
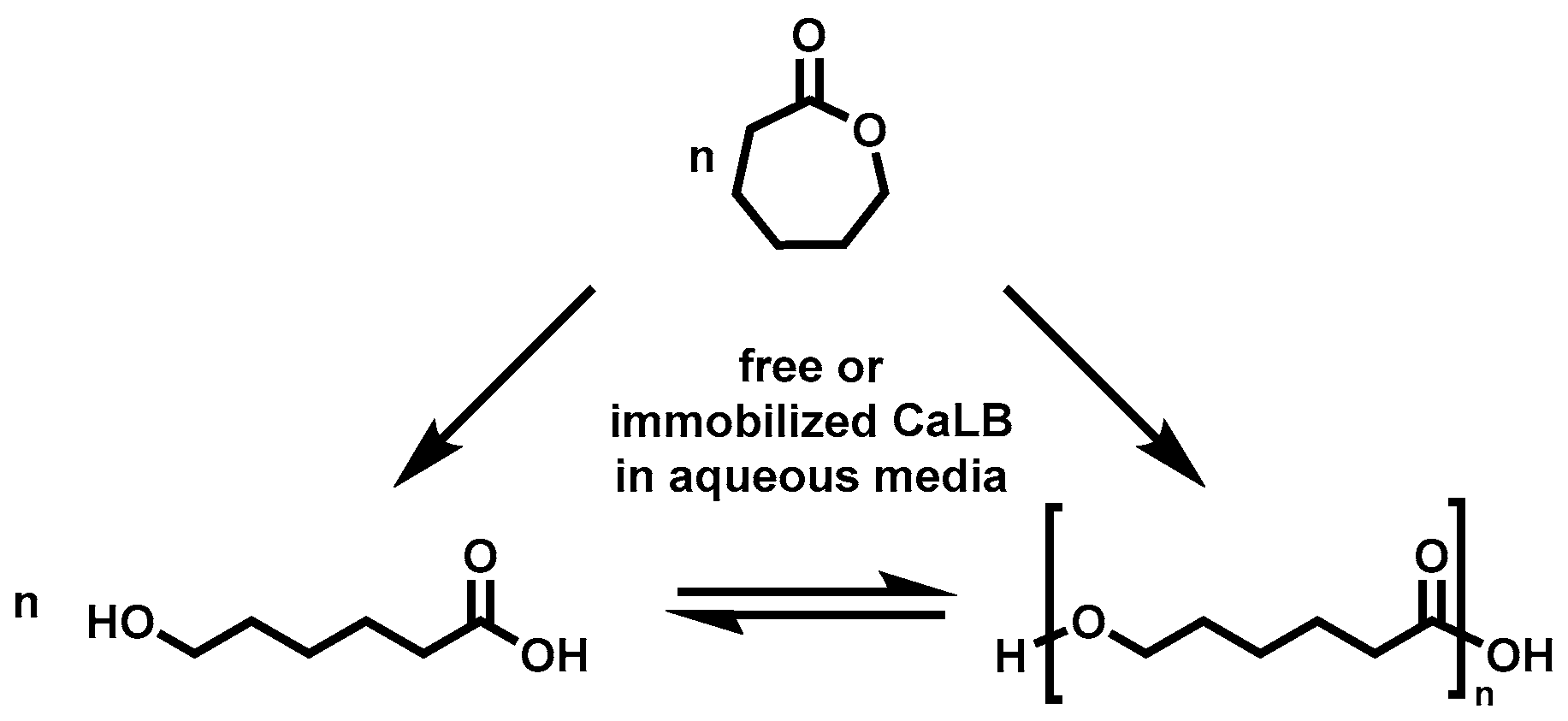
CaLB Catalyzed Conversion of ε-Caprolactone in Aqueous Medium. Part 1: Immobilization of CaLB to Microgels

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
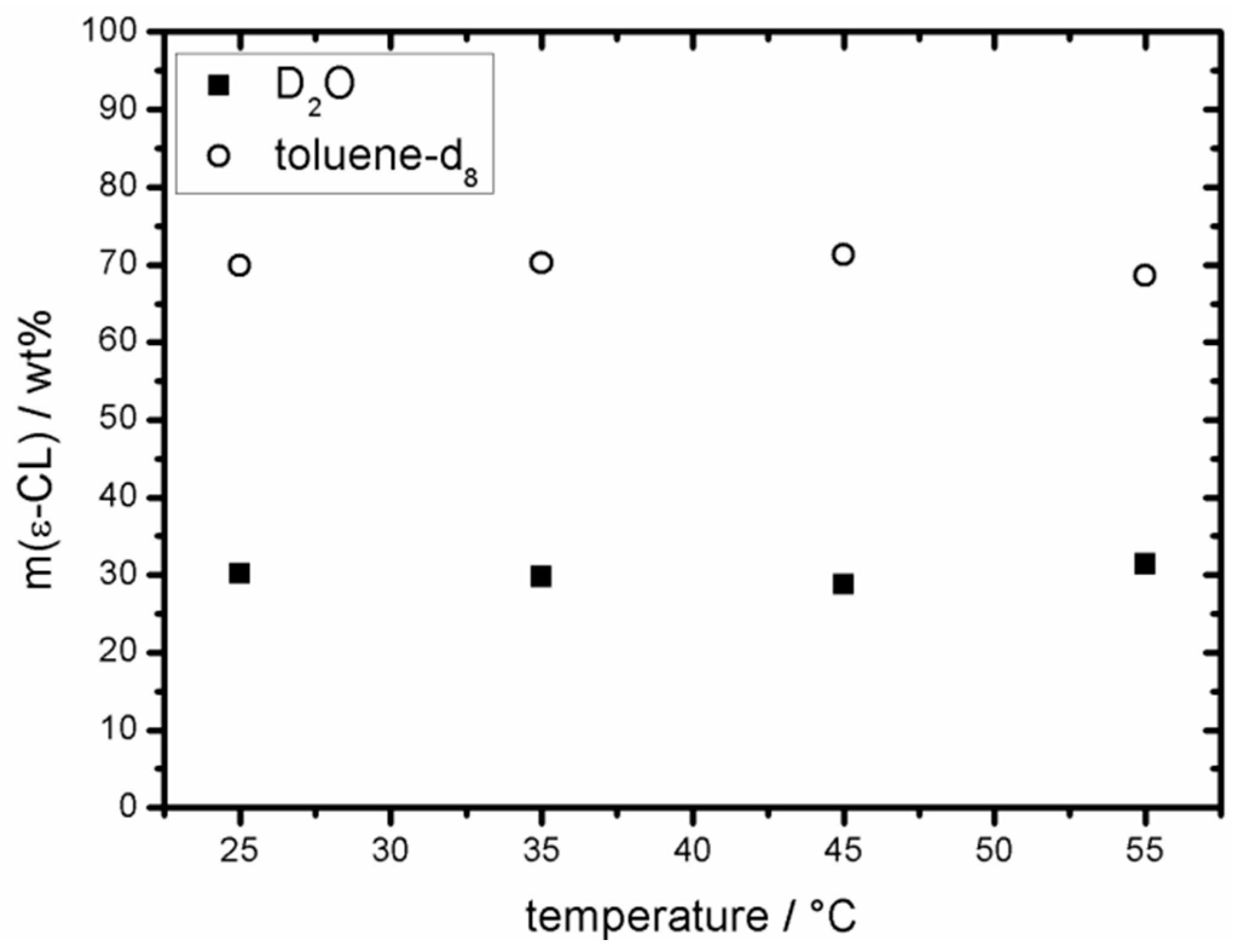
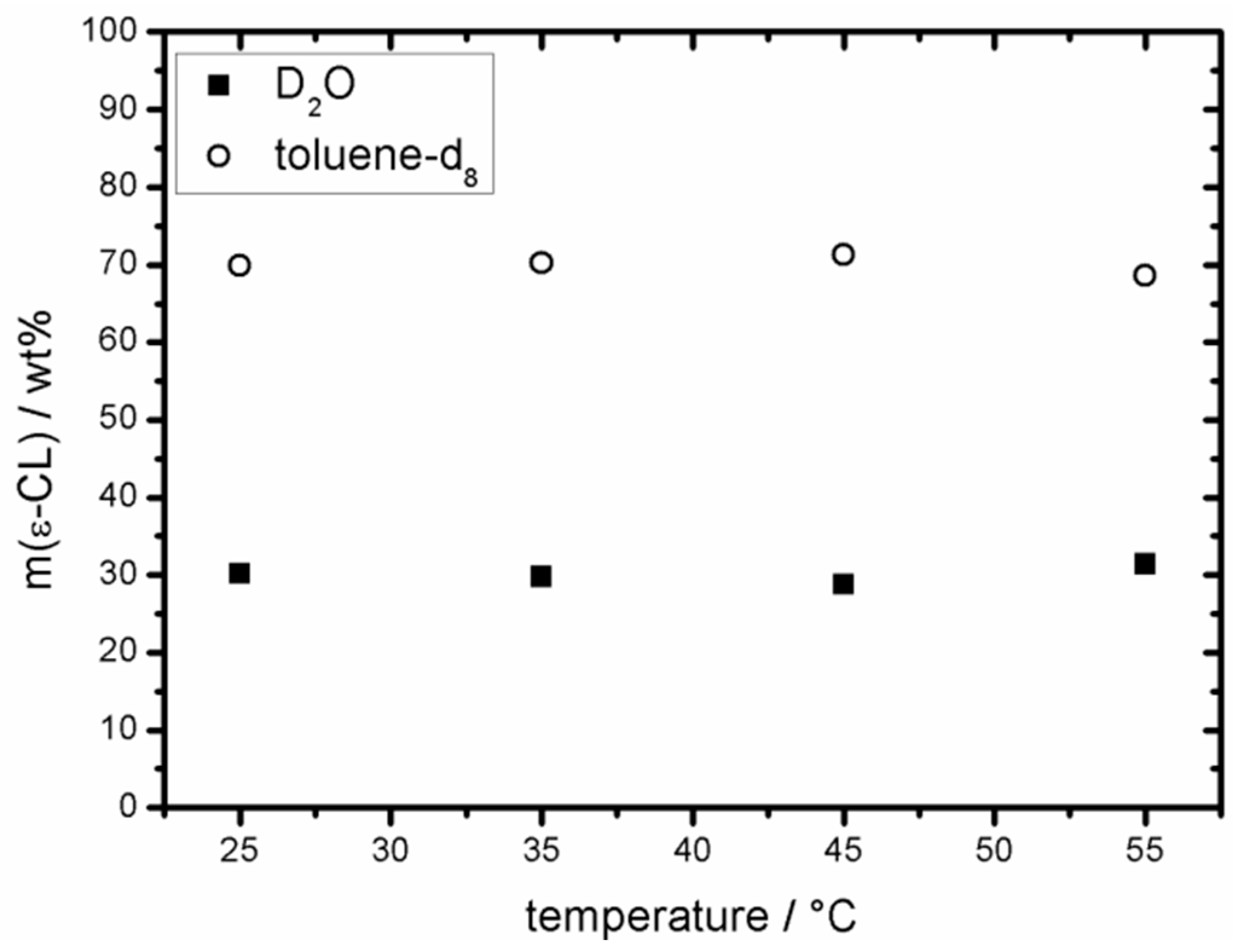
2.2. Partition Coefficient of ε-CL in D2O/Toluene-d8
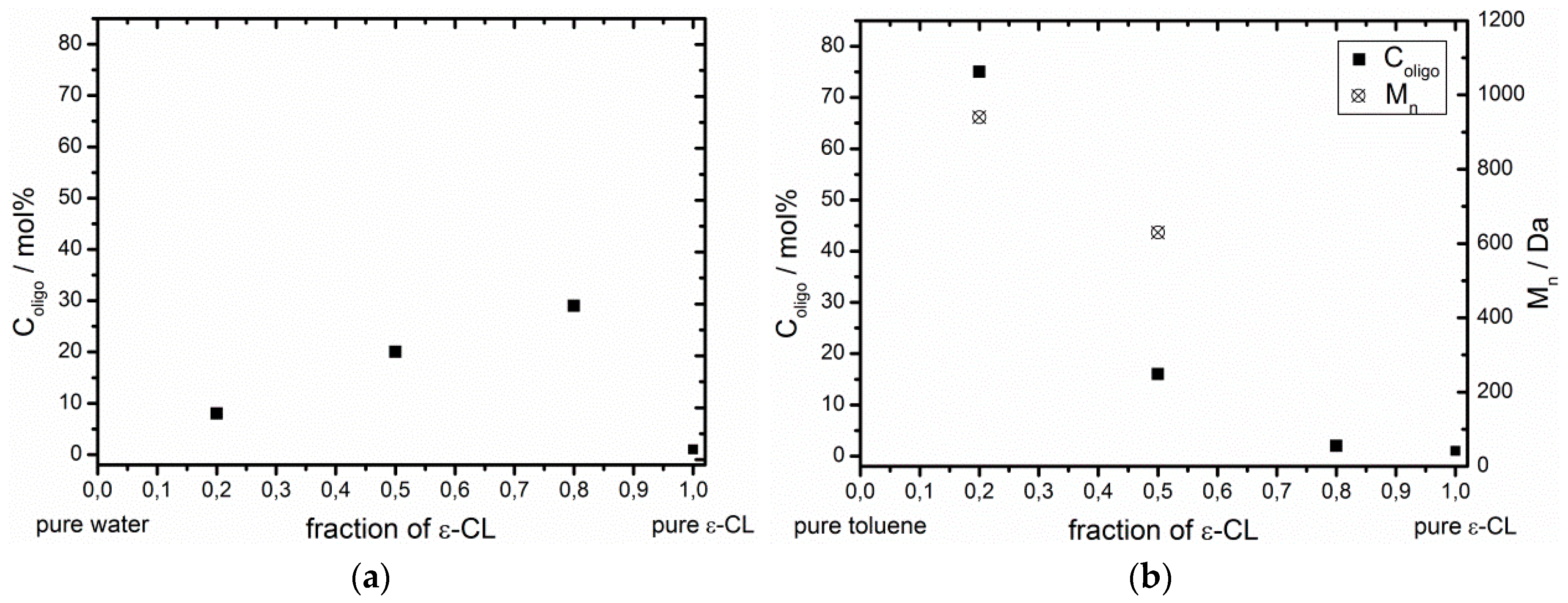
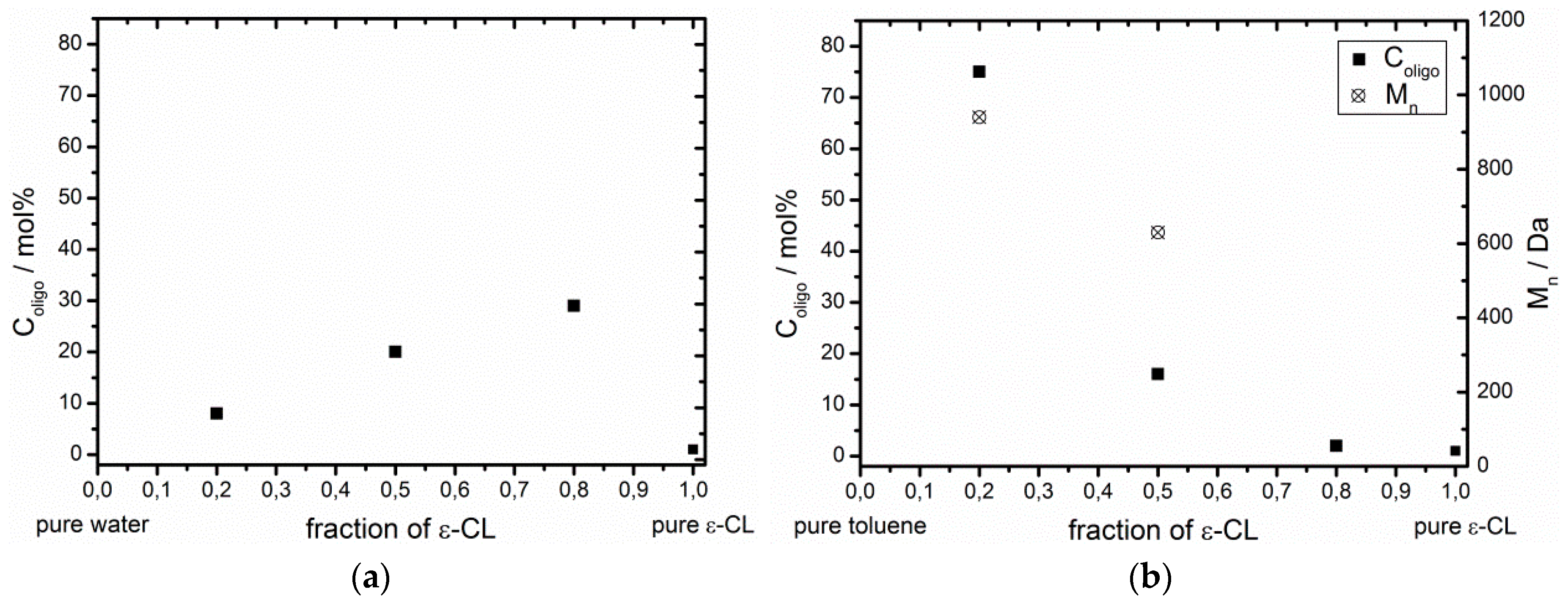
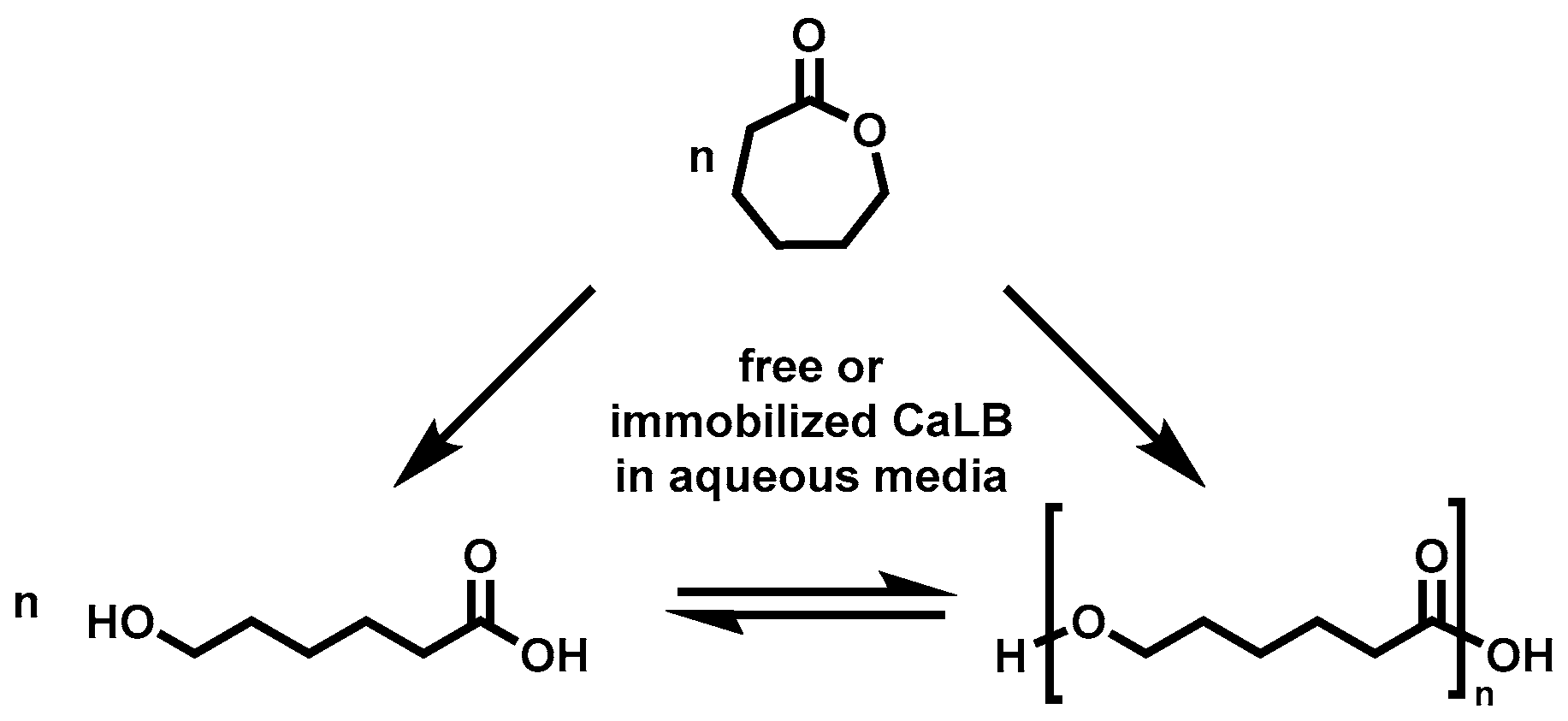
2.3. Enzymatic Polymerization of ε-CL in Water, Toluene and Bulk
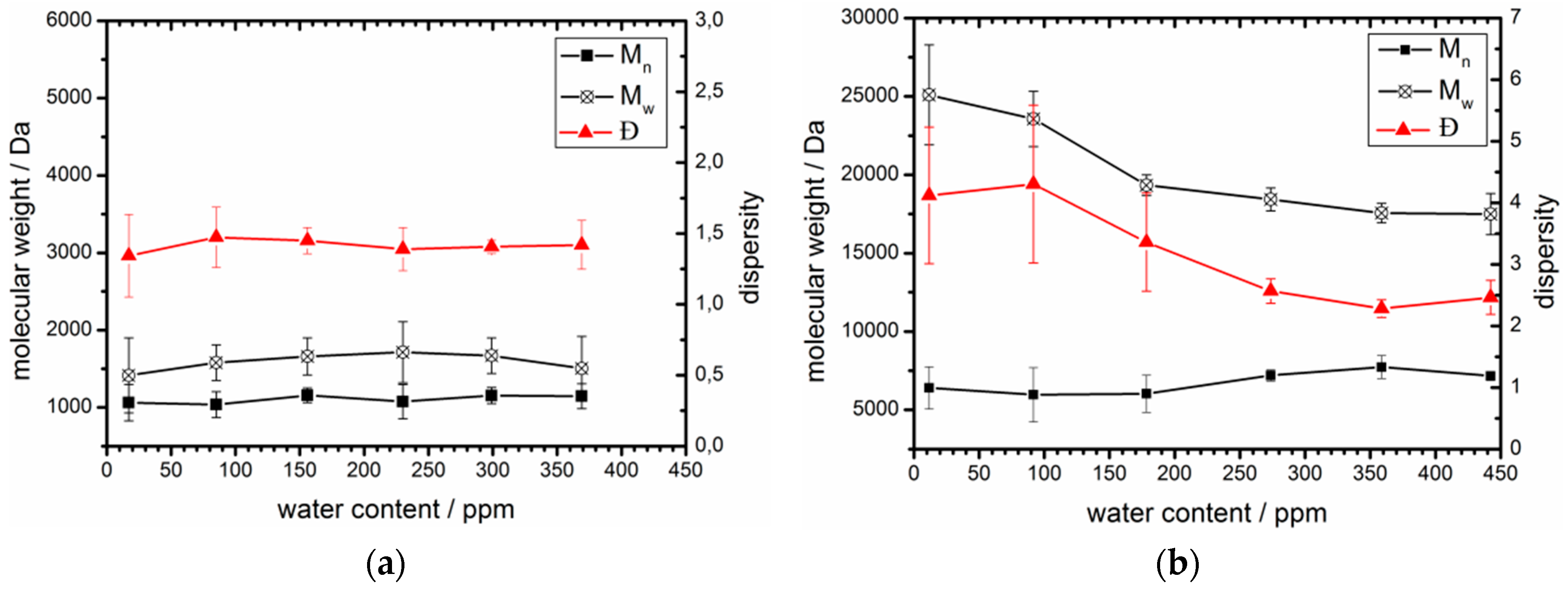
2.4. Enzymatic Polymerization of ε-CL in Toluene with Varying Water Concentrations
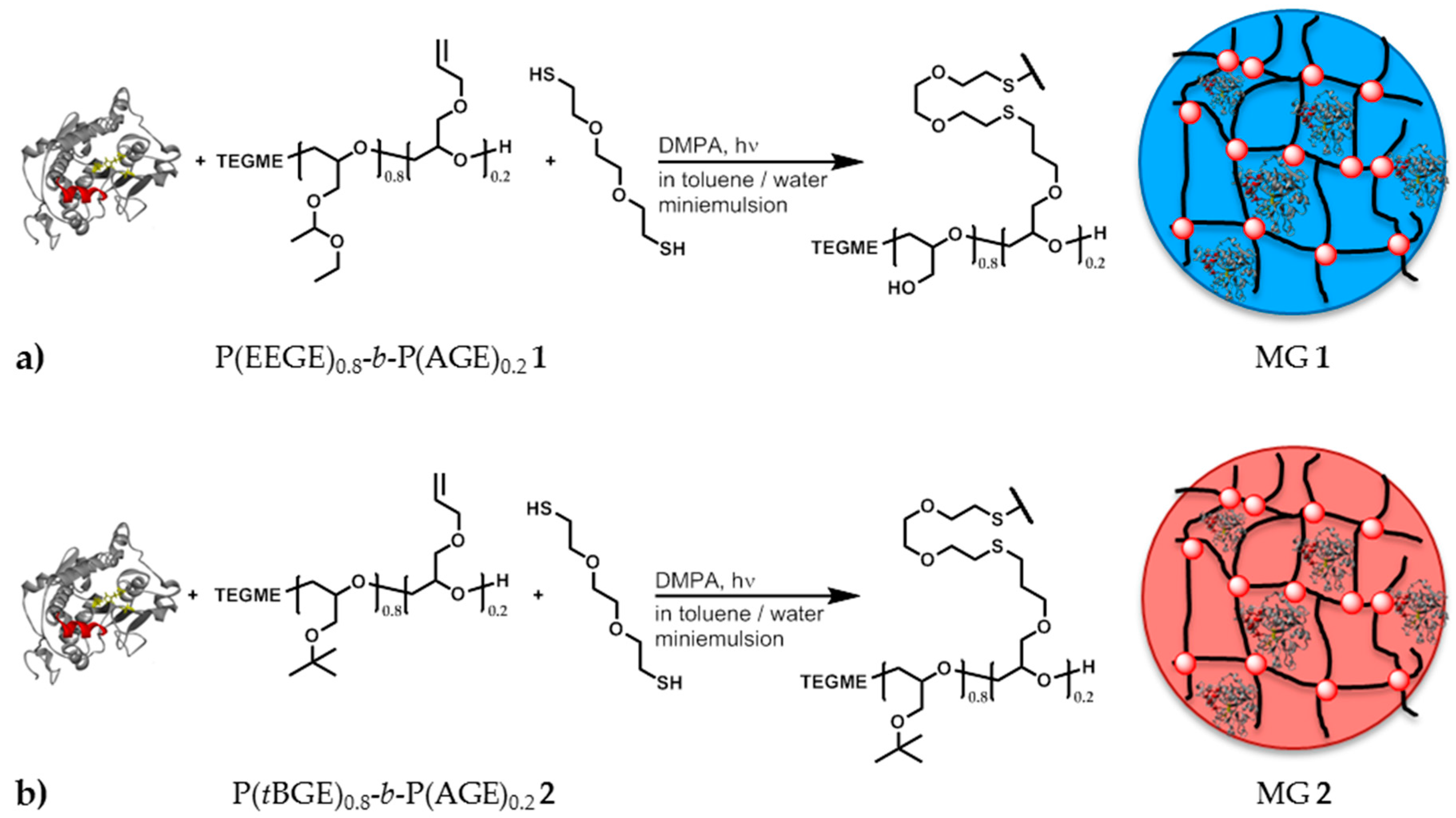
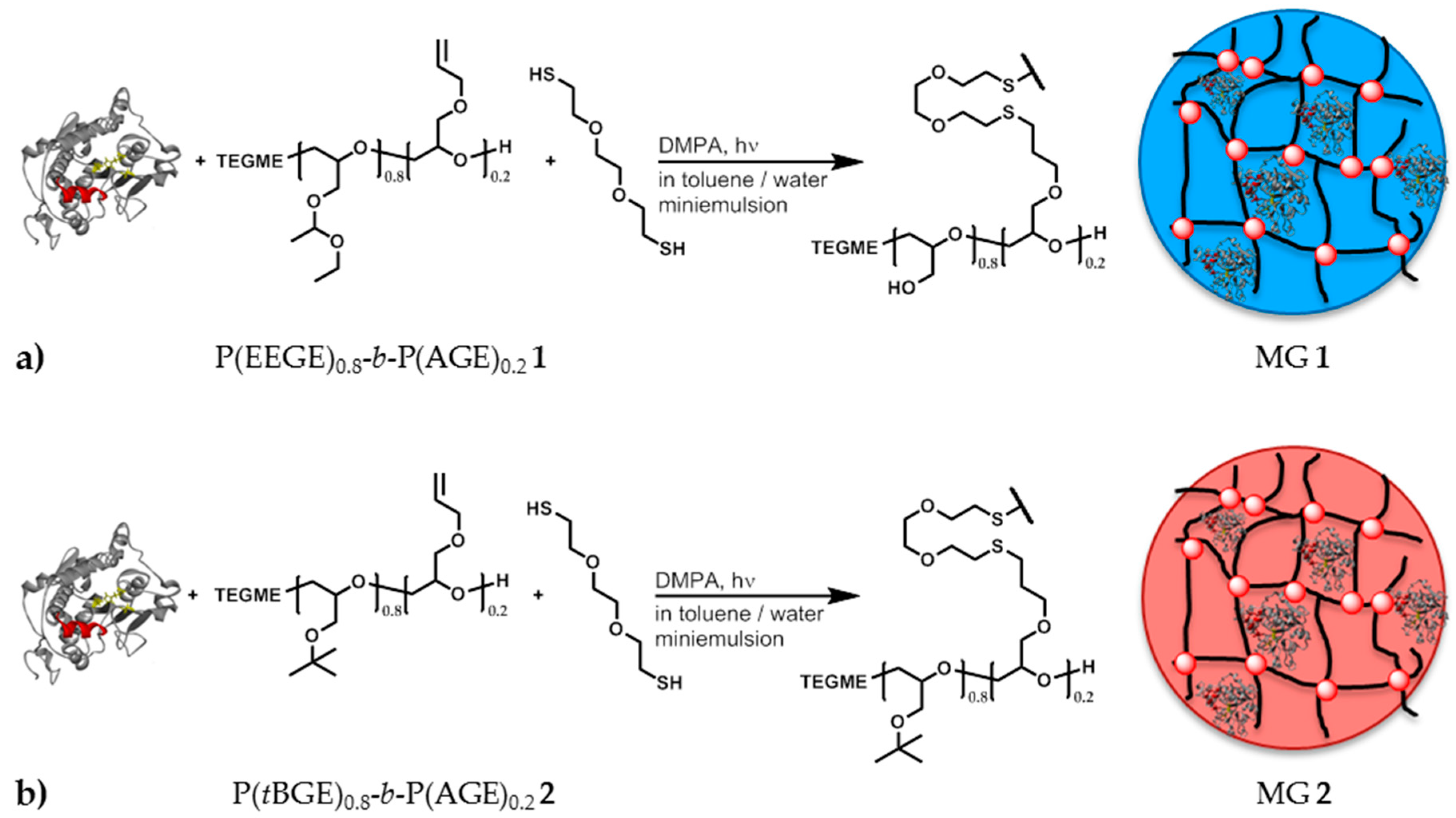
2.5. Synthesis of P(EEGE)0.8-b-P(AGE)0.2 1
2.6. Synthesis of P(tBGE)0.8-b-P(AGE)0.2 2
2.7. Synthesis of Polyglycidol Based Microgels with in Situ Entrapment of CaLB
2.8. Enzymatic Polymerization of ε-CL in Toluene with MGtol 2
3. Results and Discussion
3.1. Distribution of ε-CL in a Hydrophilic/Hydrophobic Environment
3.2. Effect of the ε-CL Concentration on the Esterification Ability of Non-Immobilized CaLB
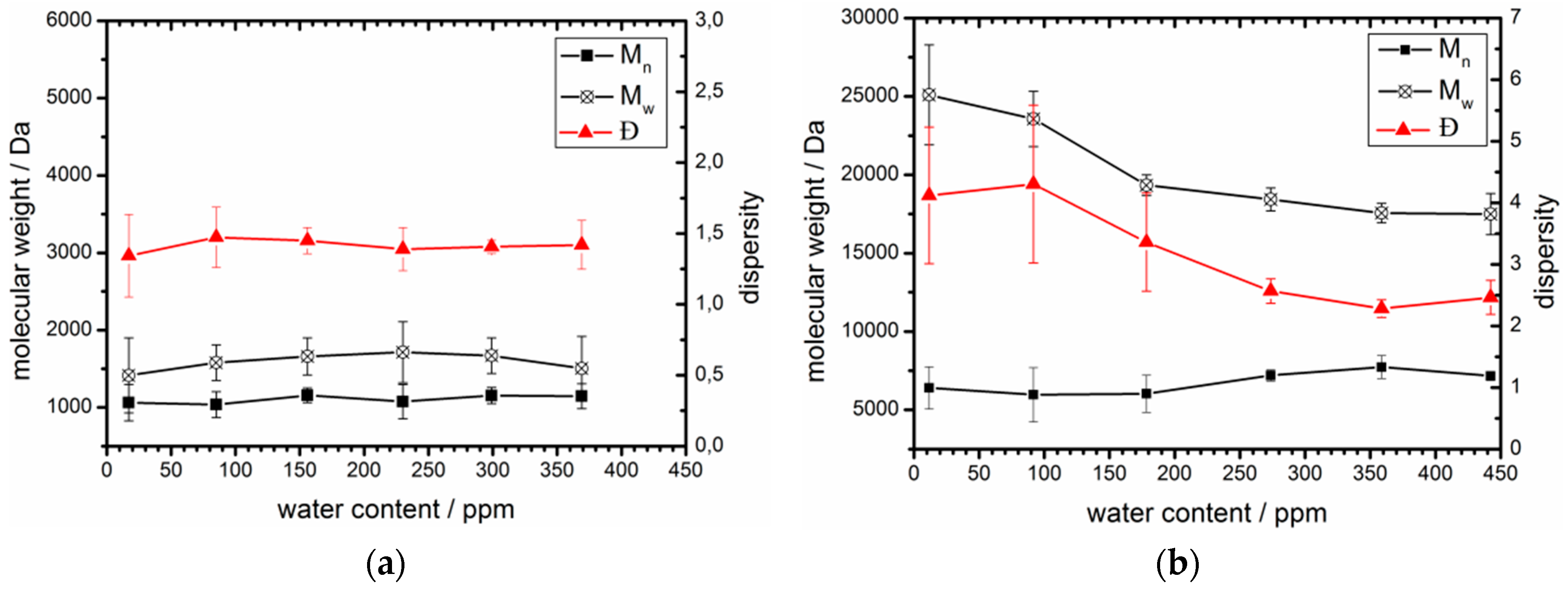
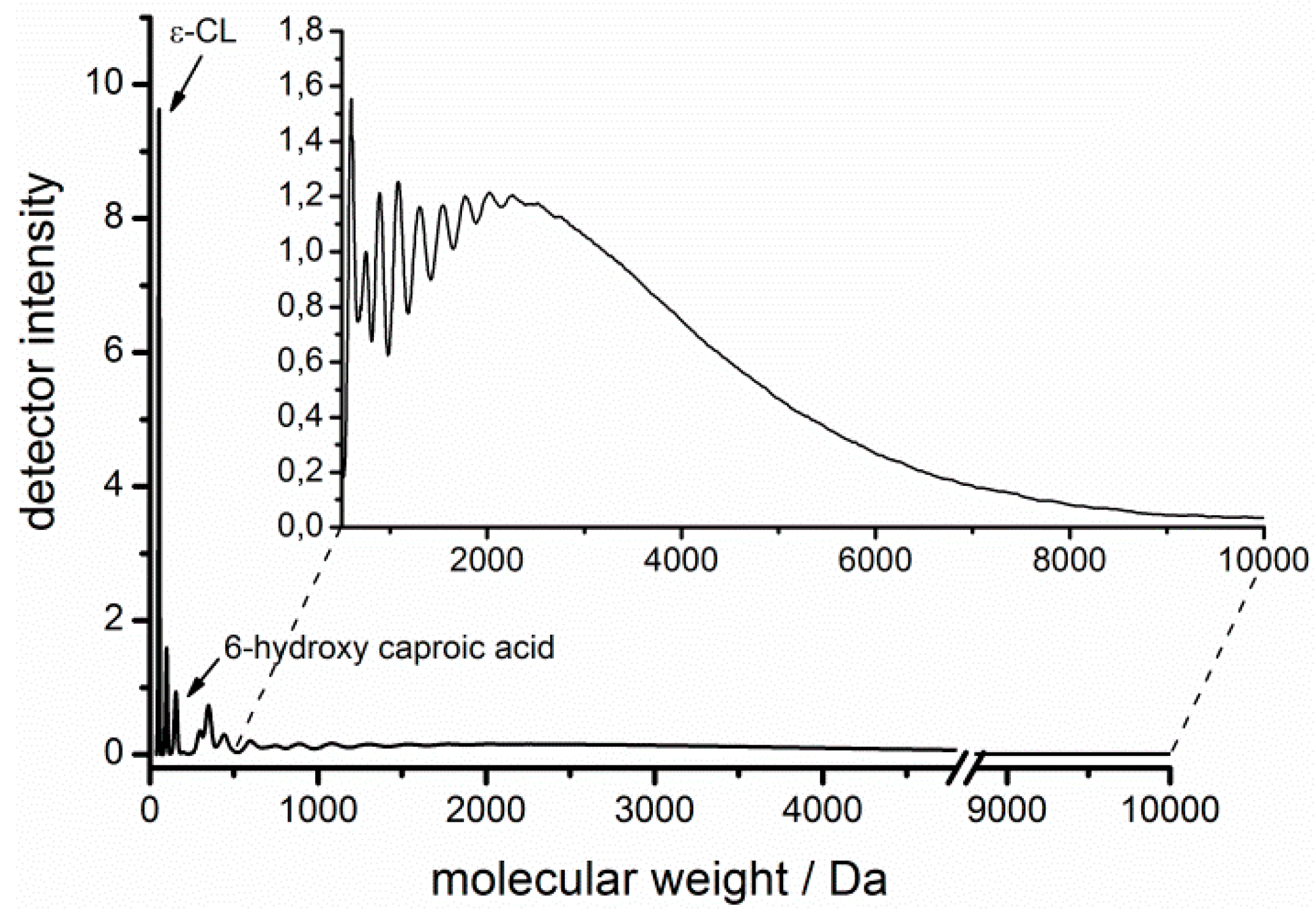
3.3. Acceptable Water Concentration in the Hydrophobic Domain
3.4. Preparation of Polyglycidol Based Microgels and Immobilization of CaLB
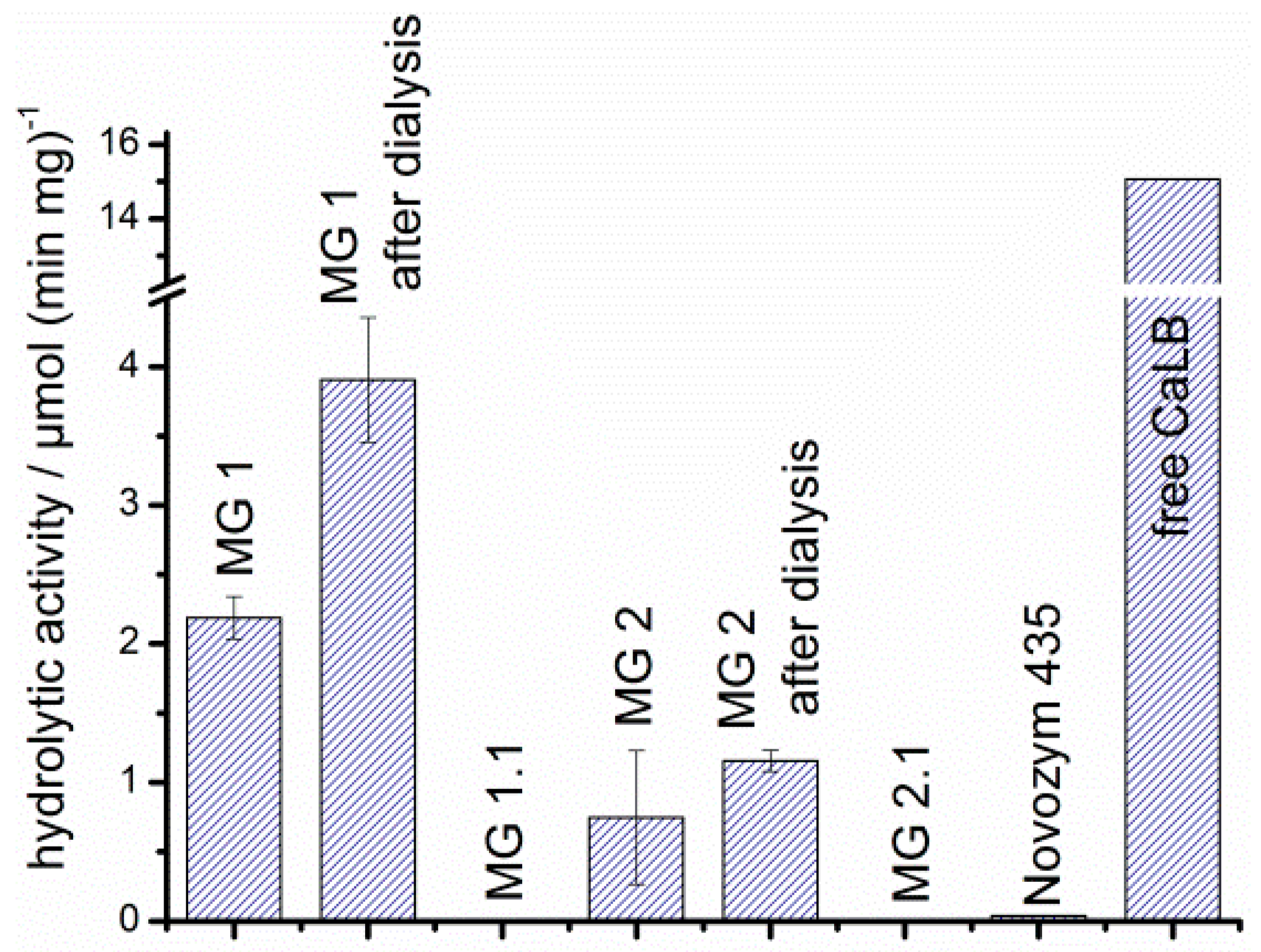
3.5. Effect of Immobilization on the Hydrolytic and Esterification Activity of CaLB
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGE | allyl glycidyl erher |
| CaLB | Candida antarctica lipase B |
| DMPA | 2,2-dimethoxy-2-phenylacetophenone |
| ε-CL | ε-caprolactone |
| EEGE | ethoxyethyl glycidyl ether |
| Ð | dispersity index |
| MBDSTFA | N-methyl-N-tert-butyldimethylsilyl-trifluoroacetamide |
| MG | microgel |
| MTP | microtiterplate |
| PDL | pentadecalactone |
| pNBP | para-nitrophenol butyrate |
| ROP | ring opening polymerization |
| SDS | sodium dodecylsulfate |
| tBGE | tert-butyl glycidyl ether |
| TEA | triethanolamine |
| TEGME | triethylene glycol monomethylether |
| UDL | 11-undecanolide |
| YPD | yeast extract peptone dextrose |
References
- Albertsson, A.C.; Varma, I.K. Aliphatic polyesters: Synthesis, properties and applications. Adv. Polym. Sci. 2002, 157, 1–40. [Google Scholar]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, H.; Wu, D.; Xing, Z.; Zhang, A.; Yang, Y.; Li, Q. Recent developments in lipase-catalyzed synthesis of polymeric materials. Process Biochem. 2014, 49, 797–806. [Google Scholar] [CrossRef]
- Reetz, M.T. Lipases as practical biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 145–150. [Google Scholar] [CrossRef]
- Schmid, R.D.; Verger, R. Lipases: Interfacial enzymes with attractive applications. Angew. Chem. Int. Ed. 1998, 37, 1608–1633. [Google Scholar] [CrossRef]
- Popescu, D.; Keul, H.; Moeller, M. Poly(meth)acrylates obtained by cascade reaction. Macromol. Rapid Commun. 2011, 32, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Vaida, C.; Keul, H.; Möller, M. Tailor-made polyesters based on pentadecalactone via enzymatic catalysis. Green Chem. 2011, 13, 889–899. [Google Scholar] [CrossRef]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of lactones in water. Polym. J. 1998, 30, 269–271. [Google Scholar] [CrossRef]
- Schmidt, S.; Scherkus, C.; Muschiol, J.; Menyes, U.; Winkler, T.; Hummel, W.; Gröger, H.; Liese, A.; Herz, H.G.; Bornscheuer, U.T. An enzyme cascade synthesis of ε-caprolactone and its oligomers. Angew. Chem. Int. Ed. 2015, 54, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nallani, M.; de Hoog, H.P.M.; Cornelissen, J.J.L.M.; Palmans, A.R.A.; van Hest, J.C.M.; Nolte, R.J.M. Polymersome nanoreactors for enzymatic ring-opening polymerization. Biomacromolecules 2007, 8, 3723–3728. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.A.; Kumar, A.; Kalra, B. Polymer synthesis by in vitro enzyme catalysis. Chem. Rev. 2001, 101, 2097–2124. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-H. Enzymatic catalysts in organic synthesis. Science 1989, 244, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B. Enzymes in organic synthesis. Tetrahedron 1986, 42, 3351–3403. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. Enzymatic ring-opening polymerization of lactones catalyzed by lipase. Chem. Lett. 1993, 22, 1149–1150. [Google Scholar] [CrossRef]
- Knani, K.; Gutman, A.L.; Kohn, D.H. Enzymatic polyesterification in organic media. Enzyme-catalyzed synthesis of linear polyesters. 1. Condensation polymerization of linear hydroxyesters. II. Ring-opening polymerization of ε-caprolactone. J. Polym. Sci. Part A 1993, 31, 1221–1232. [Google Scholar] [CrossRef]
- Uyama, H.; Takeya, K.; Kobayashi, S. Enzymatic ring-opening polymerization of lactones to polyesters by lipase catalyst: Unusually high reactivity of macrolides. Bull. Chem. Soc. Jpn. 1995, 68, 56–61. [Google Scholar] [CrossRef]
- Nomura, R.; Ueno, A.; Endo, T. Anionic ring-opening polymerization of macrocyclic esters. Macromolecules 1994, 27, 620–621. [Google Scholar] [CrossRef]
- Lecomte, P.H.; Stassin, F.; Jérôme, R. Recent developments in the ring-opening polymerization of ε-caprolactone and derivatives initiated by tin(IV) alkoxides. Macromol. Symp. 2004, 215, 325–338. [Google Scholar] [CrossRef]
- Agarwal, S.; Mast, C.; Dehnicke, K.; Greiner, A. Rare earth metal initiated ring-opening polymerization of lactones. Macromol. Rapid Commun. 2000, 21, 195–212. [Google Scholar] [CrossRef]
- De Barros, D.P.C.; Fonseca, L.P.; Cabral, J.M.S.; Aschenbrenner, E.M.; Weiss, C.K.; Landfester, K. Miniemulsion as efficient system for enzymatic synthesis of acid alkyl esters. Biotechnol. Bioeng. 2010, 106, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Ragupathya, L.; Pluhara, B.; Ziener, U.; Keller, H.; Dyllick-Brenzinger, R.; Landfester, K. Enzymatic aminolysis of lactones in aqueous miniemulsion: Catalysis through a novel pathway. J. Mol. Catal. B Enzym. 2010, 62, 270–276. [Google Scholar] [CrossRef]
- Taden, A.; Antonietti, M.; Landfester, K. Enzymatic polymerization towards biodegradable polyester nanoparticles. Macromol. Rapid Commun. 2003, 24, 512–516. [Google Scholar] [CrossRef]
- Weiss, C.K.; Landfester, K. Enzymatic catalysis at interfaces—Heterophase systems as substrates for enzymatic action. Catalysts 2013, 3, 401–417. [Google Scholar] [CrossRef]
- Gross, R.A.; Ganesh, M.; Lu, W. Enzyme-catalysis breathes new life into polyester condensation polymerizations. Trends Biotechnol. 2010, 28, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Poojari, Y.; Beemat, J.S.; Clarson, S.J. Enzymatic synthesis of poly(ε-caprolactone): Thermal properties, recovery, and reuse of lipase B from Candida Antarctica immobilized on macroporous acrylic resin particles. Polym. Bull. 2013, 70, 1543–1552. [Google Scholar] [CrossRef]
- Schulte, B.; Walther, A.; Keul, H.; Möller, M. Polyglycidol-based prepolymers to tune the nanostructure of microgels. Macromolecules 2014, 47, 1633–1645. [Google Scholar] [CrossRef]
- Schulte, B.; Rahimi, K.; Keul, H.; Demco, D.E.; Walther, A.; Möller, M. Blending of reactive prepolymers to control the morphology and polarity of polyglycidol based microgels. Soft Matter 2015, 11, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Fitton, A.O.; Hill, J.; Jane, D.E.; Millar, R. Synthesis of simple oxetanes carrying reactive 2-substituents. Synthesis 1987, 1140–1142. [Google Scholar] [CrossRef]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef]
- Kumar, A.; Gross, R.A. Candida Antartica lipase B catalyzed polycaprolactone synthesis: Effects of organic media and temperature. Biomacromolecules 2000, 1, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Song, Z. Migration of reactive trace compounds from Novozym® 435 into organic solvents and ionic liquids. Biochem. Eng. J. 2010, 49, 113–118. [Google Scholar] [CrossRef]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Idris, A.; Bukhari, A. Immobilized Candida Antarctica lipase B: Hydration, stripping off and application in ring opening polyester synthesis. Biotechnol. Adv. 2012, 30, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; dos Santos, J.C.S.; Ortiz, C.; Torres, R.; Barbosa, O.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernández-Lafuente, R. Chemical modification in the design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Chem. Rec. 2016, 16, 1436–1455. [Google Scholar] [CrossRef] [PubMed]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Frey, H. Poly(ethylene glycol-co-allyl glycidyl ether)s: A PEG-based modular synthetic platform for multiple bioconjugation. Bioconjug. Chem. 2011, 22, 436–444. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Fraction of ε-CL | Fraction of solvent | H2O Content/ppm | Coligo/mol % (1) | Mn/Da (2) |
|---|---|---|---|---|---|
| bulk ε-CL | 1 | 0 | 1,530 | 1 | - (3) |
| H2O | 0.8 | 0.2 | - | 29 | - (3) |
| H2O | 0.5 | 0.5 | - | 20 | - (3) |
| H2O | 0.2 | 0.8 | - | 8 | - (3) |
| toluene | 0.8 | 0.2 | 1,280 | 2 | - (3) |
| toluene | 0.5 | 0.5 | 900 | 16 | 630 |
| toluene | 0.2 | 0.8 | 530 | 75 | 940 |
| Toluene (Dry)/% | Toluene (water saturated)/% | Experiments with free CaLB | Experiments with Novozym® 435 | ||||
|---|---|---|---|---|---|---|---|
| Total H2O content/ppm (2) | Mn/Da (3) | Ð (3) | Total H2O content/ppm (4) | Mn/Da (3) | Ð (3) | ||
| 100 | 0 | 17 | 1,090 | 1.3 | 12 | 6,400 | 4.1 |
| 80 | 20 | 85 | 1,060 | 1.5 | 92 | 6,000 | 4.3 |
| 60 | 40 | 156 | 1,160 | 1.5 | 178 | 6,000 | 3.4 |
| 40 | 60 | 230 | 1,080 | 1.4 | 274 | 7,200 | 2.6 |
| 20 | 80 | 299 | 1,160 | 1.4 | 359 | 7,700 | 2.3 |
| 0 | 100 | 369 | 1,140 | 1.4 | 442 | 7,200 | 2.5 |
| Type of supporting material | Prepolymer | Enzyme | [CaLB]/mg·mL−1 | |
|---|---|---|---|---|
| Before dialysis (1) | After dialysis (1) | |||
| MG 1 | P(EEGE)-b-(PAGE) | CaLB | 0.58 (0.54) | 0.44 (0.48) |
| MG 1.1 | P(EEGE)-b-P(AGE) | - | 0 | 0 |
| MG 2 | P(tBGE)-b-P(AGE) | CaLB | 0.54 (0.50) | 0.34 (0.46) |
| MG 2.2 | P(tBGE)-b-P(AGE) | - | 0 | 0 |
| acrylic resin (2) | - | CaLB | 0.48 (3) | |
| Catalyst | H2O content/ppm | Coligo/mol % (1) | Mn/Da (2) | Ð (2) |
|---|---|---|---|---|
| free CaLB | 369 | 13 | 1,140 | 1.4 |
| Novozym® 435 | 359 | 96 | 7,700 | 2.3 |
| MGtol 2 | 347 | 87 | 4,900 | 2.4 |
| MGtol 2.1 | 209 | 0 | - (3) | - (3) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engel, S.; Höck, H.; Bocola, M.; Keul, H.; Schwaneberg, U.; Möller, M. CaLB Catalyzed Conversion of ε-Caprolactone in Aqueous Medium. Part 1: Immobilization of CaLB to Microgels. Polymers 2016, 8, 372. https://doi.org/10.3390/polym8100372
Engel S, Höck H, Bocola M, Keul H, Schwaneberg U, Möller M. CaLB Catalyzed Conversion of ε-Caprolactone in Aqueous Medium. Part 1: Immobilization of CaLB to Microgels. Polymers. 2016; 8(10):372. https://doi.org/10.3390/polym8100372
Chicago/Turabian StyleEngel, Stefan, Heidi Höck, Marco Bocola, Helmut Keul, Ulrich Schwaneberg, and Martin Möller. 2016. "CaLB Catalyzed Conversion of ε-Caprolactone in Aqueous Medium. Part 1: Immobilization of CaLB to Microgels" Polymers 8, no. 10: 372. https://doi.org/10.3390/polym8100372
APA StyleEngel, S., Höck, H., Bocola, M., Keul, H., Schwaneberg, U., & Möller, M. (2016). CaLB Catalyzed Conversion of ε-Caprolactone in Aqueous Medium. Part 1: Immobilization of CaLB to Microgels. Polymers, 8(10), 372. https://doi.org/10.3390/polym8100372