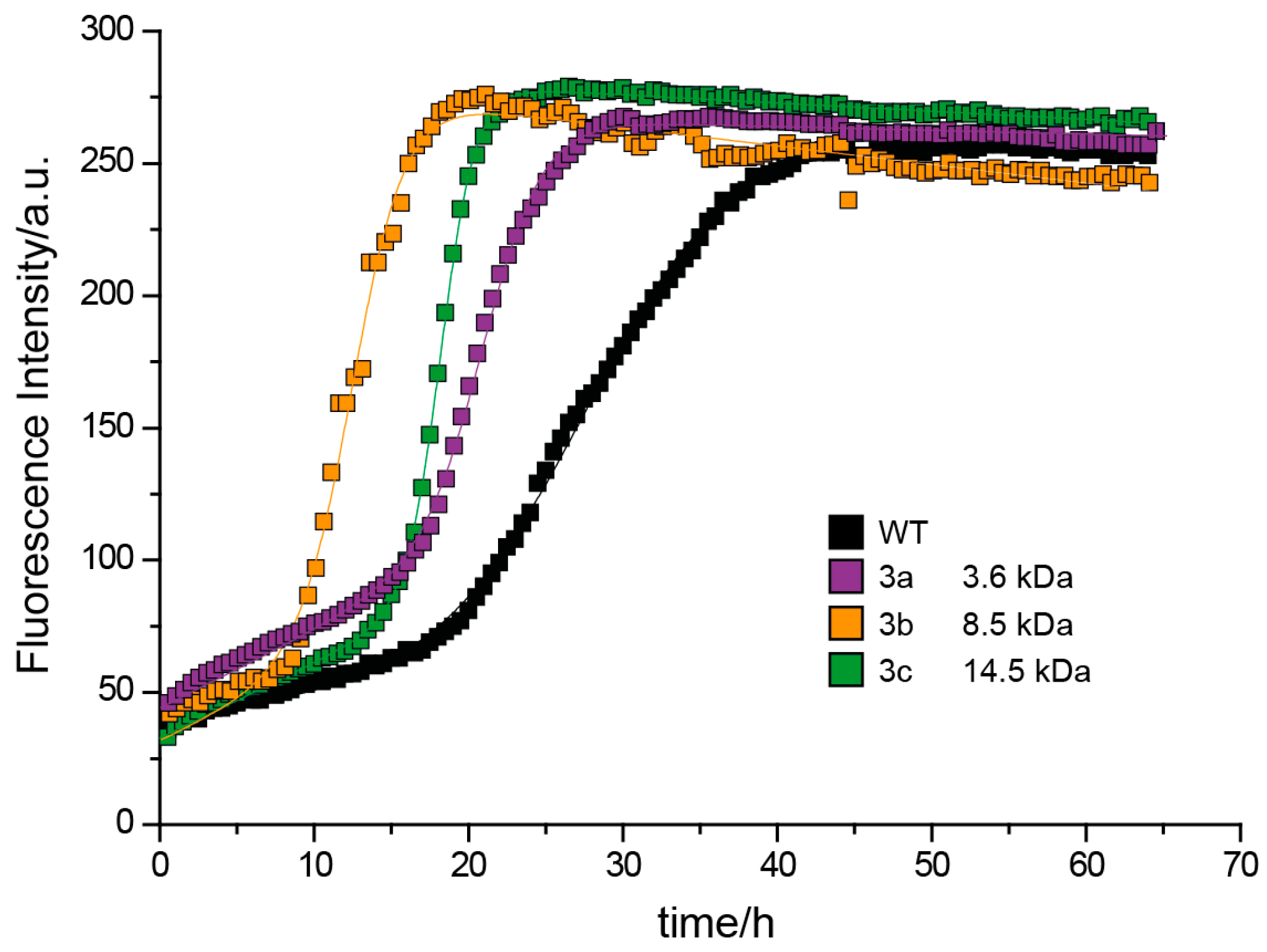
3.3. Fibrillation of Aβ40 in the Presence of Polymers 6 and 3
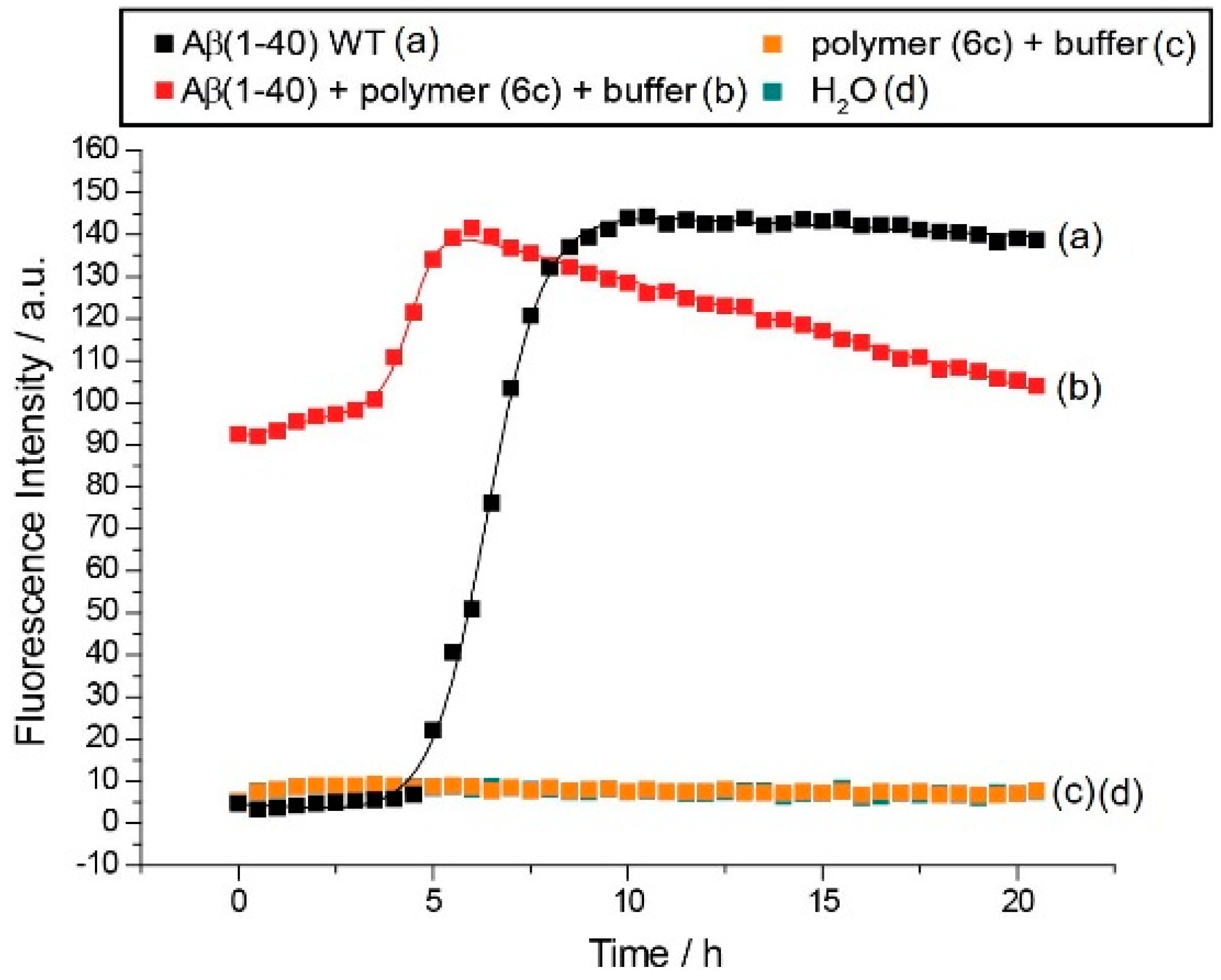
3.3.1. Effect of Poly(oxazolines) on Aβ40 Aggregation
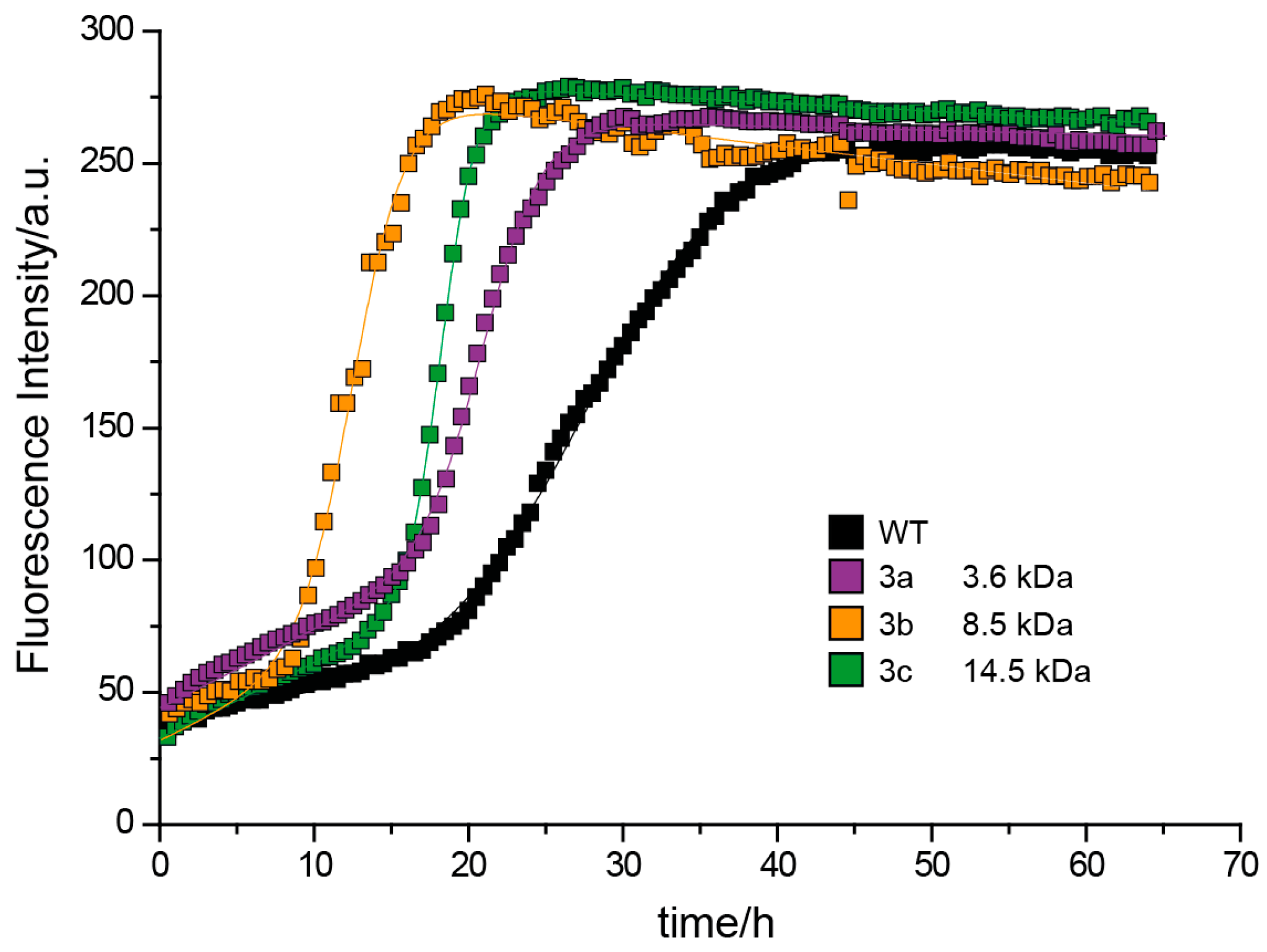
3.3.2. Effect of Poly(methoxy di(ethylene glycol)acrylates) on Aβ40 Fibrillation
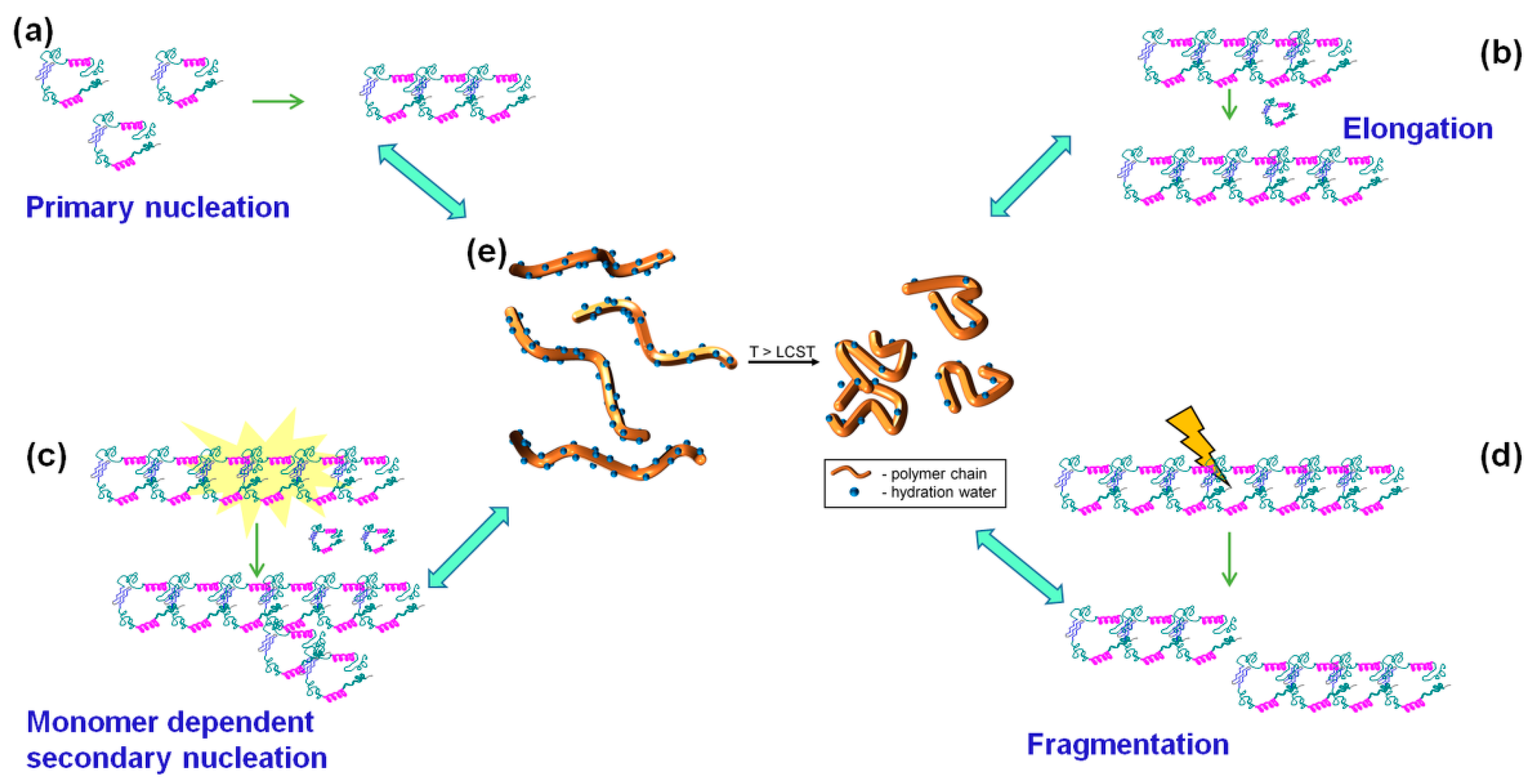
3.3.3. Discussion of Aβ40 Aggregation Experiments
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. Peptide fibrillization. Angew. Chem. Int. Ed. 2007, 46, 8128–8147. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Sgarbossa, A. Natural biomolecules and protein aggregation: Emerging strategies against amyloidogenesis. Int. J. Mol. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Cukalevski, R.; Frohm, B.; Knowles, T.P.J.; Linse, S. Quantification of the concentration of aβ42 propagons during the lag phase by an amyloid chain reaction assay. J. Am. Chem. Soc. 2014, 136, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.A.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [PubMed]
- Sackler, A.M. Self-Perpetuating Structural States in Biology, Disease, and Genetics; National Academy of Sciences: Washington, DC, USA, 2002. [Google Scholar]
- Adamcik, J.; Mezzenga, R. Proteins fibrils from a polymer physics perspective. Macromolecules 2012, 45, 1137–1150. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Morgado, I.; Rothemund, S.; Huster, D.; Fändrich, M. Solid-state NMR spectroscopic investigation of Aβ protofibrils: Implication of a β-sheet remodeling upon maturation into terminal amyloid fibrils. Angew. Chem. Int. Ed. 2011, 50, 2837–2840. [Google Scholar] [CrossRef] [PubMed]
- Chimon, S.; Shaibat, M.A.; Jones, C.R.; Calero, D.C.; Aizezi, B.; Ishii, Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007, 14, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Klajnert, B.; Cortijo-Arellano, M.; Cladera, J.; Bryszewska, M. Influence of dendrimer’s structure on its activity against amyloid fibril formation. Biochem. Biophys. Res. Commun. 2006, 345, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of sickle hemoglobin polymerization: I. Studies using temperature-jump and laser photolysis techniques. J. Mol. Biol. 1985, 183, 591–610. [Google Scholar] [CrossRef]
- Ferrone, F.A.; Hofrichter, J.; Eaton, W.A. Kinetics of sickle hemoglobin polymerization: II. A double nucleation mechanism. J. Mol. Biol. 1985, 183, 611–631. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Arosio, P.; Presto, J.; Kurudenkandy, F.R.; Biverstål, H.; Dolfe, L.; Dunning, C.; Yang, X.; Frohm, B.; Vendruscolo, M.; et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 2015, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef] [PubMed]
- Härd, T.; Lendel, C. Inhibition of amyloid formation. J. Mol. Biol. 2012, 421, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Grönwall, C.; Jonsson, A.; Ståhl, S.; Härd, T. Stabilization of a β-hairpin in monomeric alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA 2008, 105, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Madine, J.; Doig, A.J.; Middleton, D.A. Design of an n-methylated peptide inhibitor of α-synuclein aggregation guided by solid-state NMR. J. Am. Chem. Soc. 2008, 130, 7873–7881. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, H.F.; Viles, J.H. Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: Implications for Alzheimer disease. J. Biol. Chem. 2012, 287, 28163–28168. [Google Scholar] [CrossRef] [PubMed]
- Assarsson, A.; Hellstrand, E.; Cabaleiro-Lago, C.; Linse, S. Charge dependent retardation of amyloid β aggregation by hydrophilic proteins. ACS Chem. Neurosci. 2014, 5, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V.; Gendrault, J.-L.; Wolff, C.-M. Poly-l-lysine dissolves fibrillar aggregation of the alzheimer β-amyloid peptide in vitro. Biochem. Biophys. Res. Commun. 2002, 291, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Klajnert, B.; Cladera, J.; Bryszewska, M. Molecular interactions of dendrimers with amyloid peptides: pH dependence. Biomacromolecules 2006, 7, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, P.M.H.; Boas, U.; Otzen, D.E. Dendrimer effects on peptide and protein fibrillation. Macromol. Biosci. 2007, 7, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro-Lago, C.; Quinlan-Pluck, F.; Lynch, I.; Lindman, S.; Minogue, A.M.; Thulin, E.; Walsh, D.M.; Dawson, K.A.; Linse, S. Inhibition of amyloid β protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc. 2008, 130, 15437–15443. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro-Lago, C.; Quinlan-Pluck, F.; Lynch, I.; Dawson, K.A.; Linse, S. Dual effect of amino modified polystyrene nanoparticles on amyloid β protein fibrillation. ACS Chem. Neurosci. 2010, 1, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Skaat, H.; Chen, R.; Grinberg, I.; Margel, S. Engineered polymer nanoparticles containing hydrophobic dipeptide for inhibition of amyloid-β fibrillation. Biomacromolecules 2012, 13, 2662–2670. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Sparr, E.; Linse, S. Retardation of Aβ fibril formation by phospholipid vesicles depends on membrane phase behavior. Biophys. J. 2010, 98, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Jao, S.-C.; Ma, K.; Zagorski, M.G. Solution structures of micelle-bound amyloid β-(1-40) and β-(1-42) peptides of Alzheimer’s disease. J. Mol. Biol. 1999, 285, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Moore, B.D.; Reed, D.K.; Sonoda, L.K.; Bridges, A.W.; Conboy, E.; Hartigan, D.; Rosenberry, T.L. Amyloid-β(1-42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry 2007, 46, 12451–12462. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-M.; Lin, T.-L.; Jeng, U.S.; Huang, Z.-H.; Huang, Y.-S. Aggregation structure of alzheimer amyloid-β(1–40) peptide with sodium dodecyl sulfate as revealed by small-angle X-ray and neutron scattering. Soft Matter 2009, 5, 3913–3919. [Google Scholar] [CrossRef]
- Abelein, A.; Kaspersen, J.D.; Nielsen, S.B.; Jensen, G.V.; Christiansen, G.; Pedersen, J.S.; Danielsson, J.; Otzen, D.E.; Gräslund, A. Formation of dynamic soluble surfactant-induced amyloid β peptide aggregation intermediates. J. Biol. Chem. 2013, 288, 23518–23528. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro-Lago, C.; Szczepankiewicz, O.; Linse, S. The effect of nanoparticles on amyloid aggregation depends on the protein stability and intrinsic aggregation rate. Langmuir 2012, 28, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Palmal, S.; Jana, N.R.; Jana, N.R. Inhibition of amyloid fibril growth by nanoparticle coated with histidine-based polymer. J. Phys. Chem. C 2014, 118, 21630–21638. [Google Scholar]
- Assarsson, A.; Linse, S.; Cabaleiro-Lago, C. Effects of polyamino acids and polyelectrolytes on amyloid β fibril formation. Langmuir 2014, 30, 8812–8818. [Google Scholar] [CrossRef] [PubMed]
- Hocine, S.; Li, M.-H. Thermoresponsive self-assembled polymer colloids in water. Soft Matter 2013, 9, 5839–5861. [Google Scholar] [CrossRef]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Fujishige, S.; Kubota, K.; Ando, I. Phase transition of aqueous solutions of poly(N-isopropylacrylamide) and poly(N-isopropylmethacrylamide). J. Phys. Chem. 1989, 93, 3311–3313. [Google Scholar] [CrossRef]
- Graziano, G. On the temperature-induced coil to globule transition of poly-N-isopropylacrylamide in dilute aqueous solutions. Int. J. Biol. Macromol. 2000, 27, 89–97. [Google Scholar] [CrossRef]
- Lin, P.; Clash, C.; Pearce, E.M.; Kwei, T.K.; Aponte, M.A. Solubility and miscibility of poly(ethyl oxazoline). J. Polym. Sci. B Polym. Phys. 1988, 26, 603–619. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. A novel thermo-sensitive polymer. Poly(2-iso-propyl-2-oxazoline). Chem. Lett. 1992, 21, 1643–1646. [Google Scholar] [CrossRef]
- Park, J.-S.; Kataoka, K. Comprehensive and accurate control of thermosensitivity of poly(2-alkyl-2-oxazoline)s via well-defined gradient or random copolymerization. Macromolecules 2007, 40, 3599–3609. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.S.; Hudson, S.M. Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Aseyev, V.; Tenhu, H.; Winnik, F.M. Non-ionic thermoresponsive polymers in water. Adv. Polym. Sci. 2011, 242, 29–89. [Google Scholar]
- Hoogenboom, R.; Thijs, H.M.L.; Jochems, M.J.H.C.; van Lankvelt, B.M.; Fijten, M.W.M.; Schubert, U.S. Tuning the lcst of poly(2-oxazoline)s by varying composition and molecular weight: Alternatives to poly(N-isopropylacrylamide)? Chem. Commun. 2008, 5758–5760. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Jordan, R. Modulation of the lower critical solution temperature of 2-alkyl-2-oxazoline copolymers. Colloid Polym. Sci. 2008, 286, 395–402. [Google Scholar] [CrossRef]
- Glassner, M.; Lava, K.; de la Rosa, V.R.; Hoogenboom, R. Tuning the lcst of poly(2-cyclopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline. J. Polym. Sci. A Polym. Chem. 2014, 52, 3118–3122. [Google Scholar] [CrossRef]
- Xia, Y.; Burke, N.A.D.; Stöver, H.D.H. End group effect on the thermal response of narrow-disperse poly(N-isopropylacrylamide) prepared by atom transfer radical polymerization. Macromolecules 2006, 39, 2275–2283. [Google Scholar] [CrossRef]
- Huber, S.; Hutter, N.; Jordan, R. Effect of end group polarity upon the lower critical solution temperature of poly(2-isopropyl-2-oxazoline). Colloid Polym. Sci. 2008, 286, 1653–1661. [Google Scholar] [CrossRef]
- Ishizone, T.; Han, S.; Okuyama, S.; Nakahama, S. Synthesis of water-soluble polymethacrylates by living anionic polymerization of trialkylsilyl-protected oligo(ethylene glycol) methacrylates. Macromolecules 2003, 36, 42–49. [Google Scholar] [CrossRef]
- Adler, J.; Scheidt, H.A.; Kruger, M.; Thomas, L.; Huster, D. Local interactions influence the fibrillation kinetics, structure and dynamics of Aβ(1–40) but leave the general fibril structure unchanged. Phys. Chem. Chem. Phys. 2014, 16, 7461–7471. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism†. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Bolder, S.G.; Sagis, L.M.C.; Venema, P.; van der Linden, E. Thioflavin t and birefringence assays to determine the conversion of proteins into fibrils. Langmuir 2007, 23, 4144–4147. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Akiyama, Y.; Winnik, F.M.; Kataoka, K. Versatile synthesis of end-functionalized thermosensitive poly(2-isopropyl-2-oxazolines). Macromolecules 2004, 37, 6786–6792. [Google Scholar] [CrossRef]
- Witte, H.; Seeliger, W. Cyclische imidsäureester aus nitrilen und aminoalkoholen. Justus Liebigs Ann. Chem. 1974, 1974, 996–1009. [Google Scholar] [CrossRef]
- Zhong, Q.; Wang, W.; Adelsberger, J.; Golosova, A.; Bivigou Koumba, A.M.; Laschewsky, A.; Funari, S.S.; Perlich, J.; Roth, S.V.; Papadakis, C.M.; et al. Collapse transition in thin films of poly(methoxydiethylenglycol acrylate). Colloid Polym. Sci. 2011, 289, 569–581. [Google Scholar] [CrossRef]
- Vanparijs, N.; Maji, S.; Louage, B.; Voorhaar, L.; Laplace, D.; Zhang, Q.; Shi, Y.; Hennink, W.E.; Hoogenboom, R.; de Geest, B.G. Polymer-protein conjugation via a ‘grafting to’ approach—A comparative study of the performance of protein-reactive raft chain transfer agents. Polym. Chem. 2015, 6, 5602–5614. [Google Scholar] [CrossRef]
- Hua, F.; Jiang, X.; Li, D.; Zhao, B. Well-defined thermosensitive, water-soluble polyacrylates and polystyrenics with short pendant oligo(ethylene glycol) groups synthesized by nitroxide-mediated radical polymerization. J. Polym. Sci. A Polym. Chem. 2006, 44, 2454–2467. [Google Scholar] [CrossRef]
- Ferguson, C.J.; Hughes, R.J.; Nguyen, D.; Pham, B.T.T.; Gilbert, R.G.; Serelis, A.K.; Such, C.H.; Hawkett, B.S. Ab initio emulsion polymerization by raft-controlled self-assembly. Macromolecules 2005, 38, 2191–2204. [Google Scholar] [CrossRef]
- Zhong, Q.; Metwalli, E.; Rawolle, M.; Kaune, G.; Bivigou-Koumba, A.M.; Laschewsky, A.; Papadakis, C.M.; Cubitt, R.; Wang, J.; Müller-Buschbaum, P. Influence of hydrophobic polystyrene blocks on the rehydration of polystyrene-block-poly(methoxy diethylene glycol acrylate)-block-polystyrene films investigated by in situ neutron reflectivity. Macromolecules 2016, 49, 317–326. [Google Scholar] [CrossRef]
- Miasnikova, A.; Laschewsky, A. Influencing the phase transition temperature of poly(methoxy diethylene glycol acrylate) by molar mass, end groups, and polymer architecture. J. Polym. Sci. Part A 2012, 50, 3313–3323. [Google Scholar] [CrossRef]
- Christova, D.; Velichkova, R.; Loos, W.; Goethals, E.J.; Prez, F.D. New thermo-responsive polymer materials based on poly(2-ethyl-2-oxazoline) segments. Polymer 2003, 44, 2255–2261. [Google Scholar] [CrossRef]
- Calamai, M.; Kumita, J.R.; Mifsud, J.; Parrini, C.; Ramazzotti, M.; Ramponi, G.; Taddei, N.; Chiti, F.; Dobson, C.M. Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry 2006, 45, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Dutta, P.; Chakraborty, C.; Singh, P.K.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Mondal, M.; Mankar, S.; Das, S.; et al. Complexation of amyloid fibrils with charged conjugated polymers. Langmuir 2014, 30, 3775–3786. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | nNMR 1 (4) | nNMR 1 (5) | MNMR (g/mol) | MGPC (g/mol) | PDI | TCP (°C) (Sodium borate buffer) | TCP (°C) (H2O) |
|---|---|---|---|---|---|---|---|
| 3a | – | – | 3,600 | 2,700 | 1.1 | – 4,5 | 34.4 |
| 3b | – | – | 8,500 | 6,600 | 1.2 | 52.7 4 | 40.4 |
| 3c | – | – | 14,600 | 13,700 | 1.2 | 49.5 4 | 45.1 |
| 6a | 1 | – | 1,600 2 | 2,600 3 | 1.2 | 43.6 | 47.5 |
| 7 | 1 | – | 2,000 2 | 3,100 3 | 1.1 | 42.6 | 43.4 |
| 6b | 1 | – | 4,300 2 | 4,200 3 | 1.3 | 40.1 | 42.3 |
| 6c | 1 | – | 5,600 2 | 11,000 3 | 2.0 | 32.2 6 (36.2) 4 | 36.4 6 |
| 6d | 1 | – | 2,800 2 | 7,400 3 | 1.9 | 31.1/36.5 6 | 37.8 6 |
| 6e | 1 | – | 2,300 2 | 6,200 3 | 2.0 | 37.6 6 | 38.1 6 |
| 8 | 1 | – | 5,200 2 | 3,000 3 | 1.3 7 | 49.8 | 46.5 |
| 9a | 0.79 | 0.21 | 1,700 2 | 3,600 3 | 1.3 | 24.9 | 27.2 |
| 9b | 0.80 | 0.20 | 1,600 2 | 3,600 3 | 1.3 | 24.7 | 25.7 |
| 9c | 0.61 | 0.39 | 1,700 2 | 4,200 3 | 1.4 | 14.2 | 15.3 |
| Name | TCP (°C) | WT concentration 1 | Mn (kDa) | Polymer concentration 1 | tchar (h) | tlag (h) | Kinetic protocol |
|---|---|---|---|---|---|---|---|
| Aβ40 WT | – | 230 μM | 4.3 | 0 | 27.9 h | 18.3 h | II |
| 3c | 49.5 1 | 230 μM | 14.6 | 260 μM | 18.3 h | 15.9 h | II |
| 3b | 52.7 1 | 230 μM | 8.5 | 113 μM | 12.5 h | 9.1 h | II |
| 3a | – 1,2 | 230 μM | 3.6 | 196 μM | 21.1 h | 19.3 h | II |
| Aβ40 WT | – | 230 μM | 4.3 | 0 | 6.4 h | 4.9 h | I |
| 6c | 36.2 | 230 μM | 11.0 | 230 μM | 4.5 h | 3.7 h | I |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funtan, S.; Evgrafova, Z.; Adler, J.; Huster, D.; Binder, W.H. Amyloid Beta Aggregation in the Presence of Temperature-Sensitive Polymers. Polymers 2016, 8, 178. https://doi.org/10.3390/polym8050178
Funtan S, Evgrafova Z, Adler J, Huster D, Binder WH. Amyloid Beta Aggregation in the Presence of Temperature-Sensitive Polymers. Polymers. 2016; 8(5):178. https://doi.org/10.3390/polym8050178
Chicago/Turabian StyleFuntan, Sebastian, Zhanna Evgrafova, Juliane Adler, Daniel Huster, and Wolfgang H. Binder. 2016. "Amyloid Beta Aggregation in the Presence of Temperature-Sensitive Polymers" Polymers 8, no. 5: 178. https://doi.org/10.3390/polym8050178
APA StyleFuntan, S., Evgrafova, Z., Adler, J., Huster, D., & Binder, W. H. (2016). Amyloid Beta Aggregation in the Presence of Temperature-Sensitive Polymers. Polymers, 8(5), 178. https://doi.org/10.3390/polym8050178