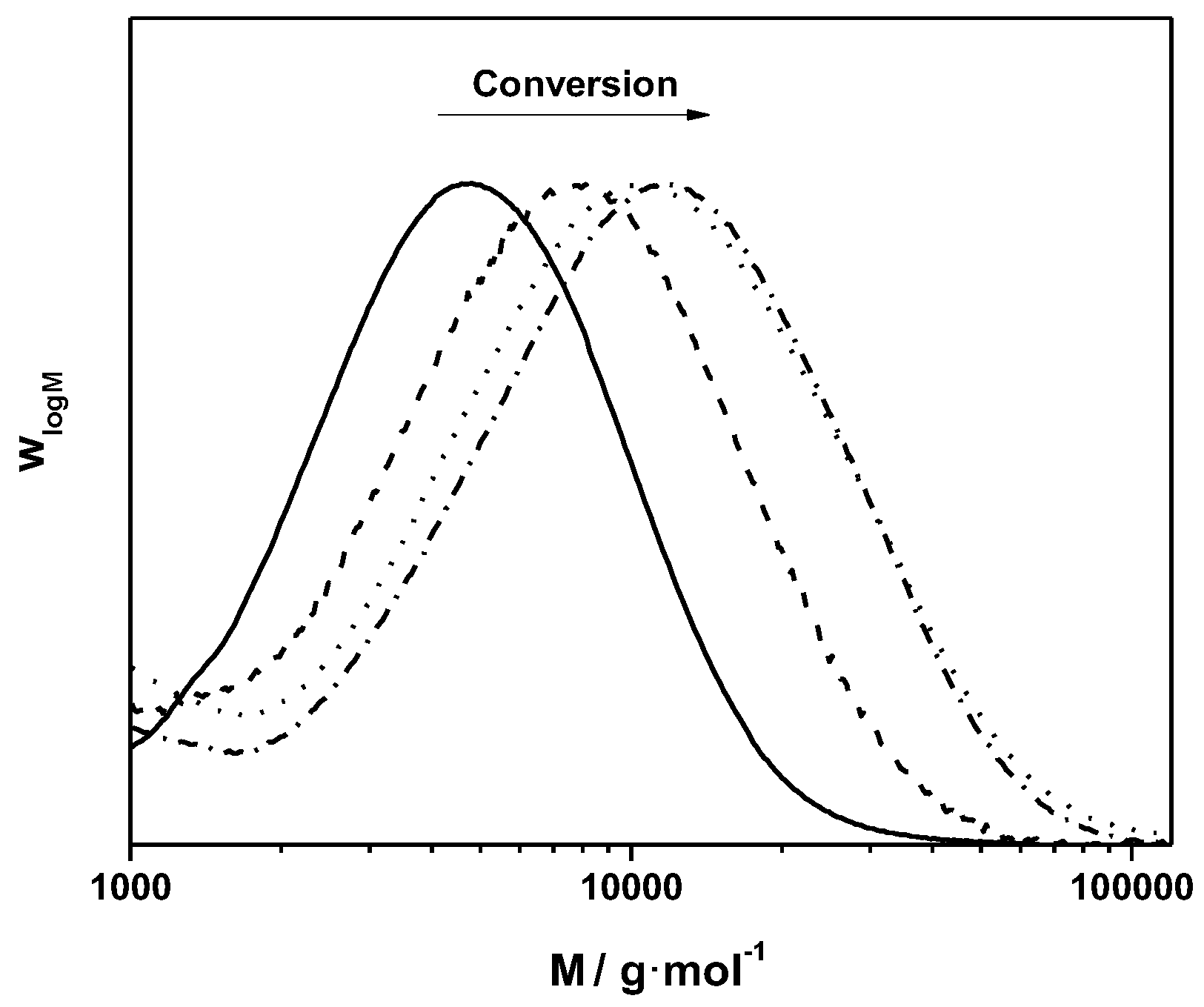
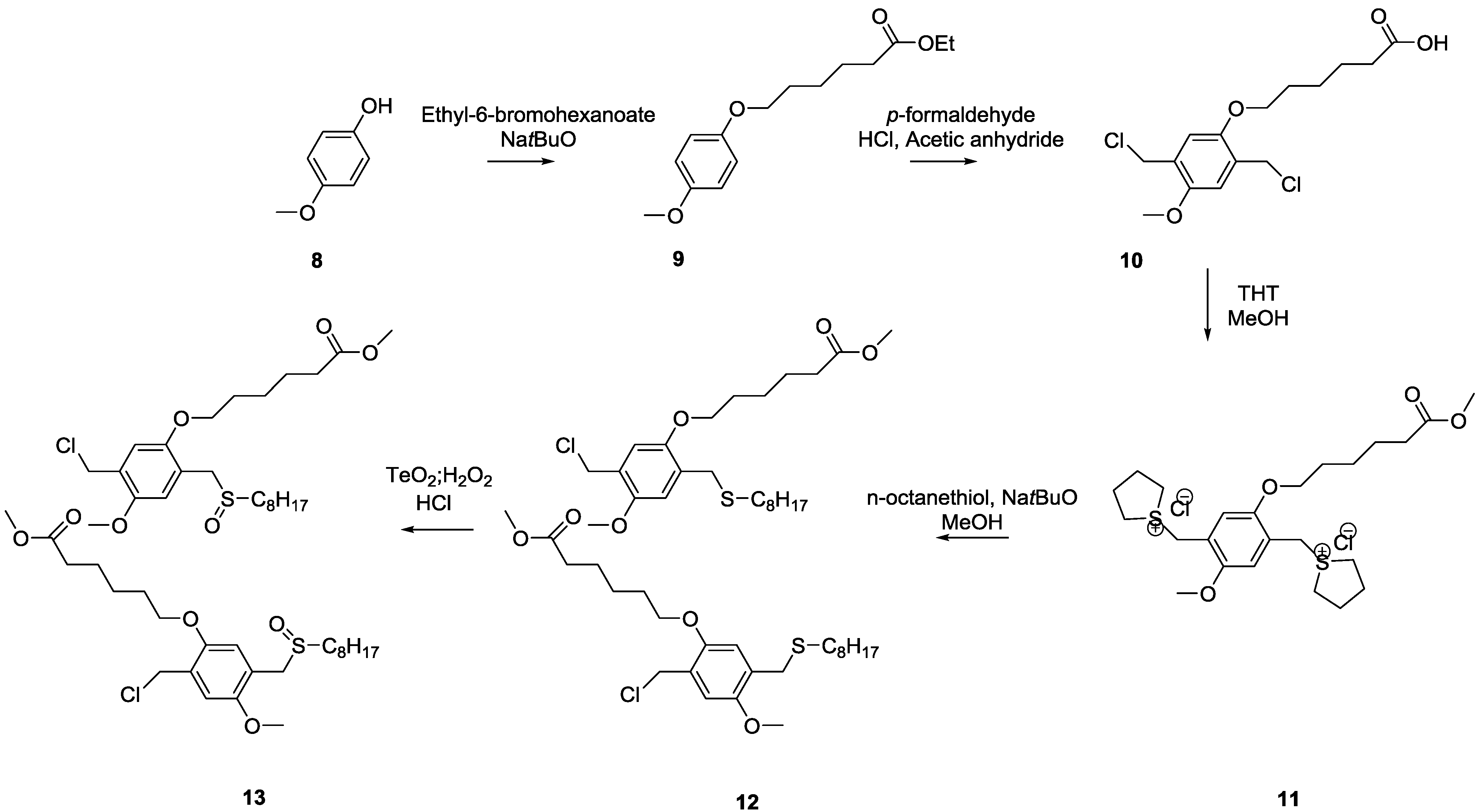
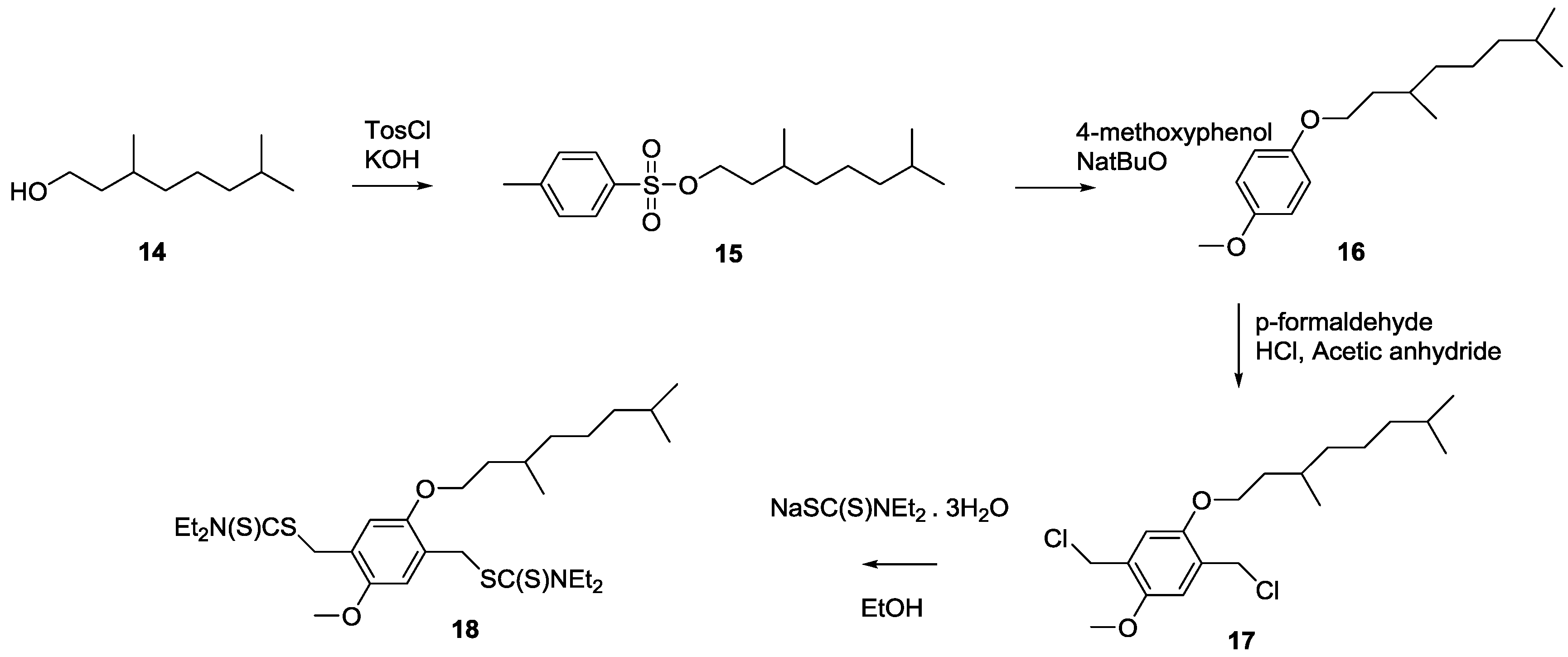
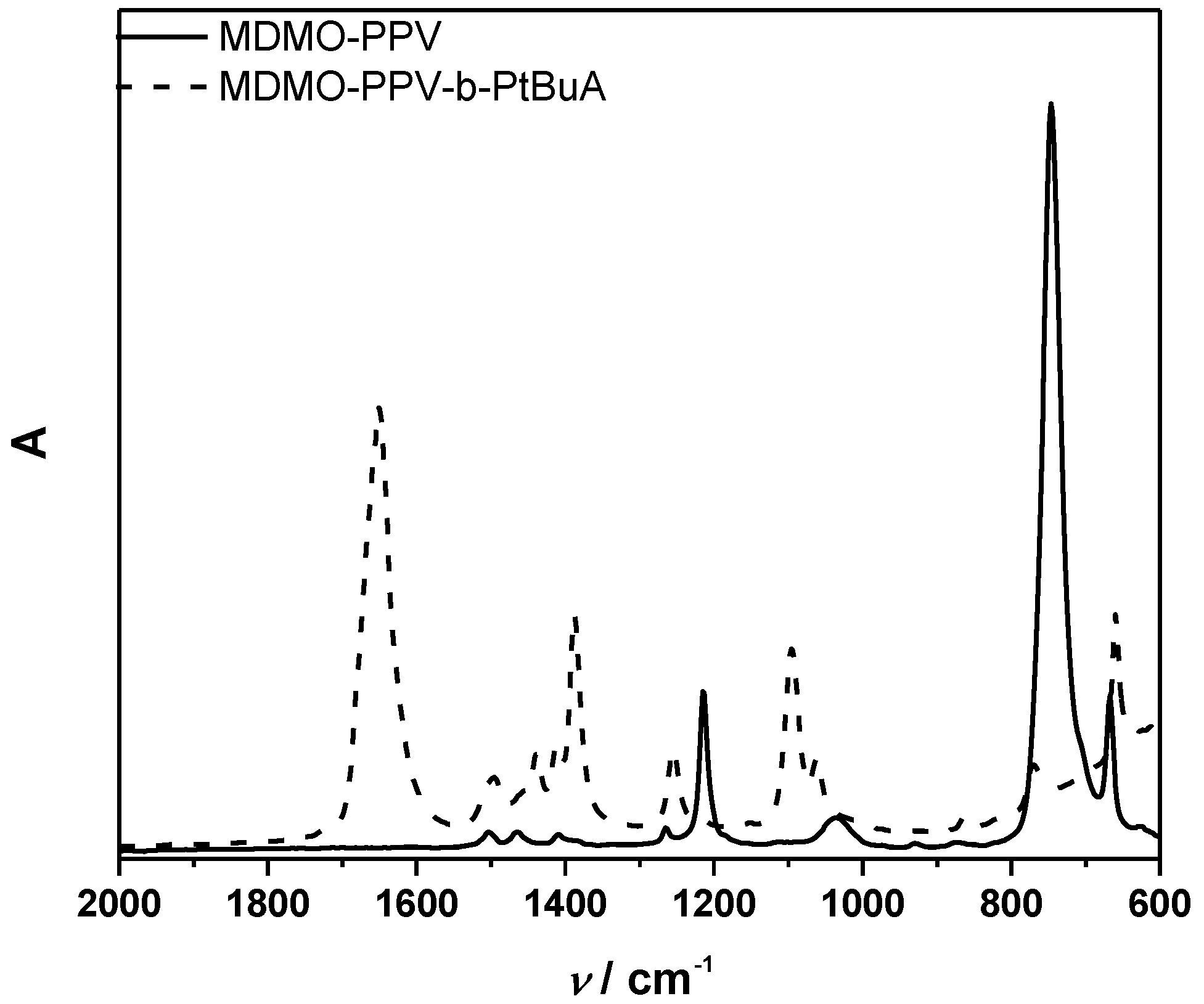
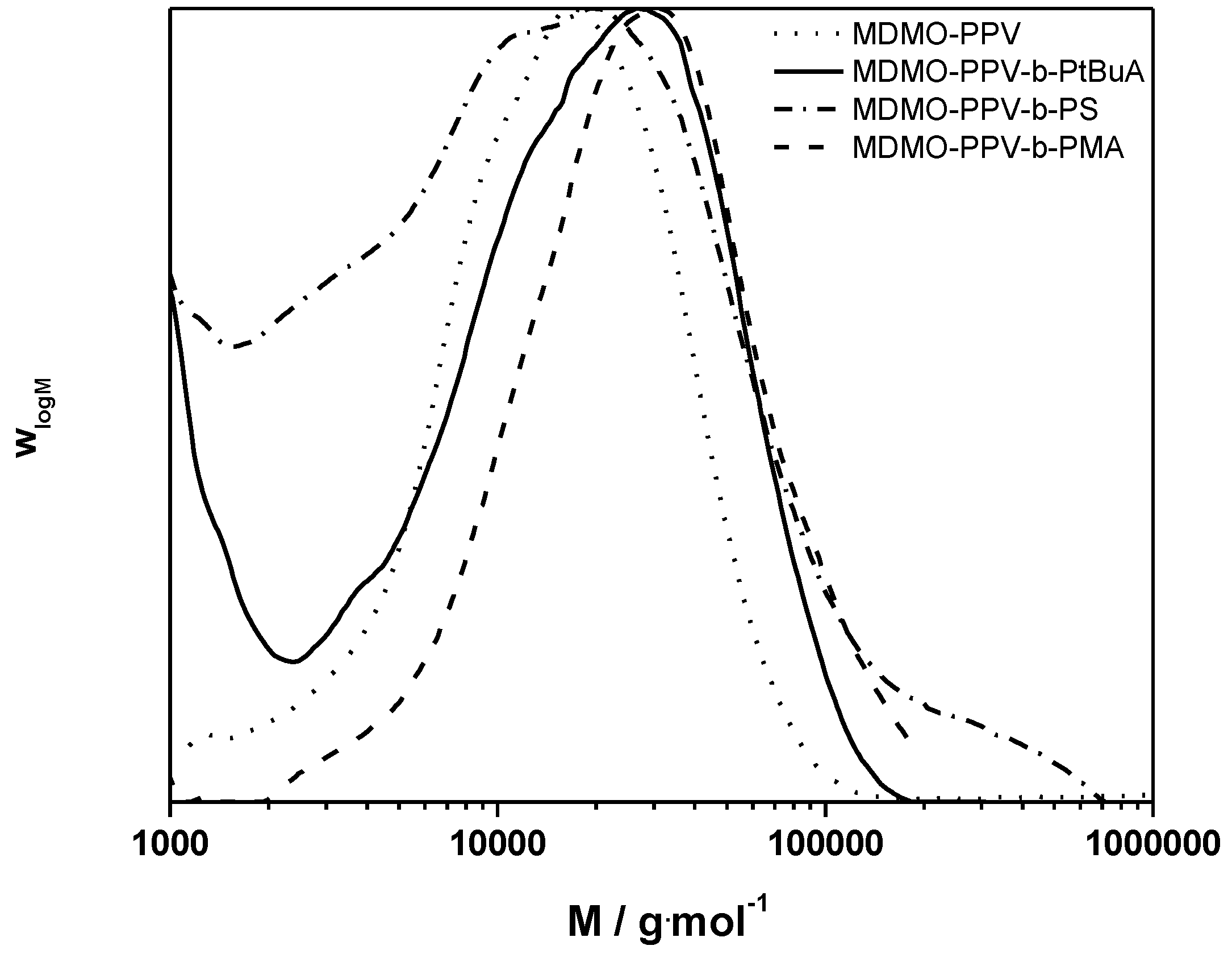
3.1. Control over the Radical PPV Polymerization by Using a CTA
A general pathway to gain control over radical polymerizations is to add control agents to the polymerization that involve the propagating radicals in reversible deactivation equilibria, leading to linear growth of chains with monomer conversion and general preselection of
Mn of the final polymer depending on control agent concentration. As mentioned above, achieving such reversible deactivation is not as straightforward for PPV polymerizations as for conventional radical polymerizations. During initiation (formation of a biradical) as well as during propagation of the
p-quinodimethane monomers, aromaticity is restored, resulting in a very high driving force for these reactions. Therefore, initiation and propagation are extremely fast and only those control agents that allow for similarly high reactivities will be able to compete. So far, no control agent from the typical RDRP methods could be identified to be effectively operational in precursor polymerizations. Only CBr
4—a classical chain transfer agent that induces molecular weight control, but not livingness of the polymerization—had been found to be active enough to have a direct (and positive) influence on the polymer product. A good correlation between the molecular weight of conjugated MDMO-PPV—synthesized via the sulfinyl route—and the transfer agent concentration is reported in a previous study [
39], resulting in polymers with molecular weights ranging from a
Mn of 12,000 to 25,000 g·mol
−1 with a dispersity (
Ð) of ~2.0.
In this study, the effect of CBr
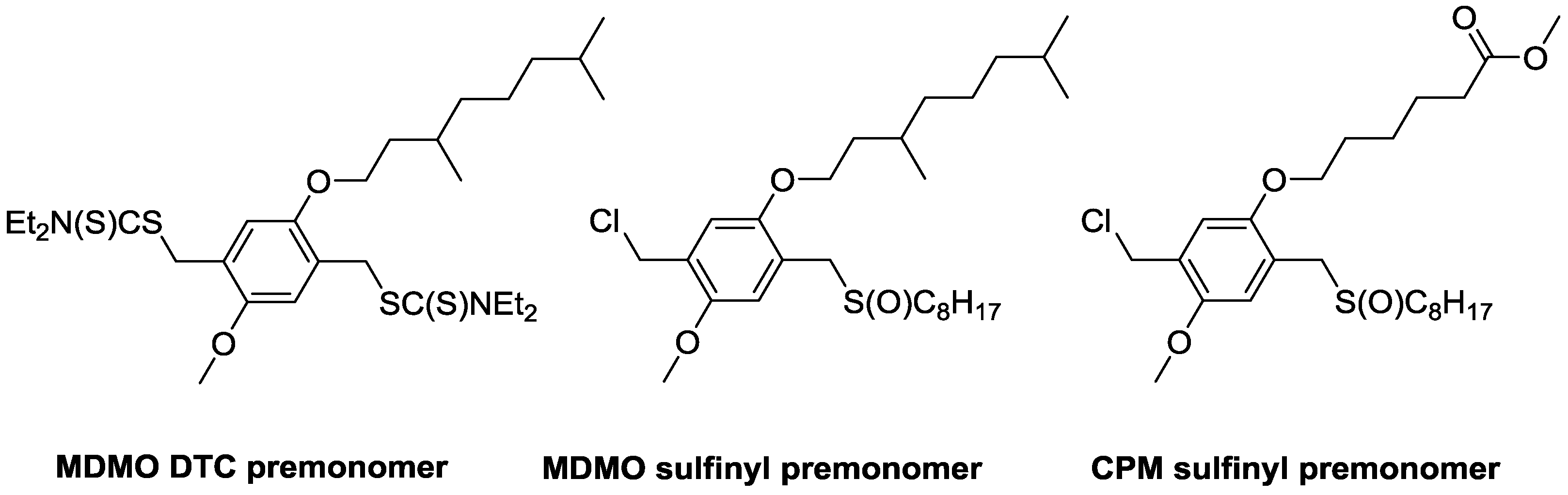
4 on the polymerization of PPV is compared between two precursor routes, e.g., the sulfinyl and the dithiocarbamate (DTC) route. Polymerizations via the sulfinyl route show very fast reactions reaching conversions of almost 100% in less than 5 min. As a result, the addition of high excesses of control agent is needed to obtain the desired effect. Polymerizations using the DTC route on the other hand result in slower polymerizations, and reactions only reach full conversion after 10–15 min. This indicates that good control over the DTC polymerization route using less control agent should be possible. Therefore, three different PPV premonomers were synthesized (see
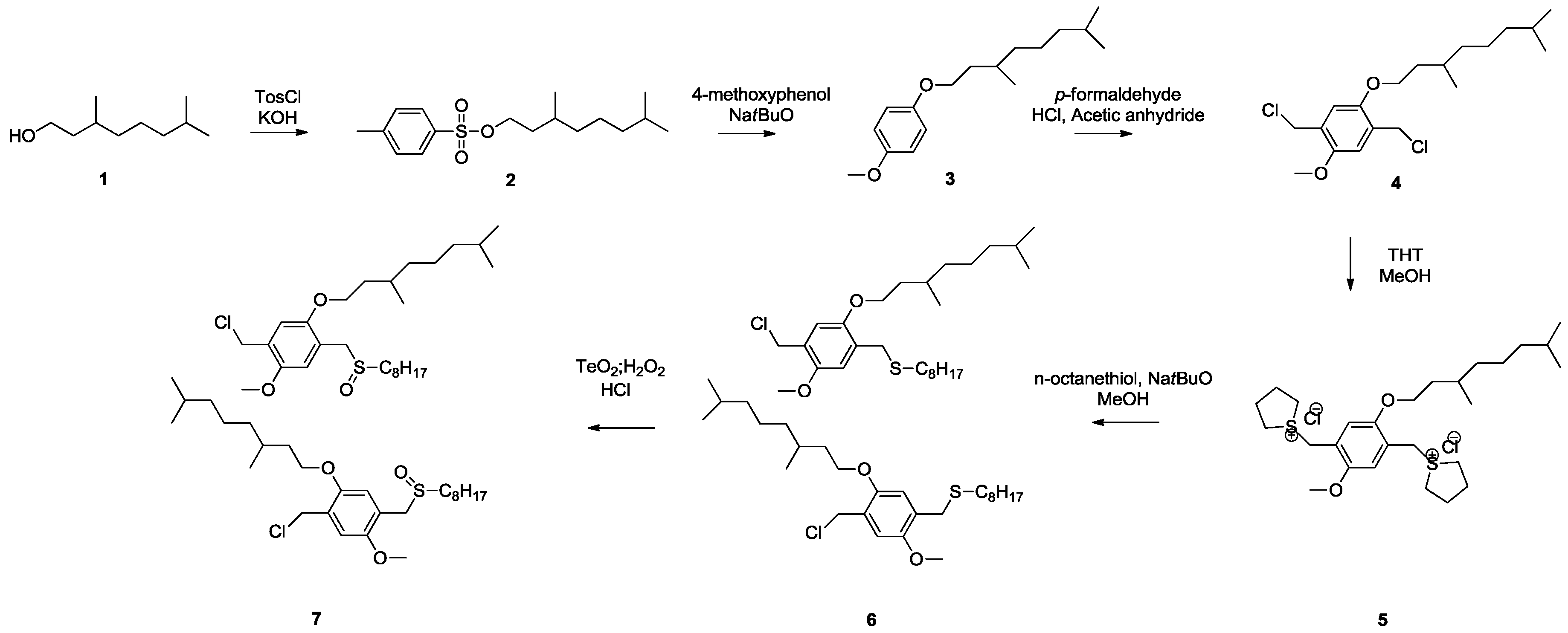
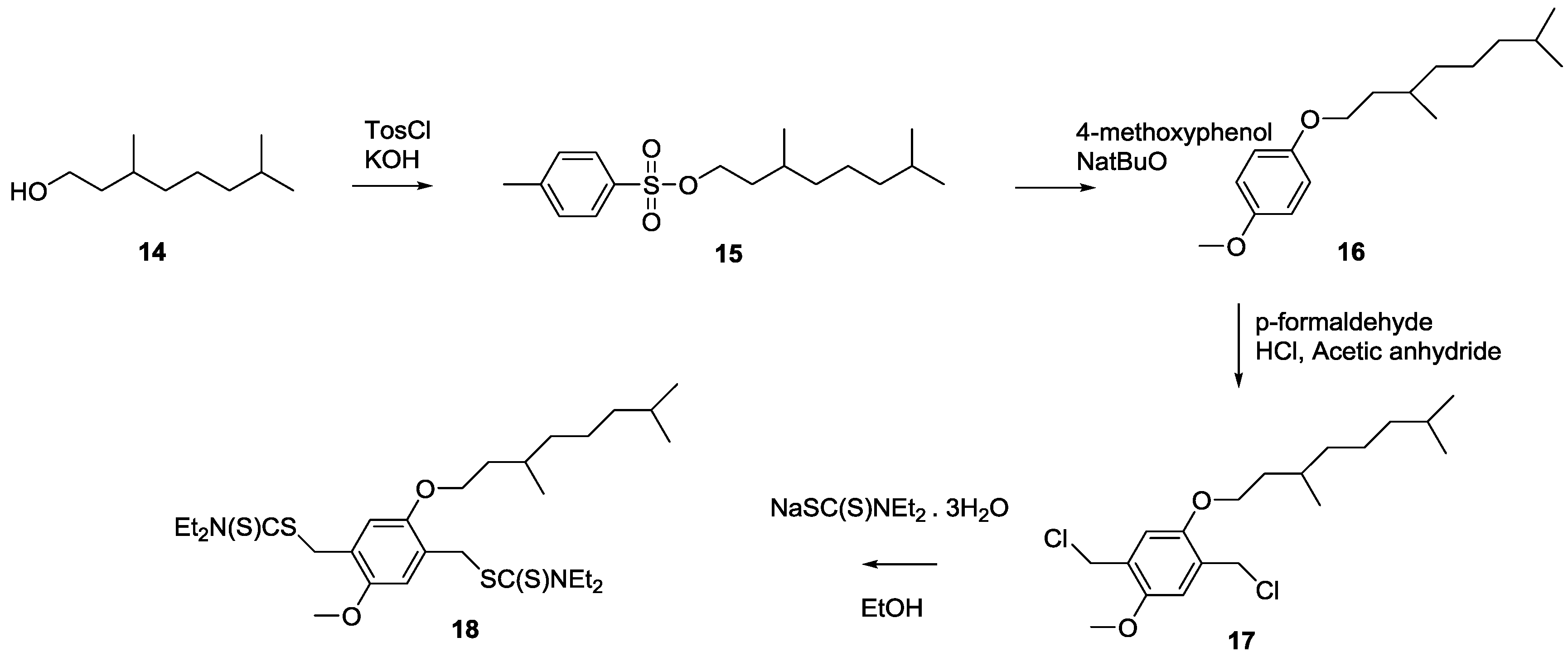
Scheme 7), namely the so-called MDMO sulfinyl premonomer, CPM sulfinyl premonomer and MDMO DTC premonomer. The two MDMO monomers lead equally to the formation of MDMO-PPV, a common conjugated material used in a multitude of applications. The CPM premonomer is used for the synthesis of a close derivative, which is interesting due to the possibility to post-functionalize the conjugated materials via trans-esterification reactions. The premonomers are polymerized using specific amounts of CBr
4 and are subsequently eliminated to result in the desired conjugated polymers that are analyzed by means of SEC, see
Table 1. It is important to note that SEC determination of molecular weights of PPVs is not straightforward. MHKS coefficients are mostly unavailable in literature and already small differences in the PPV defect structure may have a profound effect on the hydrodynamic volume of the chains. Thus, specific MHKS parameters were determined for MDMO-PPV via analysis of several broad polymer distributions using viscometry and MALLS detection. Via usage of polymer samples with precisely known mass, d
n/d
c is directly obtained from the RI detector signal and MHKS values were fitted to the light scattering
Mw data. For MDMO precursor polymer obtained via the sulfinyl route, α = 0.67605 and
k = 0.000142 mL·g
−1 and for conjugated MDMO-PPV α = 0.809 and
k = 0.00002 mL·g
−1 were obtained. Care has to be taken using MALLS detection, as the fluorescent properties of the materials lead to light emission interfering with the MALLS scatter signals. For that reason, MALLS is not easily applied and can thus not be used as a routine method for molar mass determination in this case. For the determination of MHKS values, samples were measured repeatedly at different concentrations to rule out interfering effects. For MDMO-PPV polymers obtained from the DTC route and for the CPM polymer, no specific MHKS were deduced. While parameters for the conjugated polymer should in principle be independent from the polymerization route (a concept that may be debated when going into detail), and hence MHKS for sulfinyl and DTC-made MDMO-PPV should be identical, no absolute
Mn can be given for CPM-PPV. Only apparent
Mnapp are thus given for this system.
Scheme 7.
Overview of the different PPV premonomers used in this study.
Scheme 7.
Overview of the different PPV premonomers used in this study.
Table 1.
Molecular weight data of conjugated MDMO-PPVs obtained from the sulfinyl (reactions at 30 °C using [Monomer] = 0.14 mol·L−1 and [Base] = 0.16 mol·L−1) and DTC (reactions at 35 °C using [Monomer] = 0.20 mol·L−1 and [Base] = 0.87 mol·L−1) route under variation of the amount of CBr4.
Table 1.
Molecular weight data of conjugated MDMO-PPVs obtained from the sulfinyl (reactions at 30 °C using [Monomer] = 0.14 mol·L−1 and [Base] = 0.16 mol·L−1) and DTC (reactions at 35 °C using [Monomer] = 0.20 mol·L−1 and [Base] = 0.87 mol·L−1) route under variation of the amount of CBr4.
| MDMO-PPV (DTC Route) | MDMO-PPV (Sulfinyl Route) | CPM-PPV (Sulfinyl Route) |
|---|
| CTA equiv. | Mn g∙mol−1 | Ð | CTA equiv. | Mn g∙mol−1 | Ð | CTA equiv. | Mnapp g∙mol−1 | Ð |
|---|
| 0 | 98,000 | 2.9 | 0 | 88,900 | 2.6 | 0 | 82,000 | 2.3 |
| 0.04 | 54,000 | 3.2 | 0.5 | 31,000 | 2.3 | 0.5 | 20,000 | 3.2 |
| 0.05 | 49,600 | 3.8 | 1 | 28,400 | 2.5 | 1 | 14,000 | 1.9 |
| 0.1 | 30,500 | 3.0 | 2 | 8,900 | 1.8 | 2 | 6,780 | 2.1 |
| 0.25 | 11,900 | 2.3 | 4 | 9,800 | 1.8 | 4 | 6,800 | 2.2 |
| 0.5 | 2,460 | 2.0 | 8 | 11,000 | 2.0 | 8 | 6,410 | 2.3 |
| 0.75 | 2,480 | 1.2 | 12 | 8,700 | 1.6 | - | - | - |
| 0.9 | 1,260 | 1.1 | 14 | 9,900 | 1.7 | - | - | - |
| 1 | 430 | 1.2 | 16 | 9,600 | 1.6 | - | - | - |
| 8 | 500 | 1.1 | 20 | 6,700 | 1.5 | - | - | - |
| - | - | - | 25 | 8,800 | 1.7 | - | - | - |
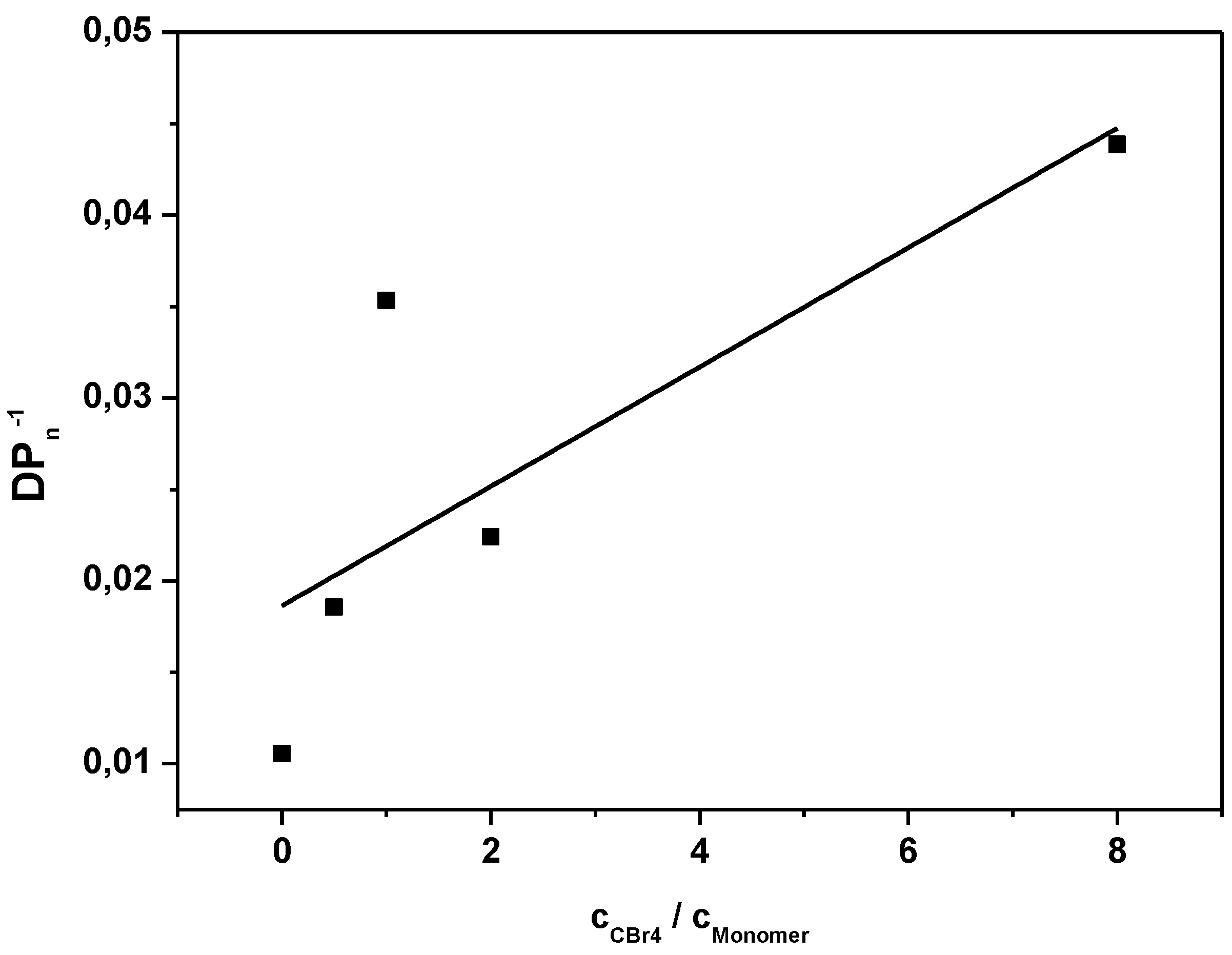
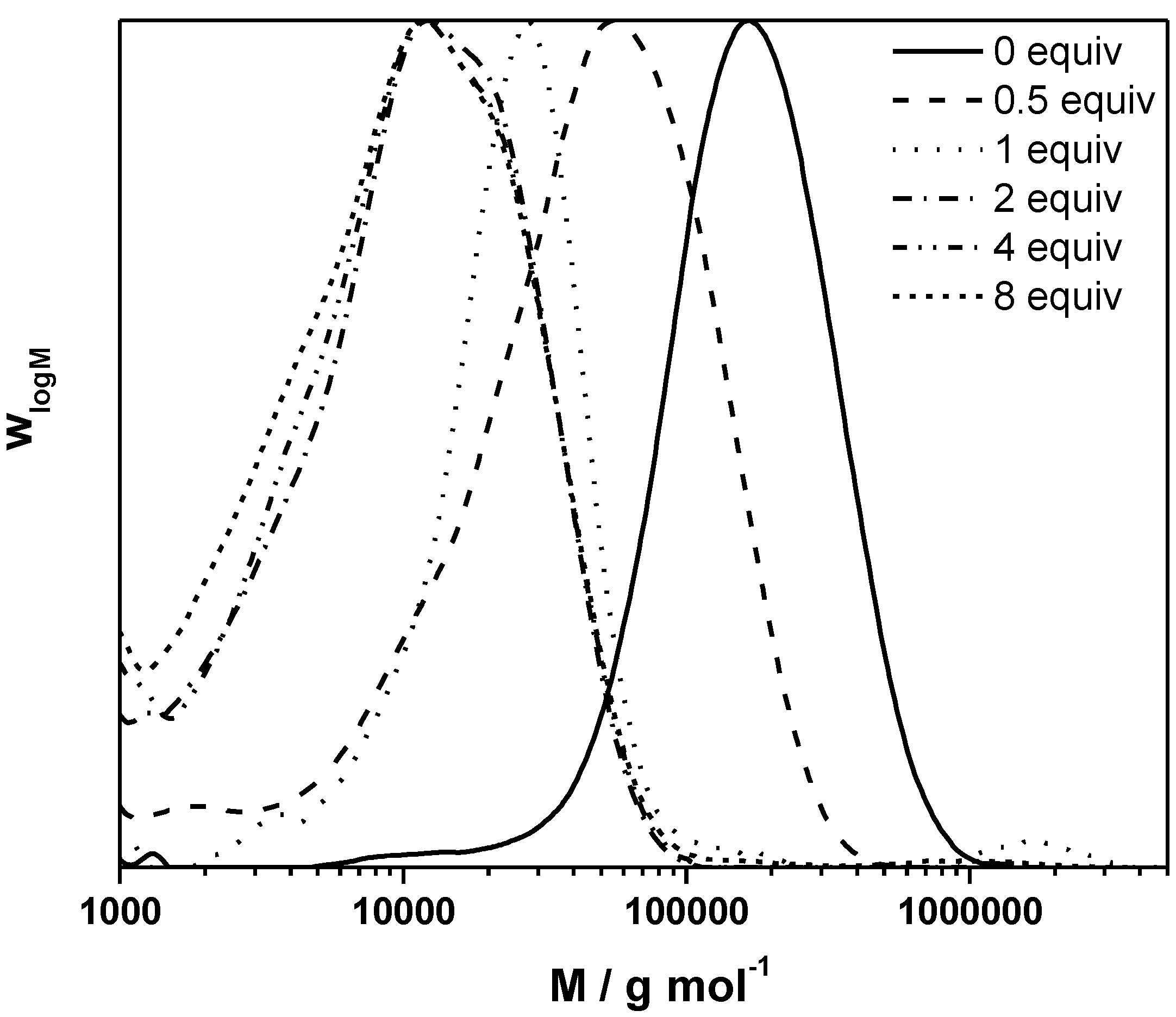
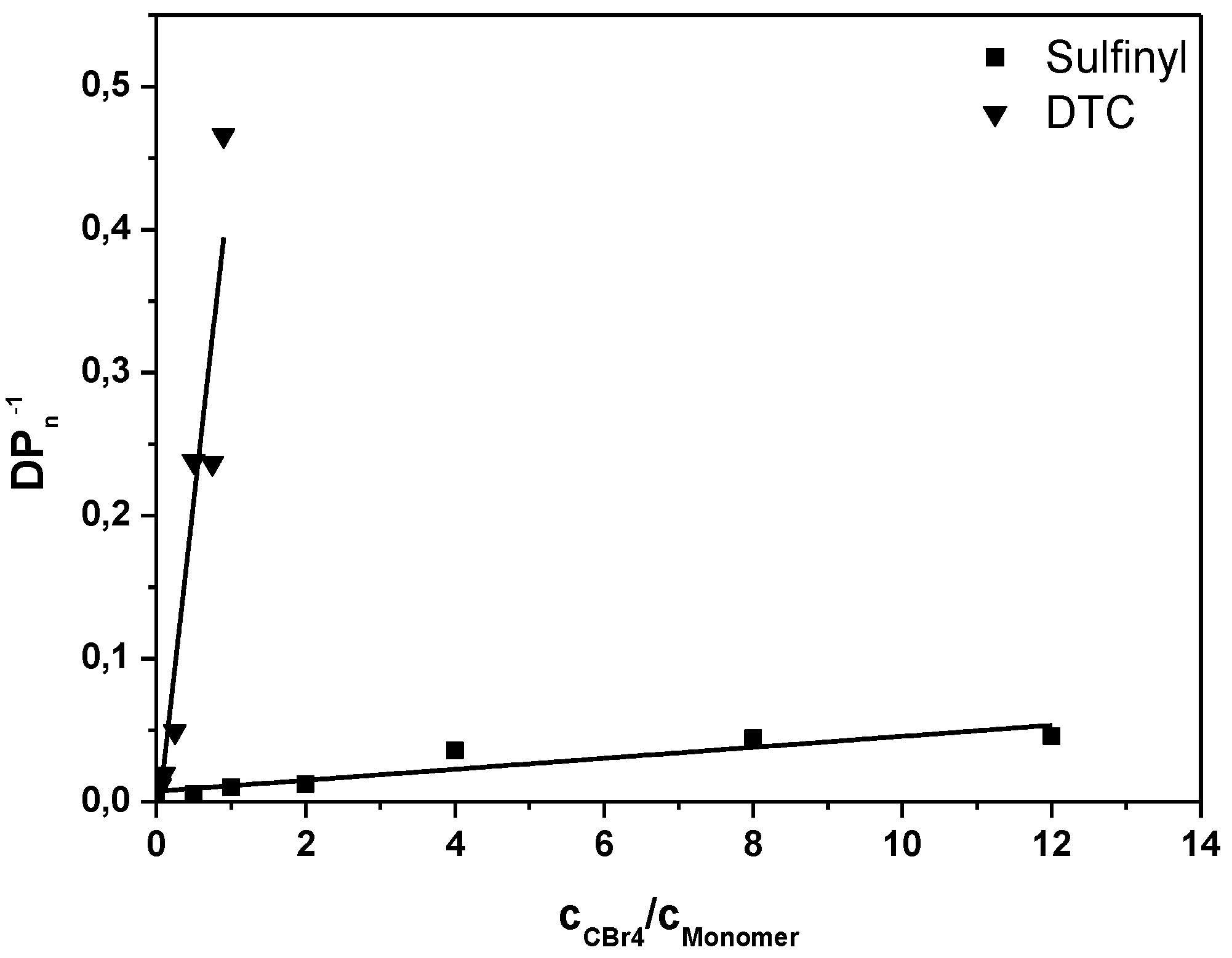
SEC analysis on the resulting conjugated MDMO-PPVs allows for chain transfer constant determination by fitting the inverse of the degree of polymerization as a function of the CTA to premonomer concentration ratio, following the well-known Mayo relation:
In this equation,
DPn represents the average degree of polymerization,
DPno the average degree of polymerization in an ideal case in absence of any transfer agent,
CCTA the transfer agent concentration and
CM the premonomer concentration.
Ctr is the chain transfer constant and is defined as the ratio of the transfer rate coefficient
ktr over the propagation rate coefficient
kp. Thus, in case
kp >>
ktr a
Ctr smaller than unity will be obtained. In
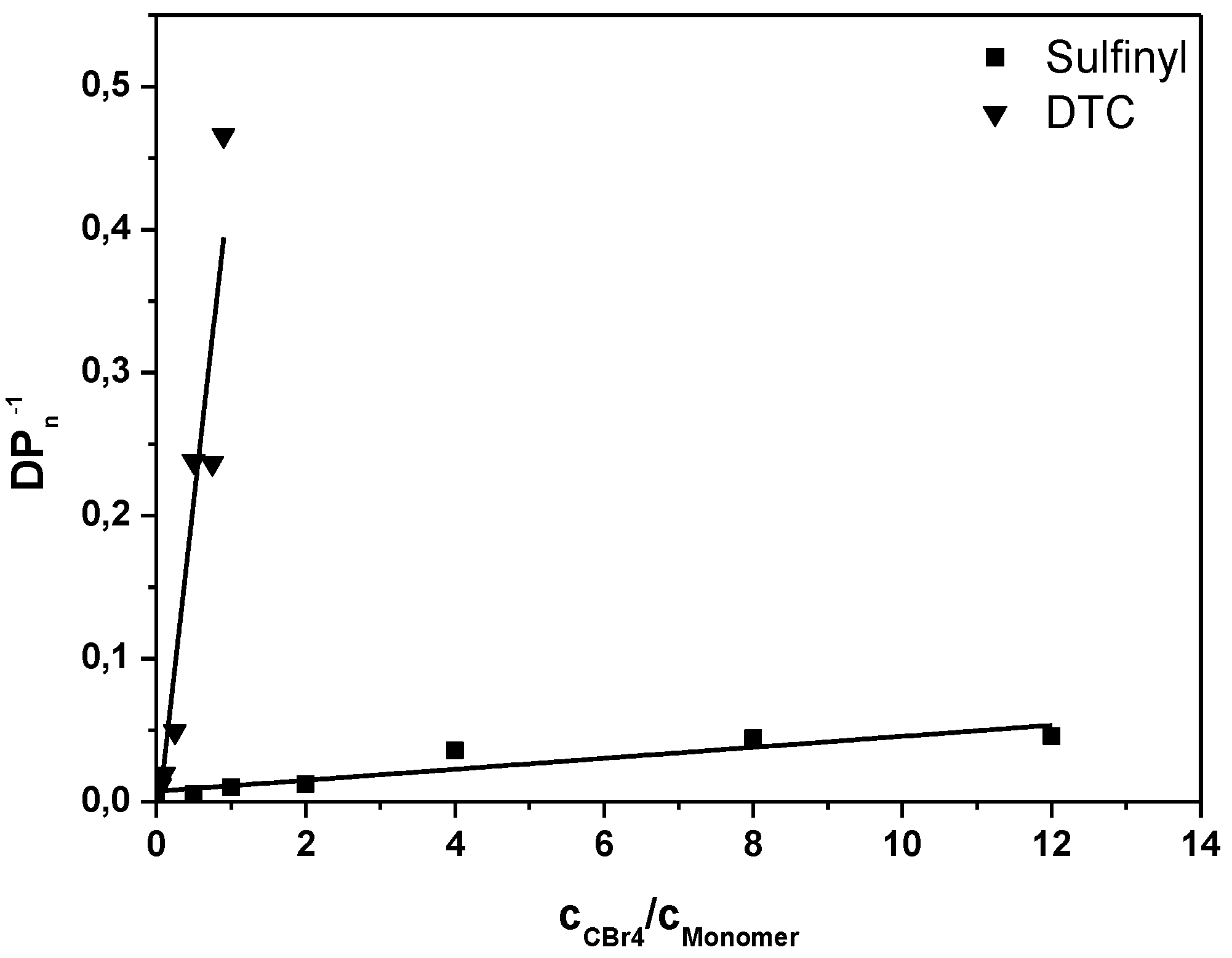
Figure 1, the Mayo plots for conjugated MDMO-PPV, polymerized via the sulfinyl and the DTC route using CBr
4 as CTA is given (see
Appendix Figure A1 for the Mayo plot of CPM-PPV and
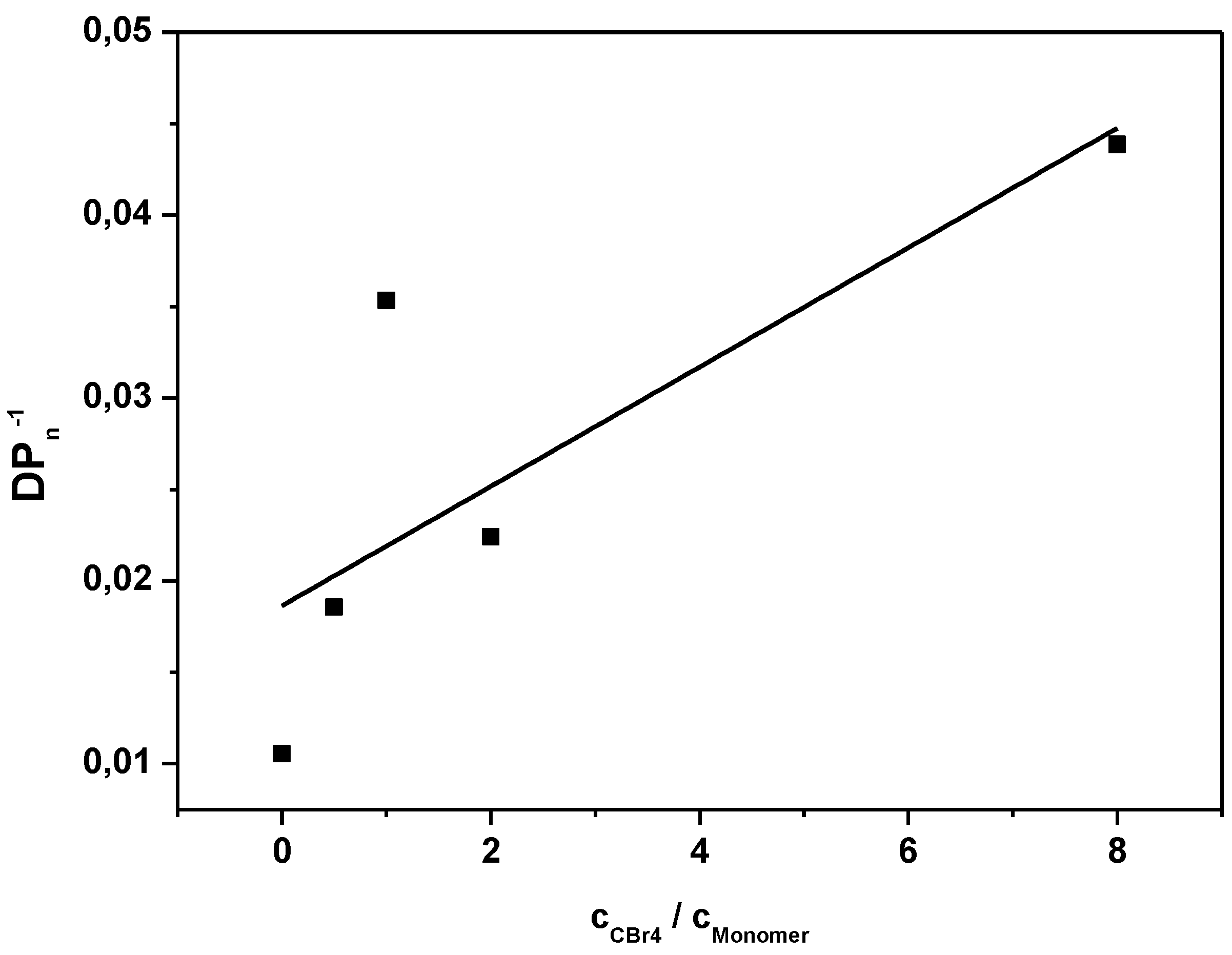
Figure A2 for the corresponding SEC elugrams). Results clearly indicate the good molecular weight control over both polymerizations upon changing the control agent concentration. Nevertheless, compared to classical vinyl polymerizations, an immense amount of CBr
4 is needed to reach this effect. Whereas in vinyl polymerizations, few mole percent of CTA are sufficient to expect good control, several equiv. of CTA are required in both precursor monomer routes. Up to 12 equiv. of control agent are needed to decrease the molecular weight of sulfinyl MDMO-PPV from 100,000 to 12,000 g·mol
−1. Standard MDMO-PPV polymerizations result in a yellow viscous oil for the precursor polymer and a red solid upon thermal elimination of the precursor to the conjugated polymer. Upon increasing amount of CBr
4, not only does the color of the conjugated MDMO-PPV changes from red to orange, also the physical appearance (from solid to viscous oil) changes, indicating the effect of the CTA on the chain length of the resulting MDMO-PPVs.
Figure 1.
Inverse of the degree of polymerization as a function of the CTA to premonomer concentration ratio for MDMO-PPV synthesized via the sulfinyl and the DTC route.
Figure 1.
Inverse of the degree of polymerization as a function of the CTA to premonomer concentration ratio for MDMO-PPV synthesized via the sulfinyl and the DTC route.
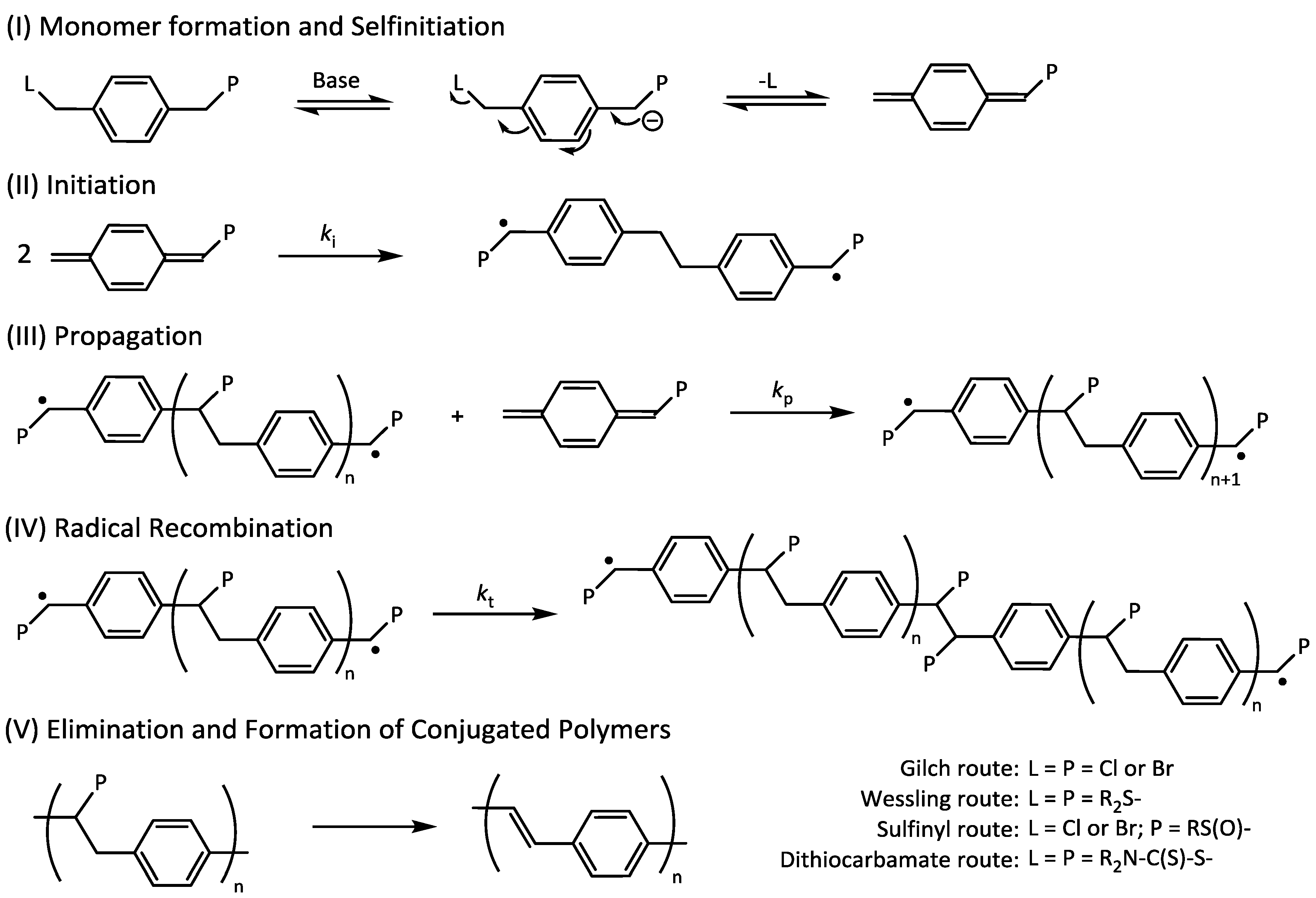
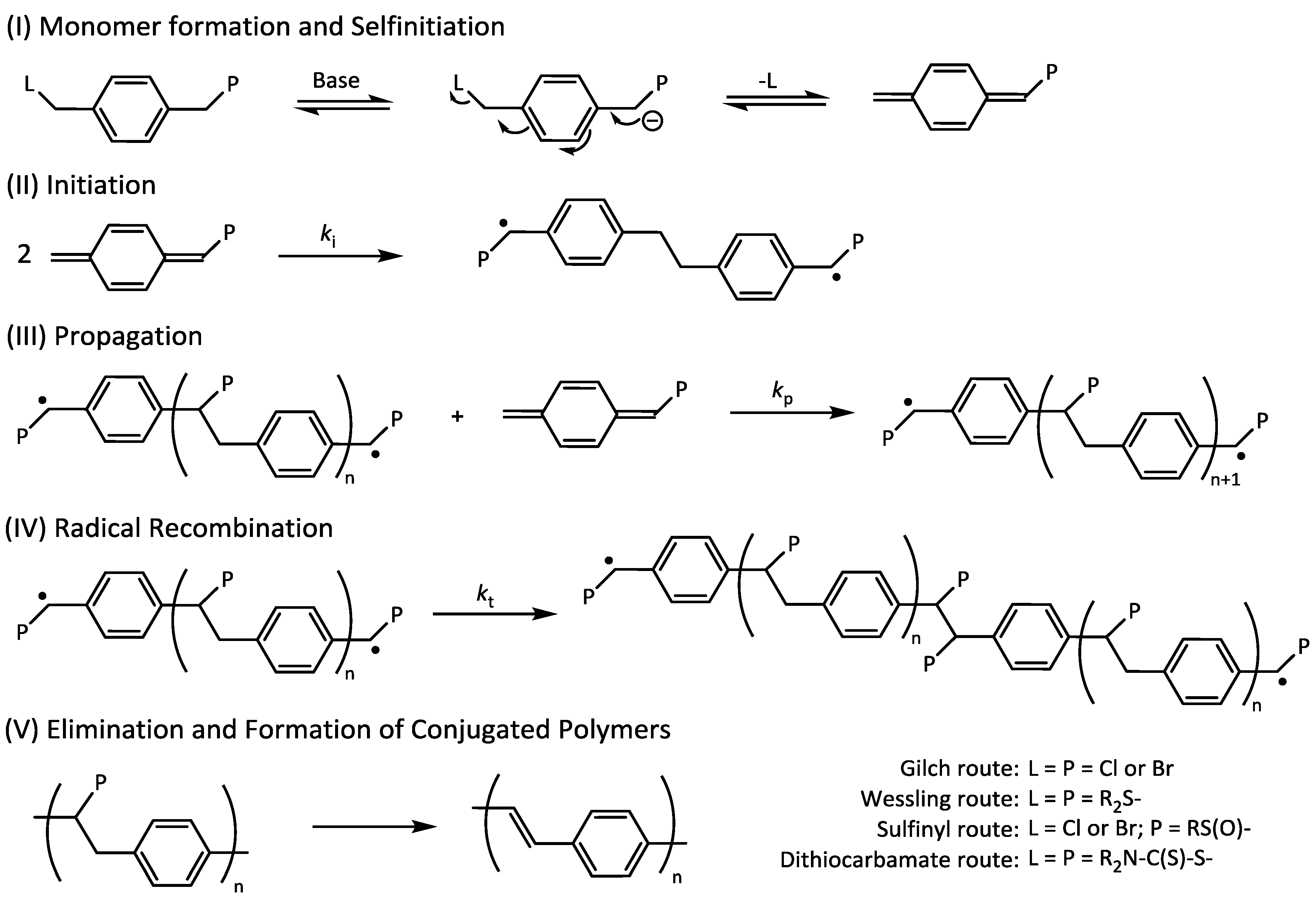
Scheme 8.
Reaction mechanism of precursor polymerizations and subsequent conversion of precursor polymers to conjugated PPV.
Scheme 8.
Reaction mechanism of precursor polymerizations and subsequent conversion of precursor polymers to conjugated PPV.
Interestingly, a further increase in control agent concentration above 8–12 equiv.—up to 25 equiv. was tested—does not decrease the molecular weight any further. This indicates that the chain transfer agent only controls molecular weight from a certain chain length on. For sulfinyl precursor MDMO-PPV, a minimum of roughly 25 repeating monomers are added to the chain before chain termination by transfer occurs. It should be noted that the precursor polymerization is not only complicated by the fact that the polymerization is rapid, but also by the fact that initiation proceeds via a biradical formation (see
Scheme 8 for full mechanism of the polymerization), hence requiring the transfer events to occur before chains are effectively dead.
Polymerizations of MDMO-PPV using the DTC precursor route show an effect of the CTA at much lower CTA concentrations. With 0.04 equiv. of CBr
4 a reduction in molecular weight is observed from 100,000 to 54,000 g·mol
−1, in comparison to 10-fold higher concentrations being required in the sulfinyl route to achieve the same effect. Increasing the CBr
4 content to one equiv. reduces chain growth to such a high extent that basically no polymerization is taking place anymore. This disparate chain transfer behavior is well represented in the individual transfer constants that are determined from the data. For both polymerizations in the sulfinyl route, for
T = 30 °C a
Ctr of 0.0034 is obtained in good agreement with our previous study, indicating that the small change in the side chain has no or only very little kinetic effect on the polymerization [
39]. For the DTC route polymerization, a
Ctr of 0.46 at
T = 35 °C is obtained (see
Table 2), thus at a value much closer to conventional vinyl polymerization. This increased transfer constant indicates that either transfer to CBr
4 is favored in this route, or that propagation is significantly slower (or in principle a combination of the two is operational). In either way, control in the DTC route is simpler to achieve compared to the sulfinyl route and further endeavors into controlling this type of polymerization should take the DTC route into account.
Table 2.
Chain transfer constant for conjugated PPVs synthesized via the sulfinyl (T = 30 °C) and DTC (T = 35 °C) precursor routes.
Table 2.
Chain transfer constant for conjugated PPVs synthesized via the sulfinyl (T = 30 °C) and DTC (T = 35 °C) precursor routes.
| Conjugated PPVs | Chain Transfer Constant Ctr |
|---|
| MDMO-PPV (DTC route) | 0.46 |
| MDMO-PPV (sulfinyl route) | 0.0038 |
| CPM-PPV (sulfinyl route) | 0.0034 |
In further investigations, a deeper look into the chain transfer kinetics of the sulfinyl route was taken. Despite the better performance of the DTC route, similarly good materials can be accessed at higher equiv. of CBr
4 in the sulfinyl route. Moreover, even though general uncertainties exist for precursor polymerizations with respect to mechanism and individual rate coefficients, the sulfinyl route is amongst the best studied precursor routes [
16,
17,
18,
19,
20]. Further systematic kinetic investigation of this reaction type is thus highly useful and thus the reason why—in spite of the better results for the DTC route—further investigation still focuses on the sulfinyl route. To date, no reliable initiation, propagation or termination coefficients are available for any precursor polymerizations, but efforts to fill this gap have been so far mostly focused on the sulfinyl route.
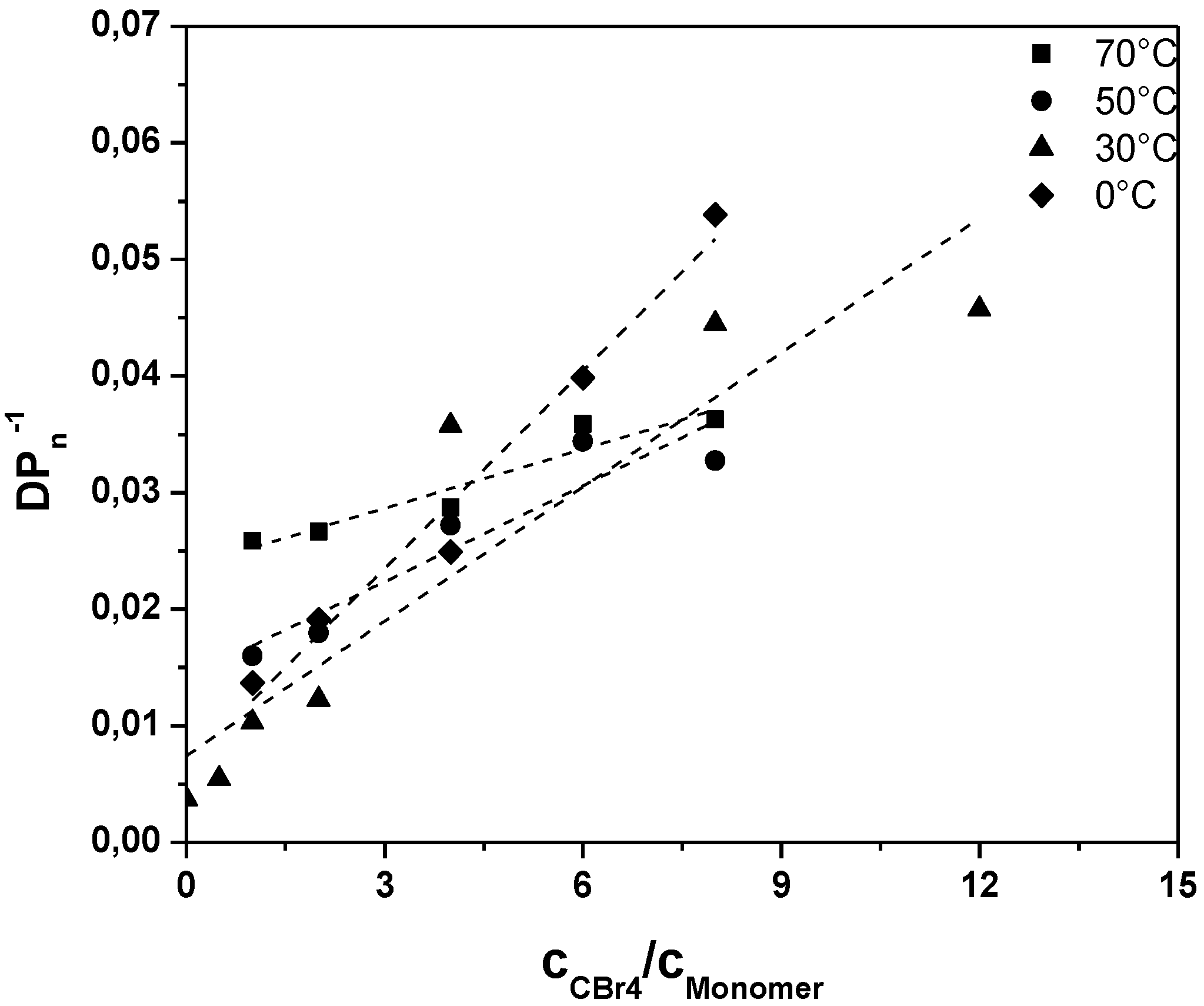
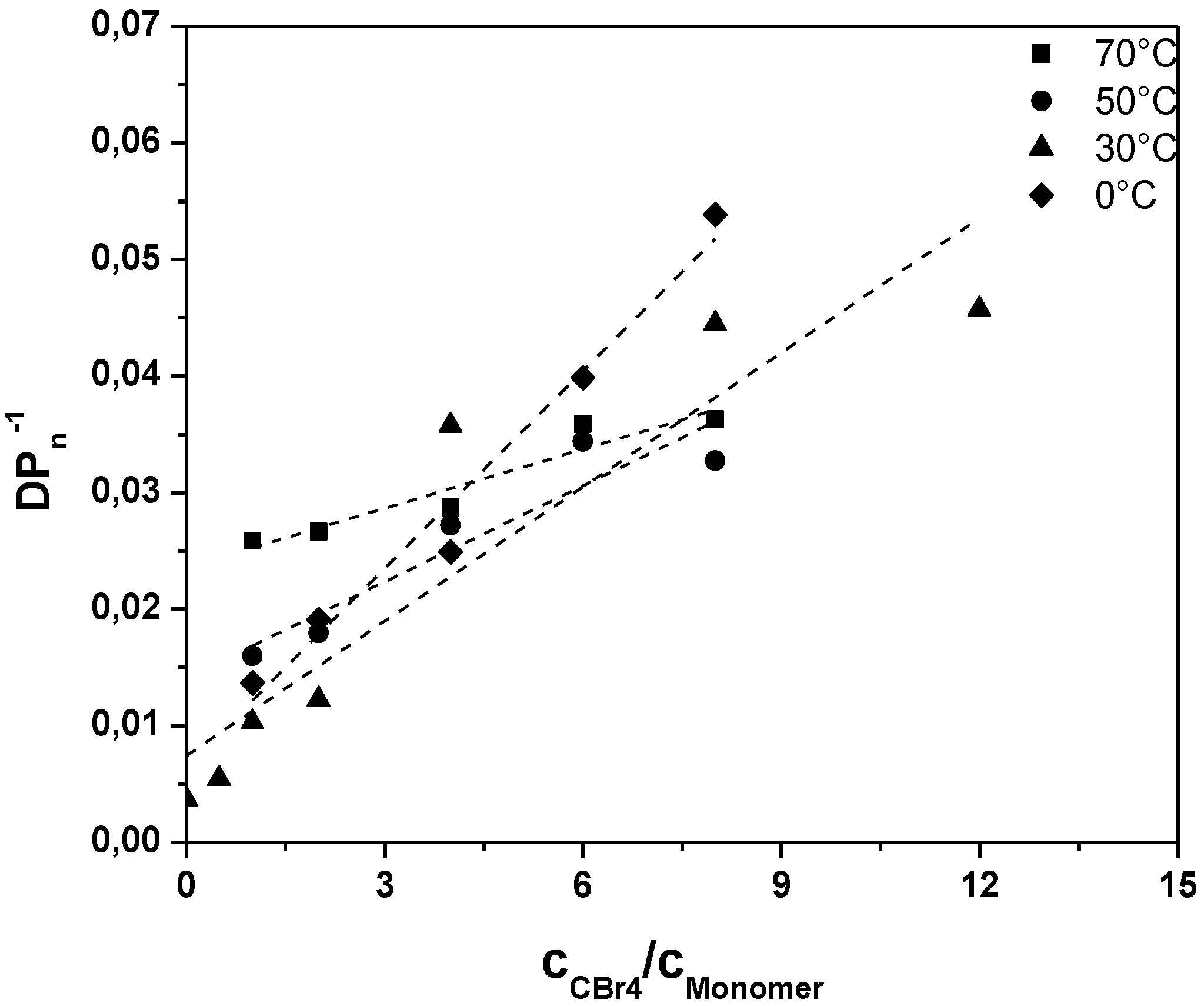
A closer look towards the activation energy of chain transfer can help in understanding the otherwise rapid sulfinyl route polymerization. Polymerizations under variation of the CTA concentration were thus conducted at different temperatures and analyzed towards the conjugated MDMO-PPV polymer (see
Figure 2,
Table 3 and
Appendix Table A1).
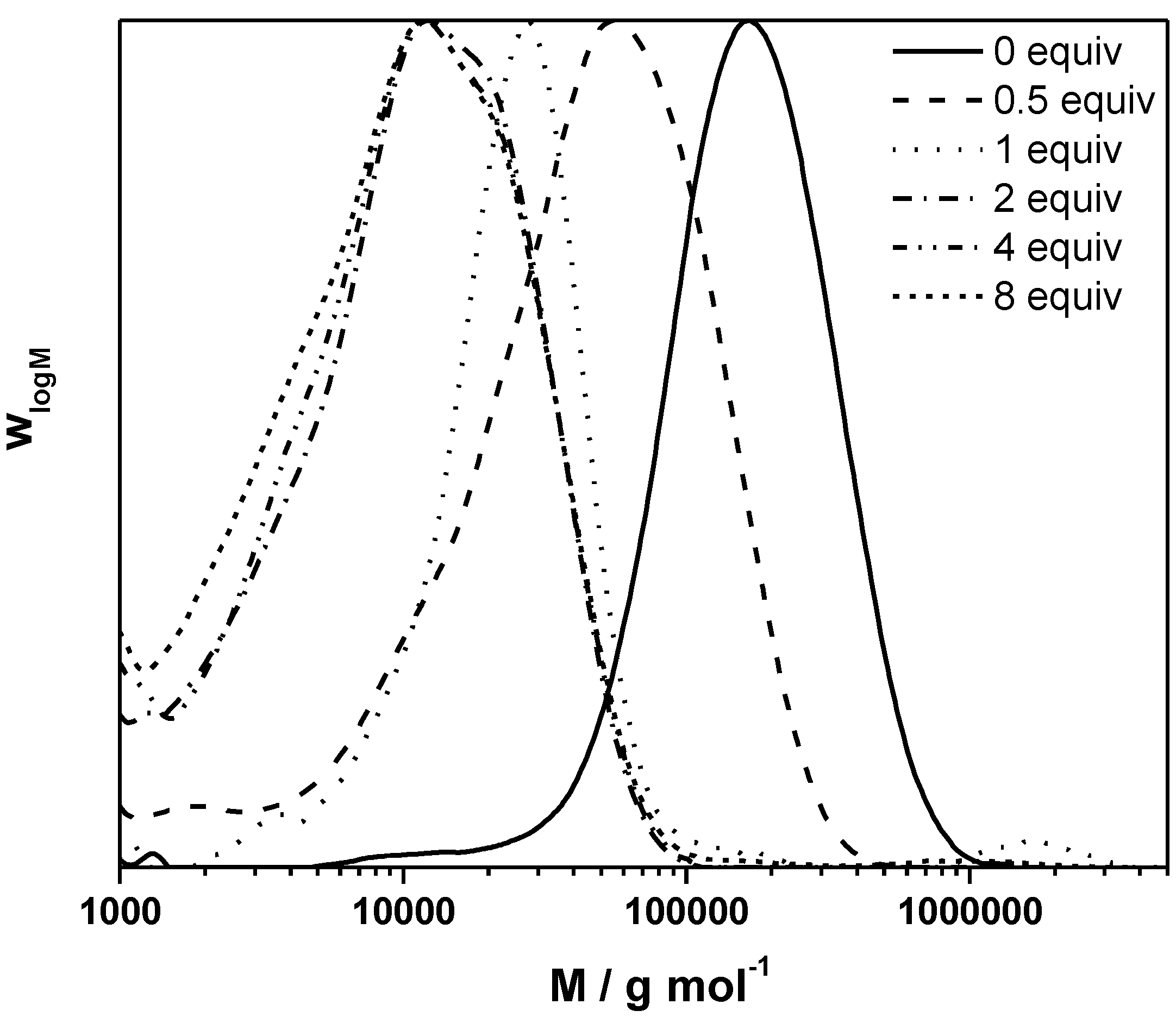
Results clearly demonstrate that upon increase in reaction temperature, a decrease in the transfer constant is followed. At the same time, lower
DPno is reached with increasing temperature, indicating that also the ratio of termination, propagation and initiation changes strongly with temperature. The decreasing
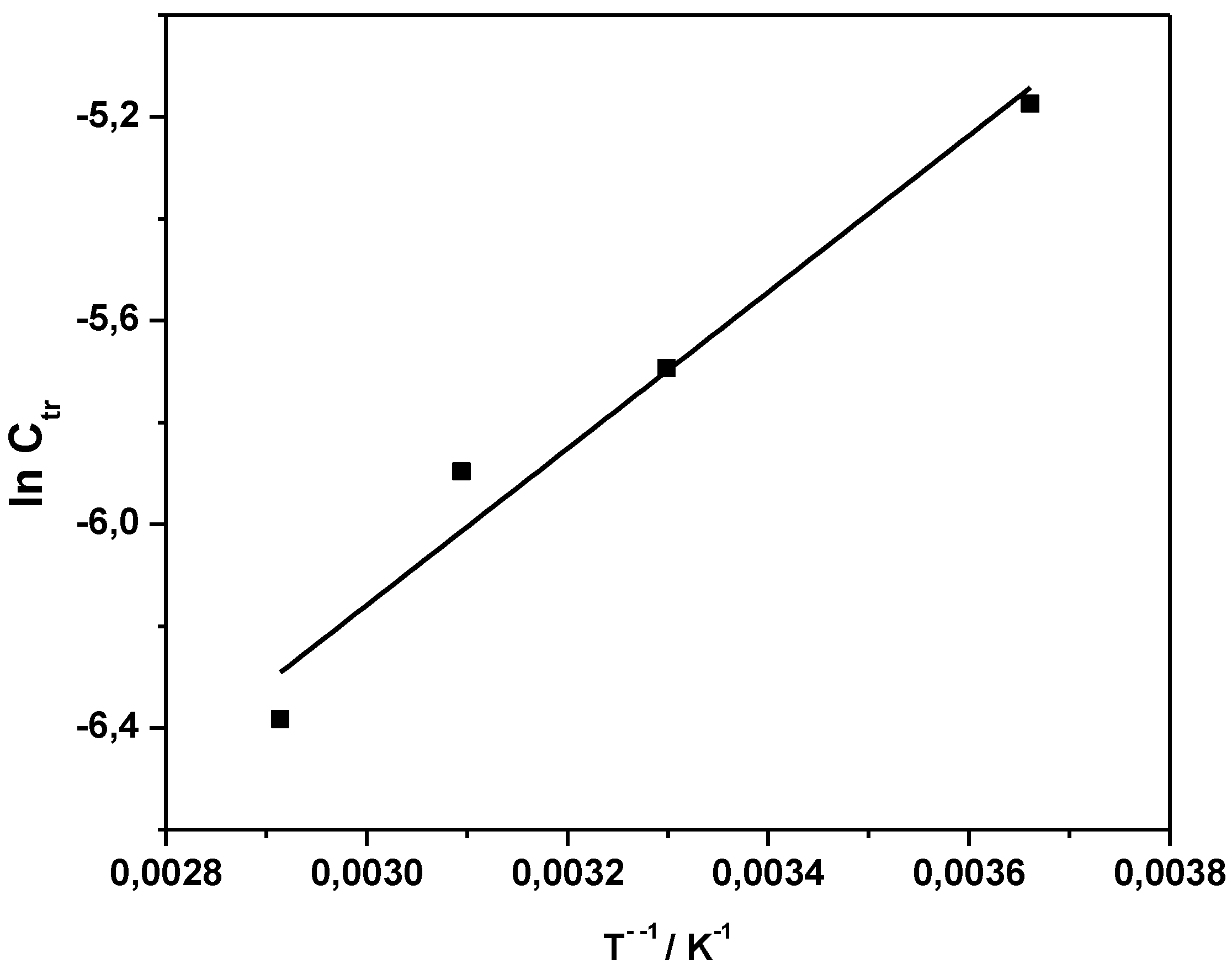
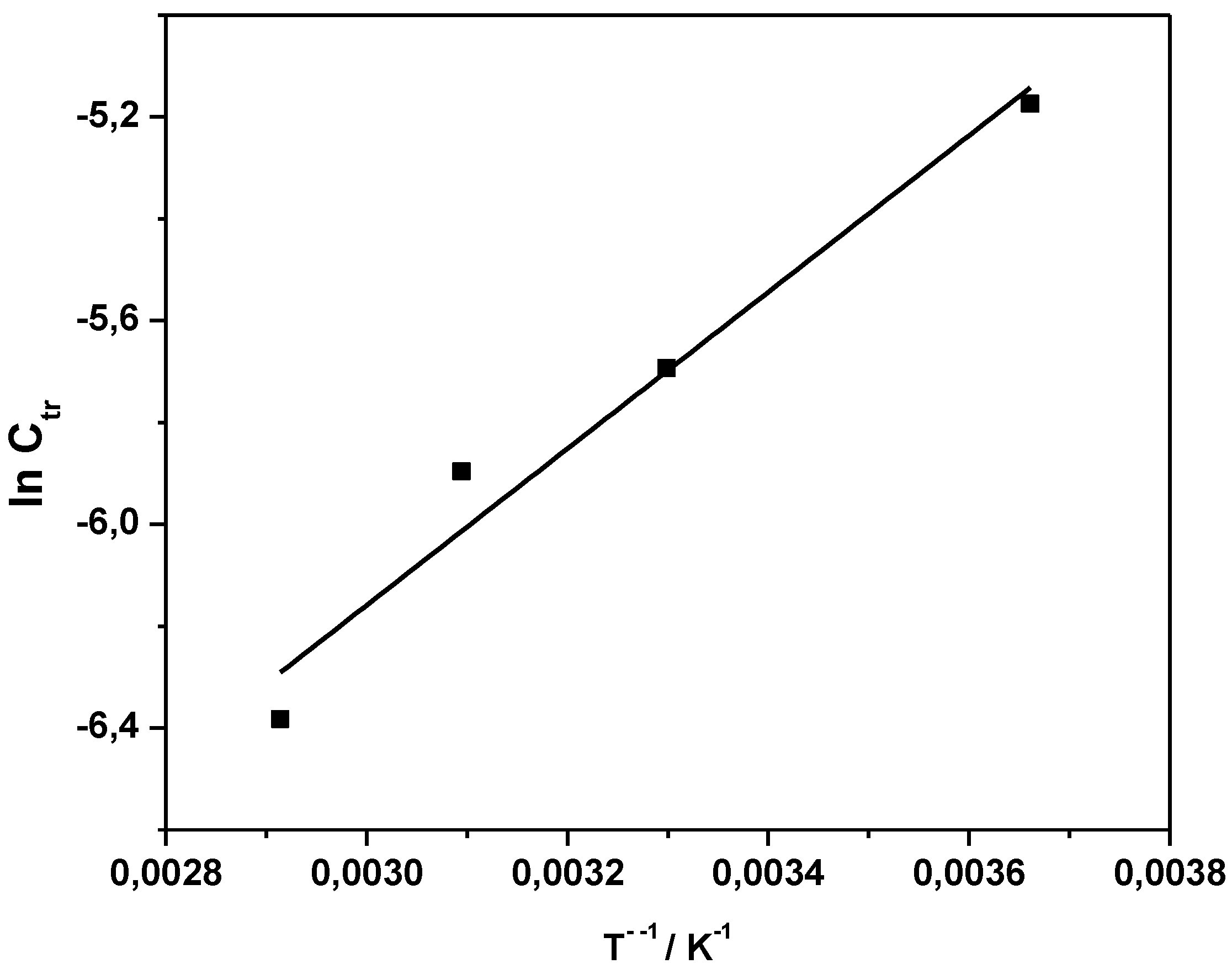
Ctr indicates that either propagation increases stronger with temperature than radical transfer, or that transfer becomes overall less effective at higher temperatures. Analysis of the Arrhenius relation for
Ctr yields an apparent activation energy
EA(
Ctr) of the coupled parameter of −12.8 kJ·mol
−1 (see
Figure 3). The negative value is explained by the relation
EA(
Ctr) =
EA(
ktr) −
EA(
kp). The negative value thus indicates that the activation energy of propagation is larger than that of transfer. Under assumption that radical transfer is usually associated with activation energies in the range of 20 kJ·mol
−1, the conclusion can be drawn that the activation energy of propagation is in the range of 30 kJ·mol
−1 or higher, a value that is common for radical propagation reactions.
Figure 2.
Inverse of the degree of polymerization as a function of the CTA to premonomer concentration ratio for MDMO-PPV synthesized via the sulfinyl “precursor” route at different polymerization temperatures.
Figure 2.
Inverse of the degree of polymerization as a function of the CTA to premonomer concentration ratio for MDMO-PPV synthesized via the sulfinyl “precursor” route at different polymerization temperatures.
Table 3.
Chain transfer constants determined from the plots given in
Figure 2 for conjugated MDMO-PPV synthesized via the sulfinyl “precursor” route at different polymerization temperatures.
Table 3.
Chain transfer constants determined from the plots given in Figure 2 for conjugated MDMO-PPV synthesized via the sulfinyl “precursor” route at different polymerization temperatures.
| T | Ctr |
|---|
| 0 °C | 0.0056 |
| 30 °C | 0.0034 |
| 50 °C | 0.0028 |
| 70 °C | 0.0017 |
Figure 3.
Arrhenius plot of Ctr obtained in the temperature range of 0–70 °C.
Figure 3.
Arrhenius plot of Ctr obtained in the temperature range of 0–70 °C.
The above kinetic experiments nicely demonstrate the general difficulty to carry out controlled radical polymerizations to obtain PPV materials. The sulfinyl route is less susceptible to interfering transfer reactions, but allows for facile access of chain-length controlled polymers when several equiv. of transfer agent are employed. The dithiocarbamate route is simpler to control, which is reflected by a close to 100-fold difference in the specific chain transfer constant. Determination of the activation energy of the transfer constant for the sulfinyl route shows that less control is achieved with increasing reaction temperatures due to the high activation energy of the propagation reaction. This information may seem trivial on first glance, yet for a polymerization system for which to date only very few kinetic parameters are known, this is a significant advance in knowledge. Any additional rate parameter (or at least activation energy) that becomes available will aid in future modelling studies and a paramount understanding of the polymerization. Only with such models at hand, rational selection of reaction conditions will be possible and true product control achieved.
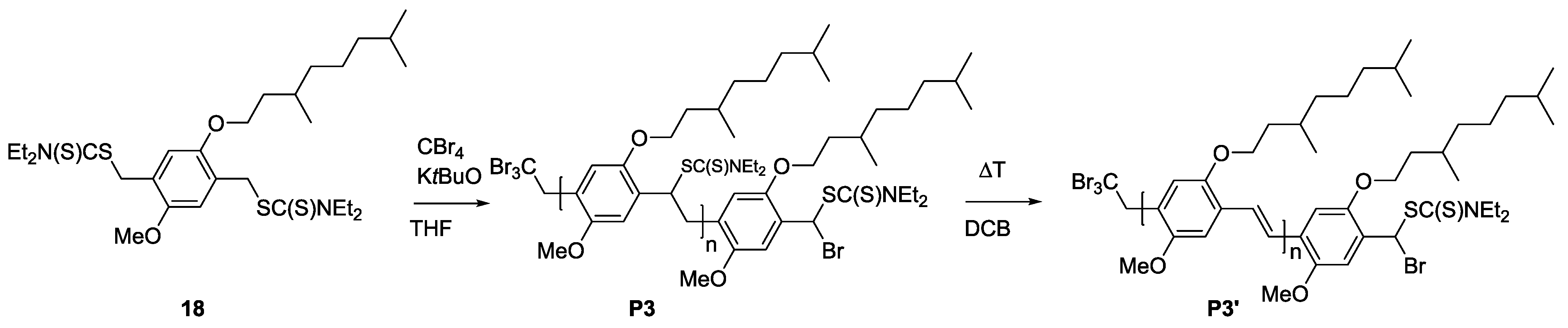
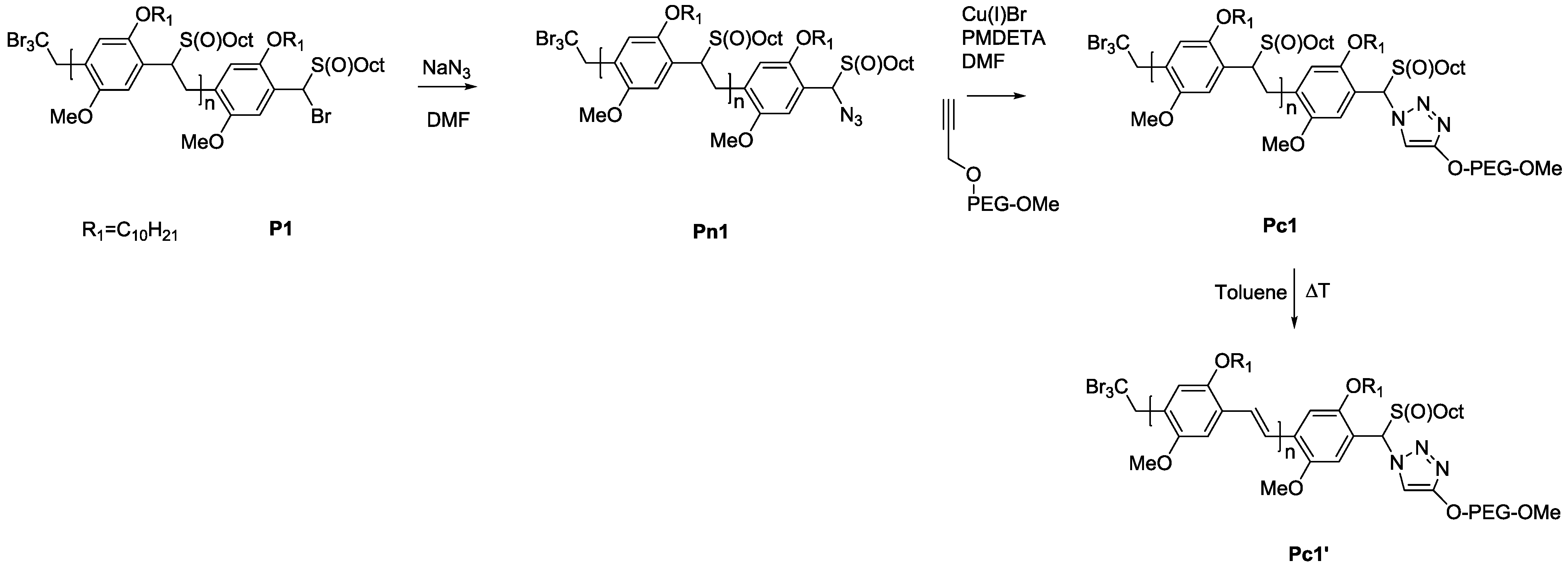
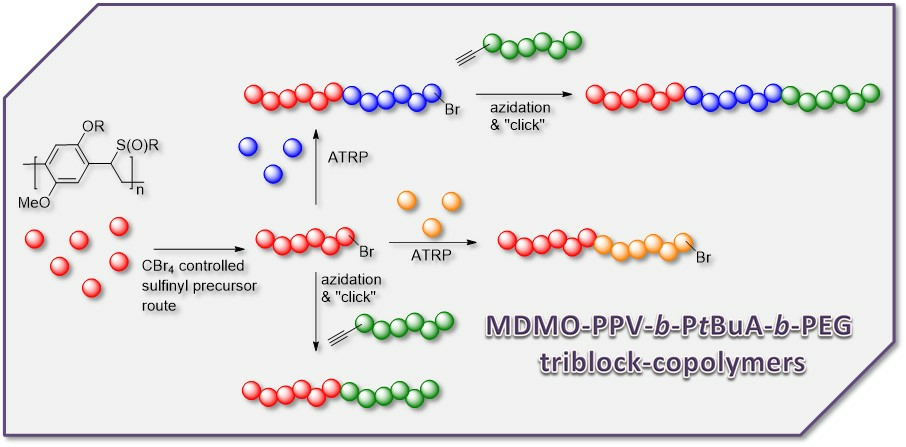
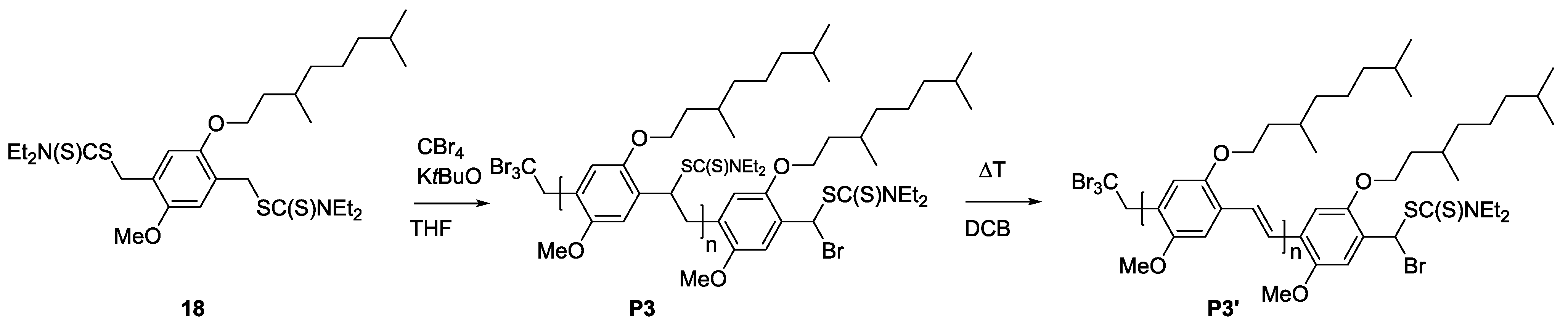
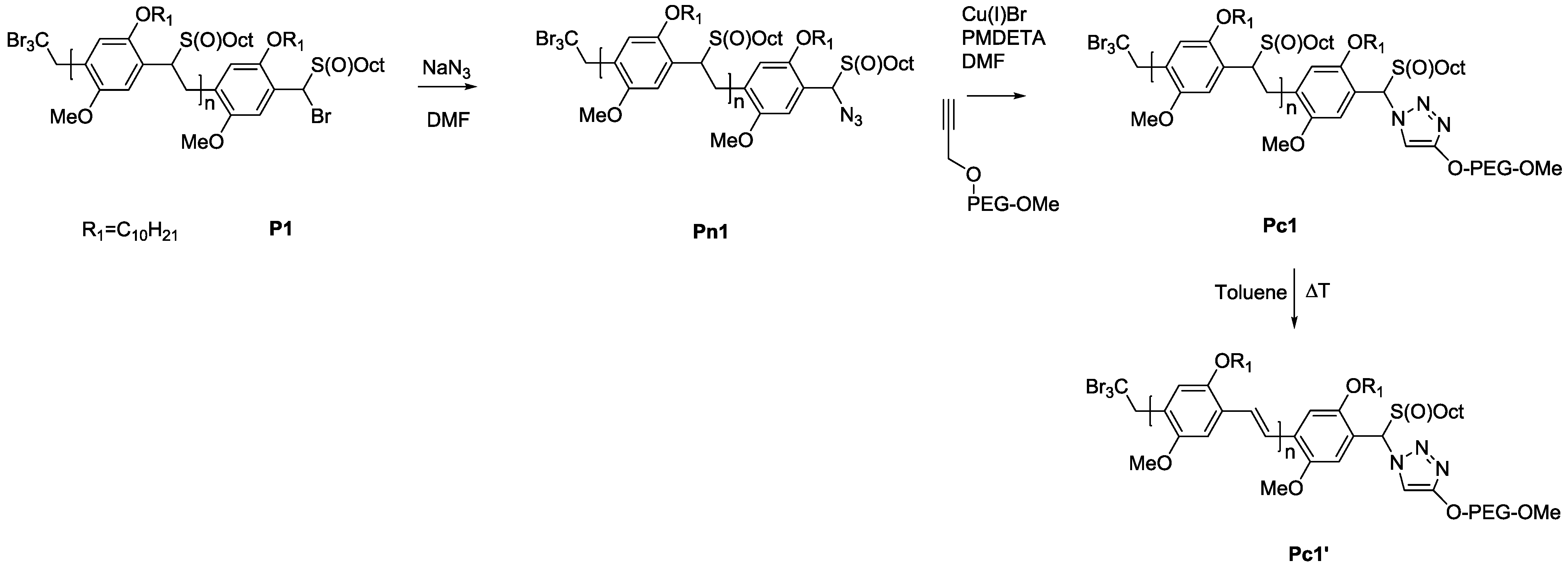
3.3. Conjugation of PPV Block Copolymers Using CuAAC Conditions
Sequential approaches towards successful block copolymerization (e.g., ATRP or SET-LRP) restrict the choice of the second block to vinyl-type monomers that are able to undergo (controlled) radical polymerization. Thus, the development of a modular approach allowing any combination of building blocks is also highly attractive. Therefore, copper-catalyzed alkyne-azide cycloaddition (CuAAC) is employed for the synthesis of PPV block copolymers. As a first attempt, MDMO-PPV-
b-PEG block copolymers were targeted using such a modular approach. Therefore, bromine endcapped precursor sulfinyl MDMO-PPV was first functionalized at elevated temperatures with an azide group, using an excess amount of sodium azide (10 equiv. to polymer) in DMF. In a second step, the obtained precursor MDMO-PPV-azide is then clicked to a PEG-alkyne using standard CuAAC reaction conditions. Reactions are carried out at 25 °C, using DMF as solvent and Cu(I)Br and PMDETA as catalyst. Both precursors PPV and PEG were added in equimolar amounts to avoid excesses of homopolymer being left after reaction. The reaction mixture was allowed to stir for an extended time (up to three days) after which copper was removed from the product mixture by passing the solution over a short alumina column. In a second step, all precursor block copolymers are eliminated, resulting in conjugated MDMO-PPV-
b-PEG block copolymer. Results regarding molecular weight and dispersity for the starting MDMO-PPV homopolymer and the resulting MDMDO-PPV-
b-PEG block copolymer can be found in
Table 6 and molecular weight profiles are plotted in
Figure 7.
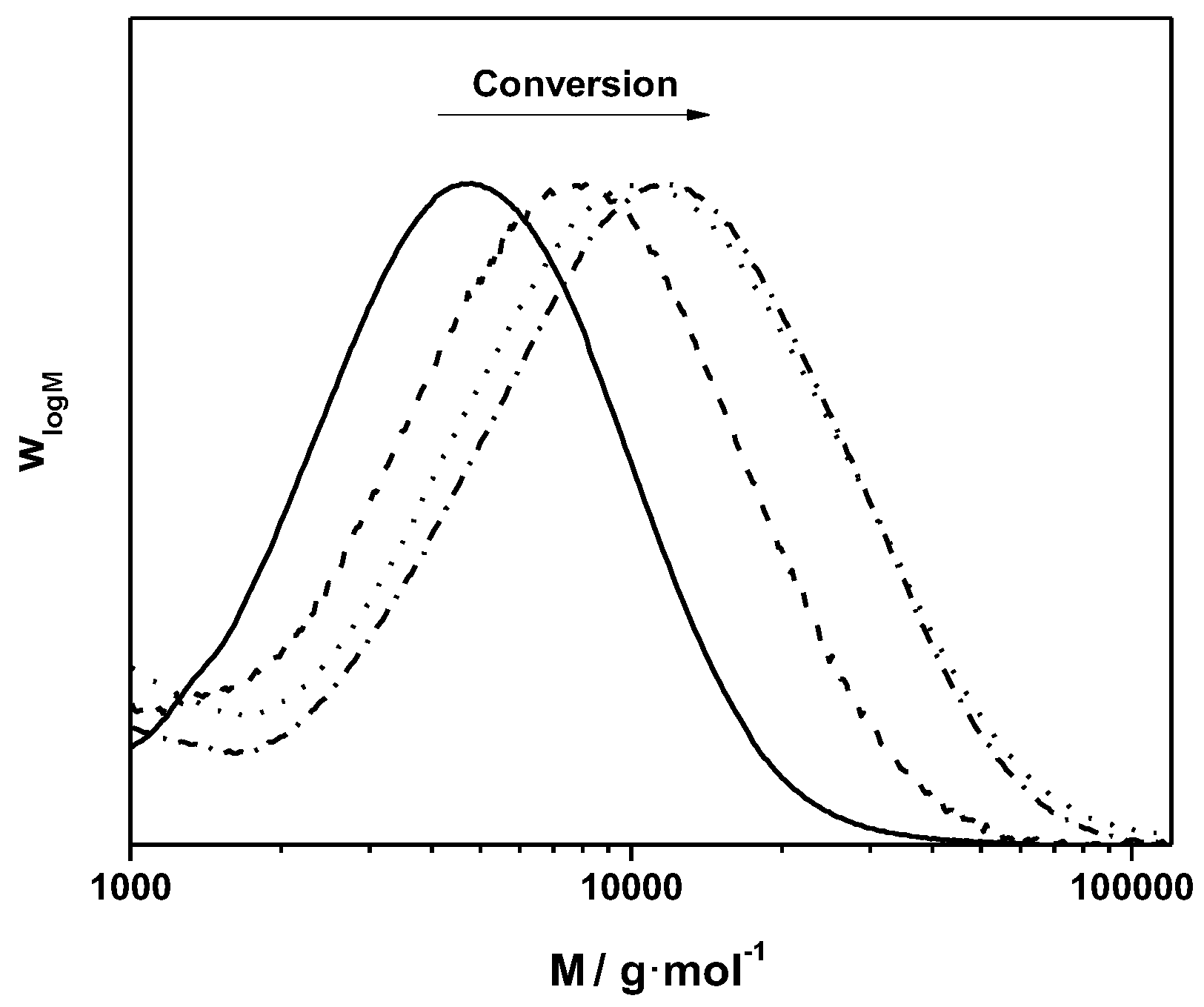
As can be seen from the molecular weight distributions in
Figure 7, a bimodal SEC profile is obtained after reaction, representing a mixture of two separate distributions—MDMO-PPV homopolymer—and the block copolymer MDMO-PPV-
b-PEG, indicating a partly successful ligation. Comparison of the starting materials
Mp indicated that the click reaction by itself was successful, but that pure PPV remained—whether due to reaction inefficiencies or due to insufficient functionalization of the homopolymer. Closer inspection and repetitions of the CuAAC reaction leads to the conclusion that the partial success (no improvement is seen even after three days’ reaction time under varying conditions) is due to solubility issues. As mentioned before, the azide functionalized MDMO-PPV does not dissolve well in DMF, which can make the azide partially inaccessible for reaction. Purification of the MDMO-PPV-b-PEG block copolymer from the homopolymer leftovers is far from trivial. Residual PEG can relatively easily be removed by washing the mixture with water, residual MDMO-PPV can only be removed by preparative recycling SEC, as was done before [
44].
Table 6.
Molecular weights and Đ for the homopolymers and conjugated MDMO-PPV-b-PEG block copolymer.
Table 6.
Molecular weights and Đ for the homopolymers and conjugated MDMO-PPV-b-PEG block copolymer.
| MDMO-PPV homo- and di-block copolymer | Mnapp g∙mol−1 | Mwapp g∙mol−1 | Mpapp g∙mol−1 | Đ |
|---|
| MDMO-PPV-N3 | 7,800 | 12,400 | 8,300 | 1.60 |
| PEG-alkyne | 6,400 | 6,600 | 7,300 | 1.10 |
| MDMO-PPV-b-PEG | - | - | 18,400 | - |
Figure 7.
SEC profile for the direct coupling of alkyne-functionalized PEG to azide functionalized eliminated MDMO-PPV using click conditions without purification of the homopolymer leftovers.
Figure 7.
SEC profile for the direct coupling of alkyne-functionalized PEG to azide functionalized eliminated MDMO-PPV using click conditions without purification of the homopolymer leftovers.
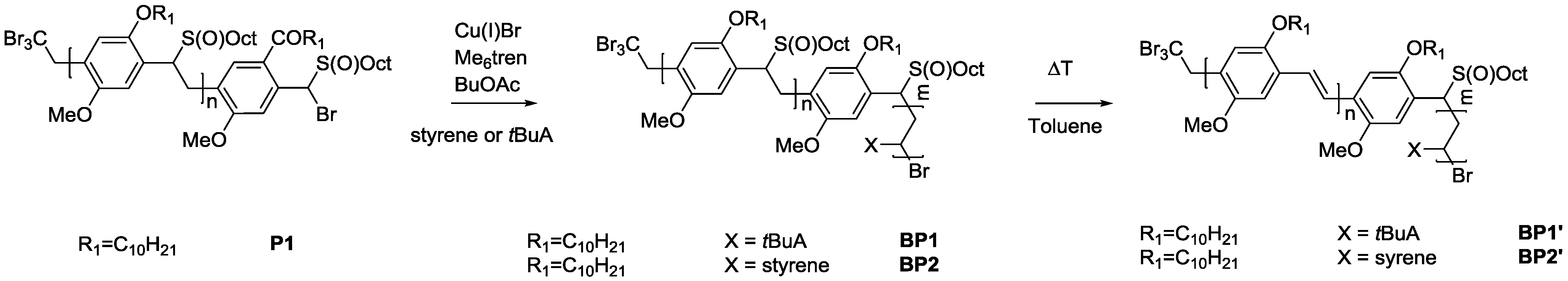
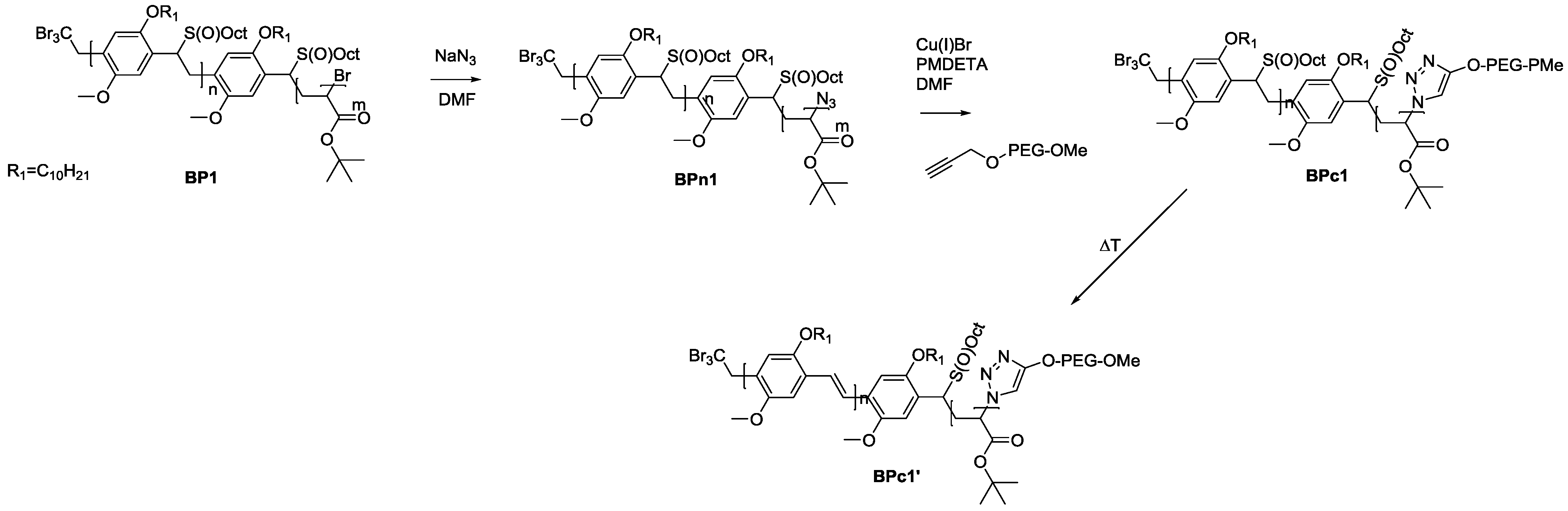
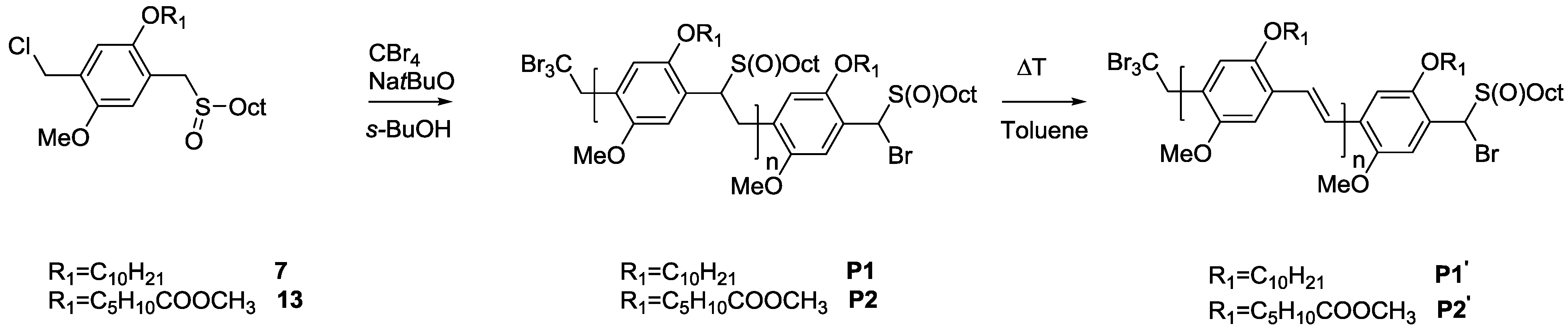
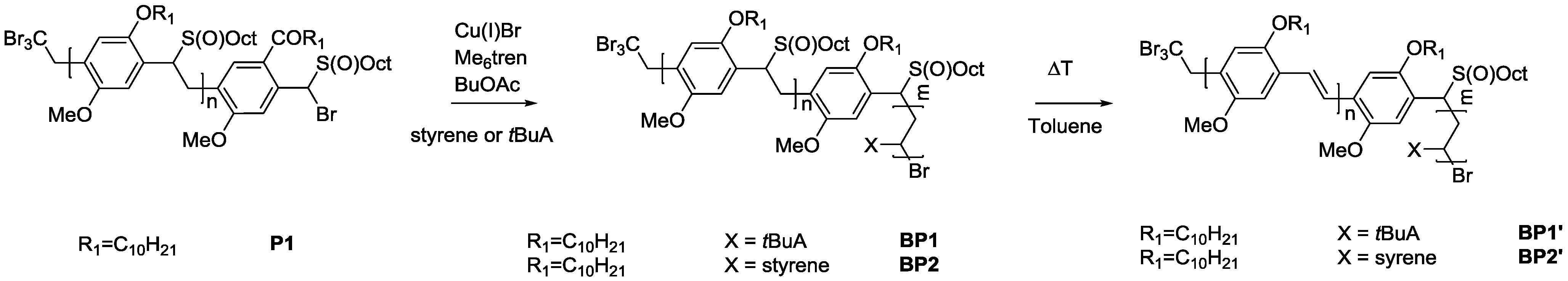
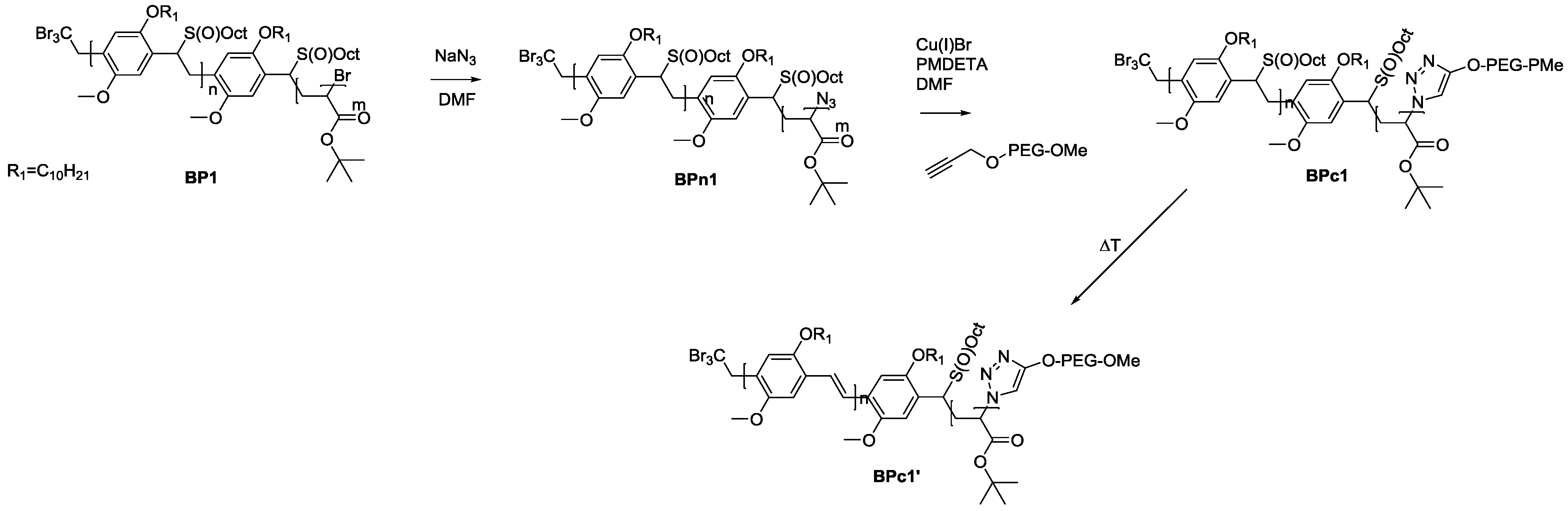
As solubility of the polymers may play a significant role in the somewhat hindered click reaction, synthesis of triblock copolymers from successive chain transfer polymerization, ATRP chain extension with an acrylate and CuAAC conjugation was tested, yielding material with the desired structure MDMO-PPV-
b-P
tBuA-
b-PEG. MDMO-PPV-
b-P
tBuA-Br polymers as discussed above were modified with NaN
3 to obtain azide-functional material. The generally better solubility of the acrylate block and the higher chain flexibility of the second block should aid in the click reaction and improve the system. Furthermore, P
tBuA can at a later stage be eliminated to yield poly(acrylic acid) blocks and thus give access to pH-responsive triblock copolymer structures. The CuAAC reaction with PEG-alkyne was then again allowed to react for three days under the same conditions of the diblock CuAAC conjugation reaction, after which the reaction is stopped by precipitation of the resulting triblock copolymers in ice-cold hexane followed by removal of the copper on a short alumina column. After thermal elimination of the material and precipitation, an orange colored conjugated MDMO-PPV-
b-P
tBuA-
b-PEG was obtained. Molecular weight distributions of the individual homo-, di- and resulting tri-block copolymers as well as corresponding molecular weights are presented in
Table 7 and
Figure 8. The CuAAC conjugation shows in this case good success.
Table 7.
Average molecular weights and Đ for the MDMO-PPV-b-PtBuA-b-PEG triblock copolymers and its precursors.
Table 7.
Average molecular weights and Đ for the MDMO-PPV-b-PtBuA-b-PEG triblock copolymers and its precursors.
| MDMO-PPV di- and tri-block copolymers | Mnapp g∙mol−1 | Mwapp g∙mol−1 | Mpapp g∙mol−1 | Đ |
|---|
| MDMO-PPV | 6,100 | 9,300 | 8,060 | 1.50 |
| MDMO-PPV-b-PtBuA | 7,300 | 11,800 | 10,500 | 1.60 |
| PEG-alkyne | 6,400 | 6,600 | 6,500 | 1.10 |
| MDMO-PPV-b-PtBuA-b-PEG | - | - | 19,200 | - |
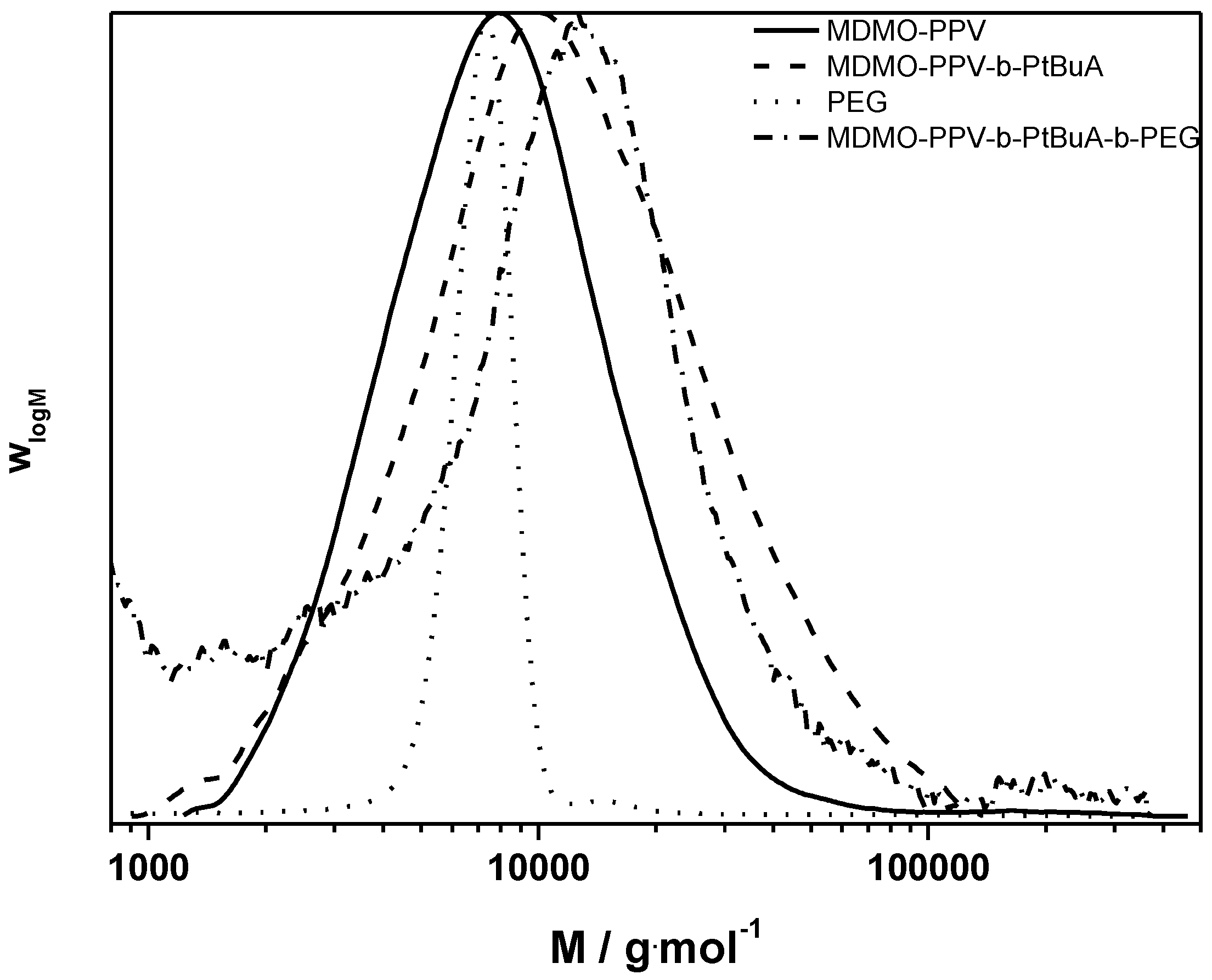
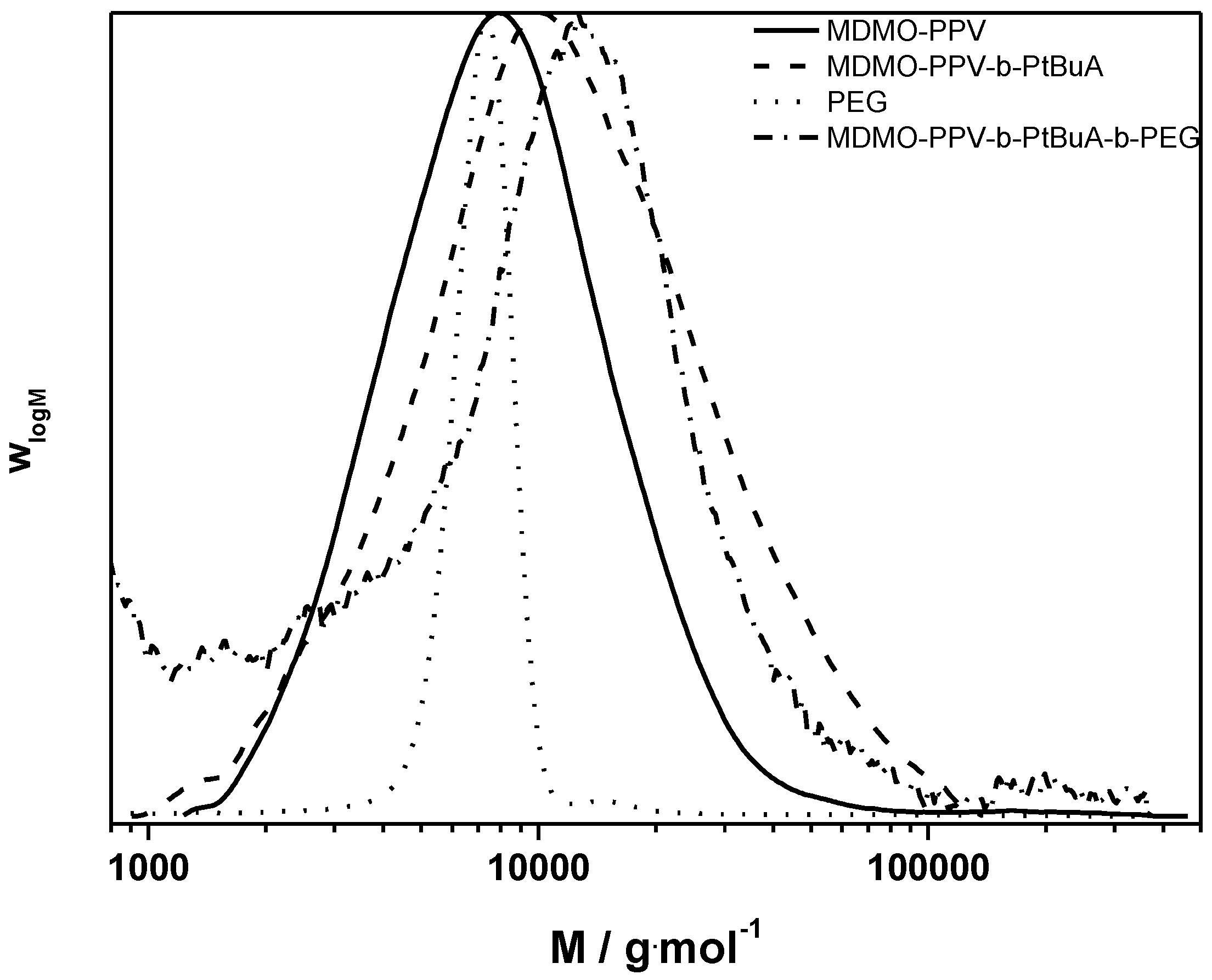
Figure 8.
Molecular weight distributions of the clicked MDMO-PPV-b-PtBuA-b-PEG triblock copolymer and its precursors.
Figure 8.
Molecular weight distributions of the clicked MDMO-PPV-b-PtBuA-b-PEG triblock copolymer and its precursors.
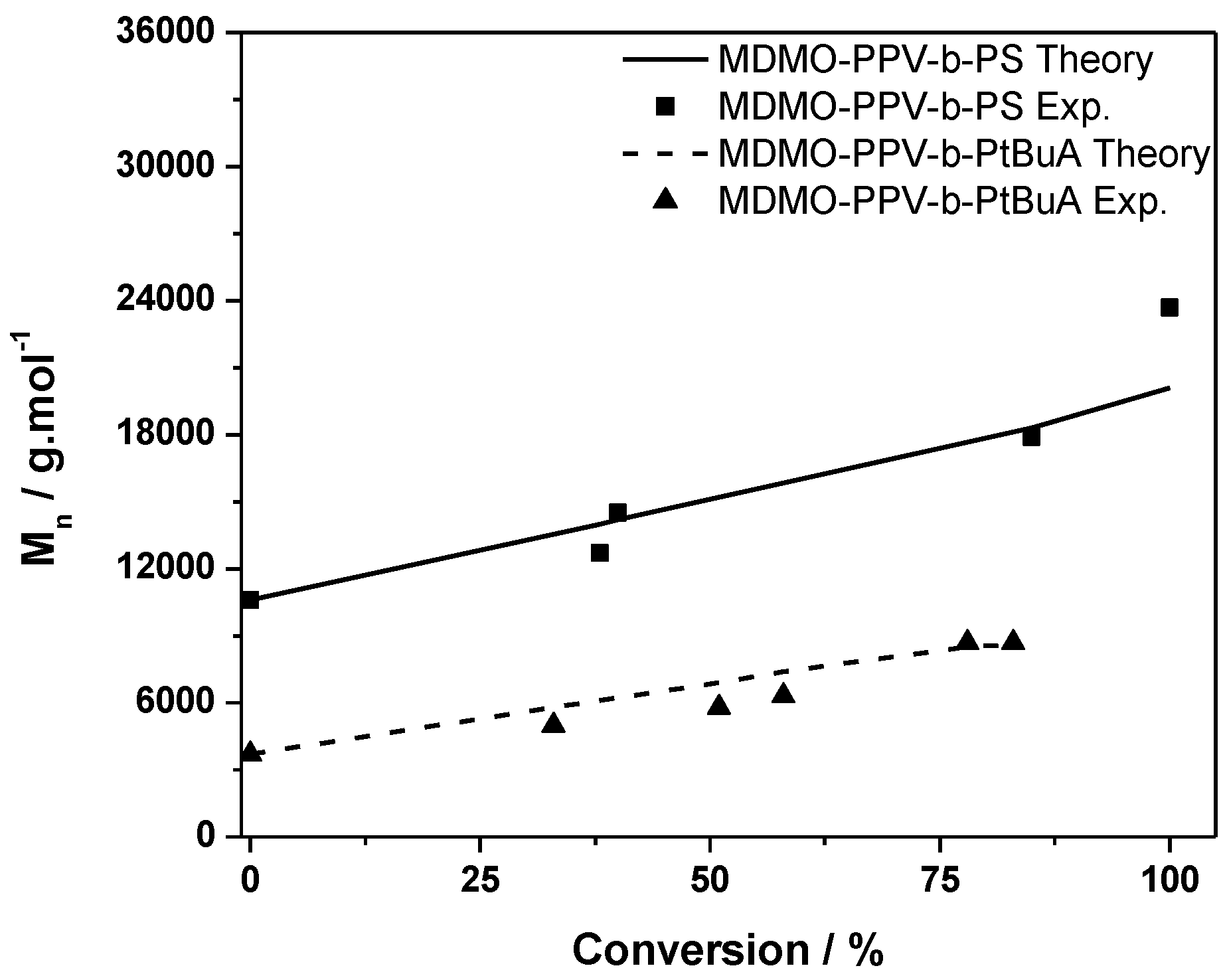
The ATRP chain extension yields similar good results as in the case described above. The CuAAC reaction proceeds well. The triblock copolymer product distribution still shows some material at the lower molecular weight side of the distribution, which could not be removed by precipitation or washing. Thus, no average molecular weights could be determined and again only Mp is discussed. The MDMO-PPV-b-PtBuA copolymer has a peak molecular weight of 10,500 g·mol−1. The PEG-alkyne block has a molecular weight of 6500 g·mol−1. In the sum, the triblock structure features a Mp of 19,200 g·mol−1, which can be regarded as a good match—taking into account that no calibration exists for these materials and large variations in the MHKS parameters of the chains must be expected with each consecutive chain extension.



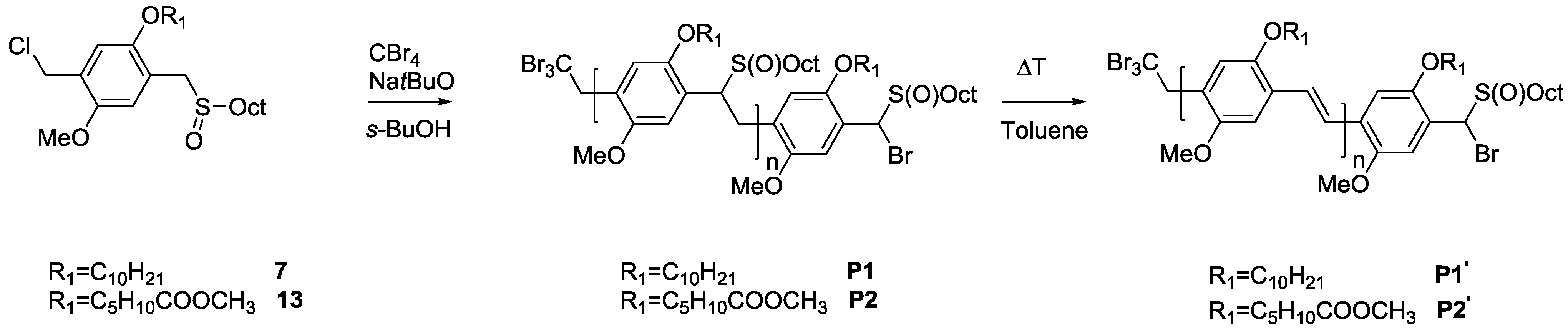


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}