1. Introduction
Natural polysaccharides such as chitosan (CS) comprise a class of very important polymers that have been widely utilized in a variety of fields [
1]. The most important feature of chitosan is its low toxicity compared with other natural polysaccharides. It is safety in terms of inertness, and low or no toxicity has been demonstrated by
in vivo toxicity studies, in which it’s oral lethal dose 50 (LD
50) in mice was found to be in excess of 16 g/day/kg body weight, which is higher than that of sucrose [
2,
3]. Additionally, chitosan is well tolerated by living tissues, including the skin, ocular membranes, as well as the nasal epithelium. For these reasons, chitosan is very valuable for a wide range of biomedical applications [
4,
5,
6].
Chitosan has a variety of applications in pharmaceutical, medicinal, and agricultural fields as well as wastewater treatment, food, cosmetics, and so on [
7,
8,
9,
10]. Also, being a natural polymer, chitosan can be used in nucleic acid delivery and tissue engineering applications. Chitosan is a biocompatible material that interacts with living cells without being cytotoxic [
11]. Chitosan has various biological properties including antimicrobial properties [
12], antioxidant properties [
13], and anti-inflammatory properties [
14]. Chitosan is also mucoadhesive, making it highly suitable for gene delivery to epithelium including the lungs and gastrointestinal tract [
15,
16,
17]. Chitosan has found use in novel applications such as vaccine and peptide delivery, in addition to its use in tissue engineering [
2,
6,
18]. In fact, a number of commercial applications of chitosan benefit from its antimicrobial properties, including its use in food preservation [
19,
20], in dentistry and ophthalmology, in the manufacture of wound dressings, and antimicrobial finished textiles. Therefore, investigations of the. antimicrobial potential of chitosan and its derivatives has recently gained momentum. However, the unsatisfactory performance of naturally available polymers usually fails to meet the needs of different fields. In order to expand the range of applications, structure modification is considered to be the effective ways in improving the performance of natural polymers [
21].
Accordingly, in this work we try to synthesize some new derivatives of chitosan by its reaction with a number of aromatic aldehydes and study their structures using different physical and chemical methods, as well as their antimicrobial and anticancer properties hoping to be more active.
2. Materials and Methods
2.1. Materials
Chitosan was purchased from Acros Organics, Morris Plains, NJ, USA. Its deacetylation degree is 88% and its average molecular weight is 100,000–300,000 Da. Acetic acid, methanol, were of analytical grade from Aldrich and were used as received. Dimethyl sulfoxide (DMSO), crystal violet and trypan blue dye were purchased from Sigma (St. Louis, MO, USA). Fetal Bovine serum, DMEM (Dulbecco Modified Eagle’s Medium), RPMI-1640, HEPES buffer solution, l-glutamine, gentamycin, and 0.25% Trypsin-EDAT were purchased from Lonza (Basel, Switzerland). Crystal violet (1%) was composed of 0.5% (w/v) crystal violet and 50% methanol, then made up to volume dd H2O and filtered through a whatmann No. 1 filter paper. Antimicrobial analysis and anti-cancer activity screening were done by the regional center for mycology and biotechnology, Al-Azhar University.
2.2. Characterization of Chitosan
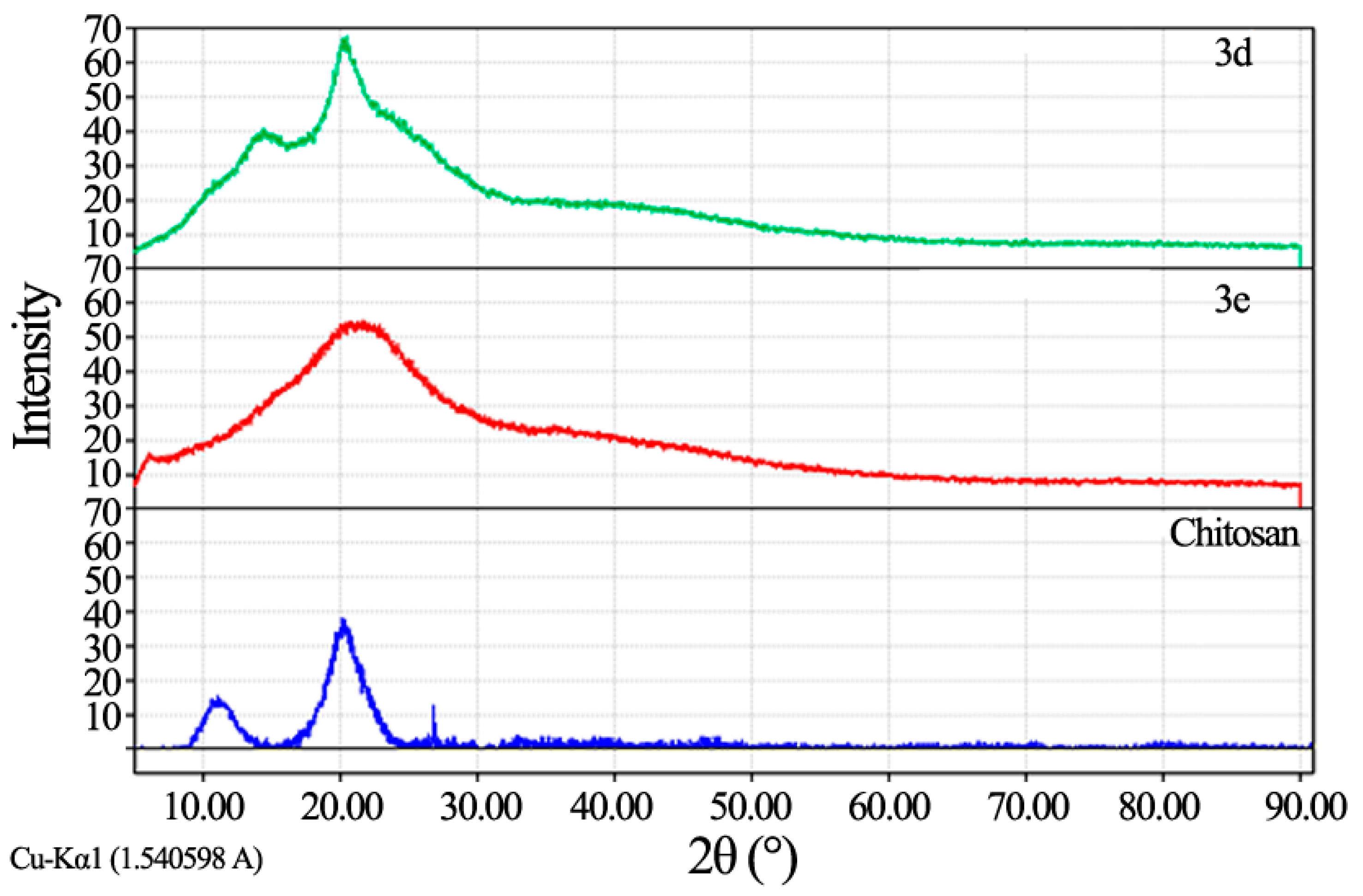
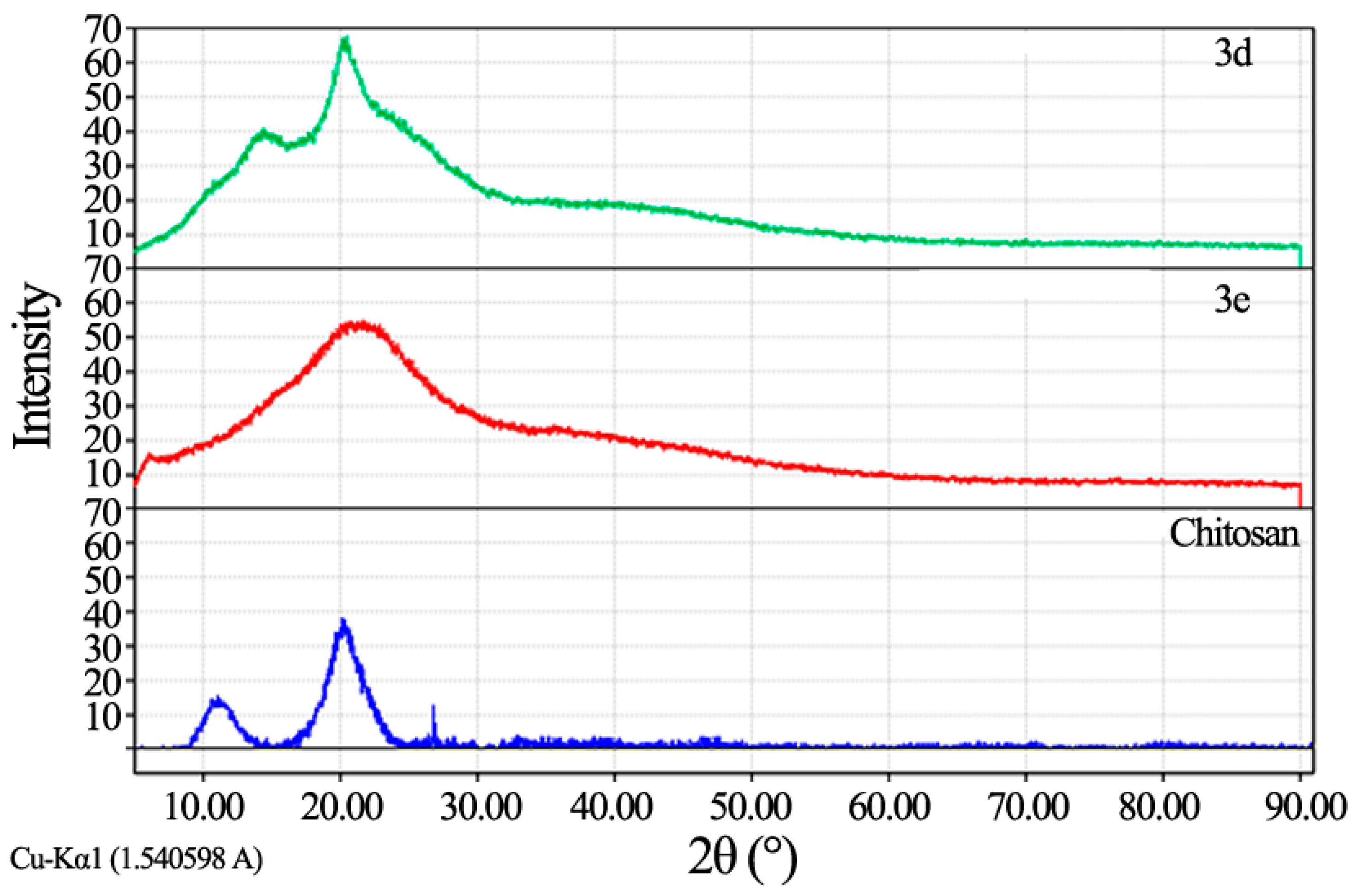
Fourier transforms infrared spectroscopy (FT-IR) analysis: FT-IR spectra were recorded using KBr discs on Perkin Elmer- USA Spectrometer at room temperature within the wave number range of 4000–400 cm−1; Proton Nuclear Magnetic Resonance (1H NMR): 1H NMR spectra were recorded using a Gemini-300 MHz instrument in DMSO–d6 as a solvent at 25 °C. Chemical shifts (δ) are expressed in part per million (ppm) using tetramethylsilane as an internal standard; X-ray diffraction (XRD) analysis: In X-ray diffraction technique (XRD), X-ray diffraction profiles of chitosan and chitosan derivatives were recorded by Bruker, Germany powder X-ray diffractometer, model D8 Advance, source 2.2 kW Cu anode. The relative intensities were recorded within the range of 10°–90° (2θ) at a scanning rate of 5°·min−1.
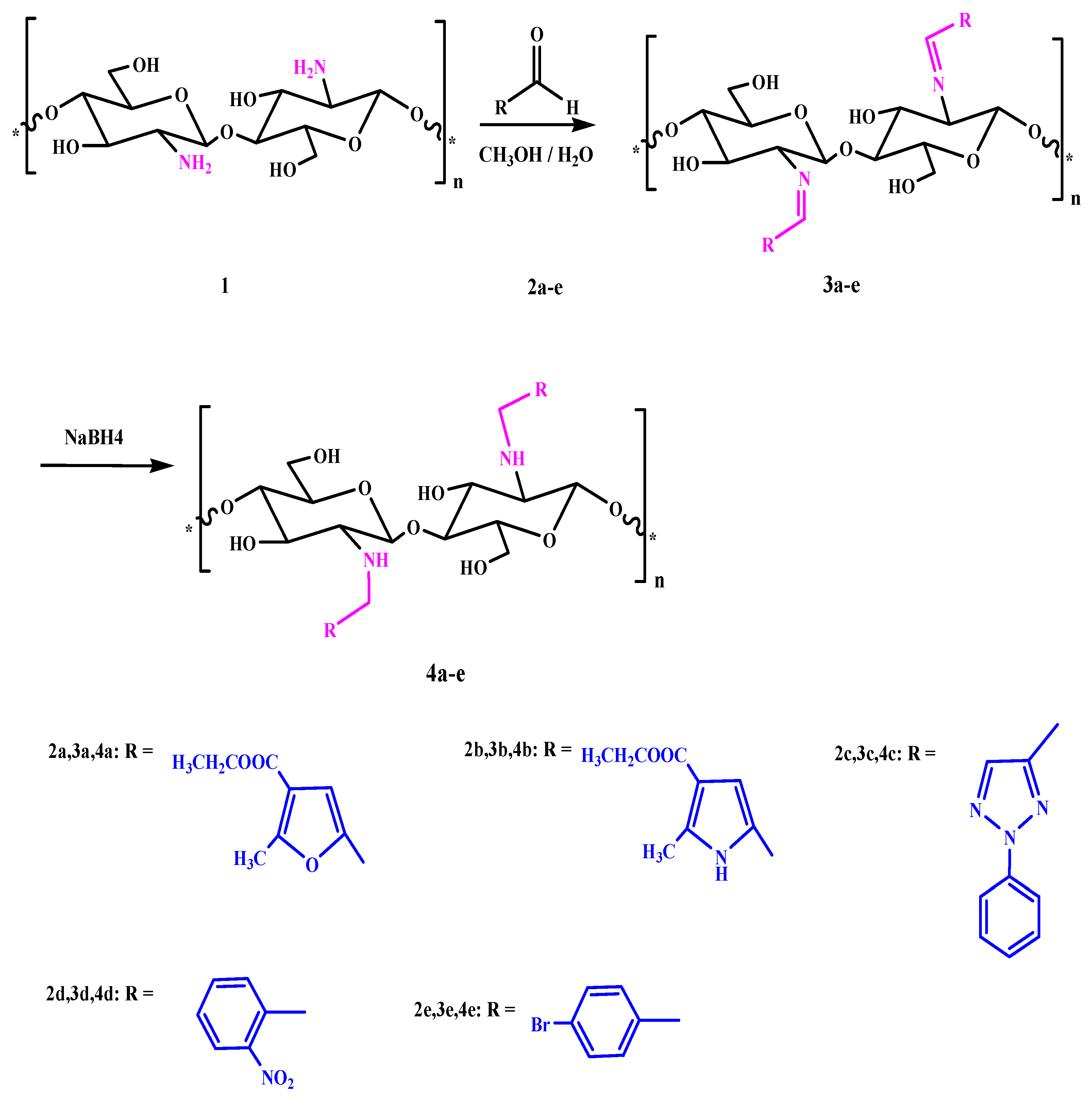
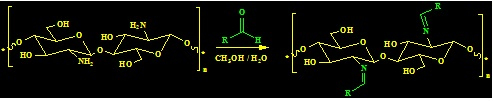
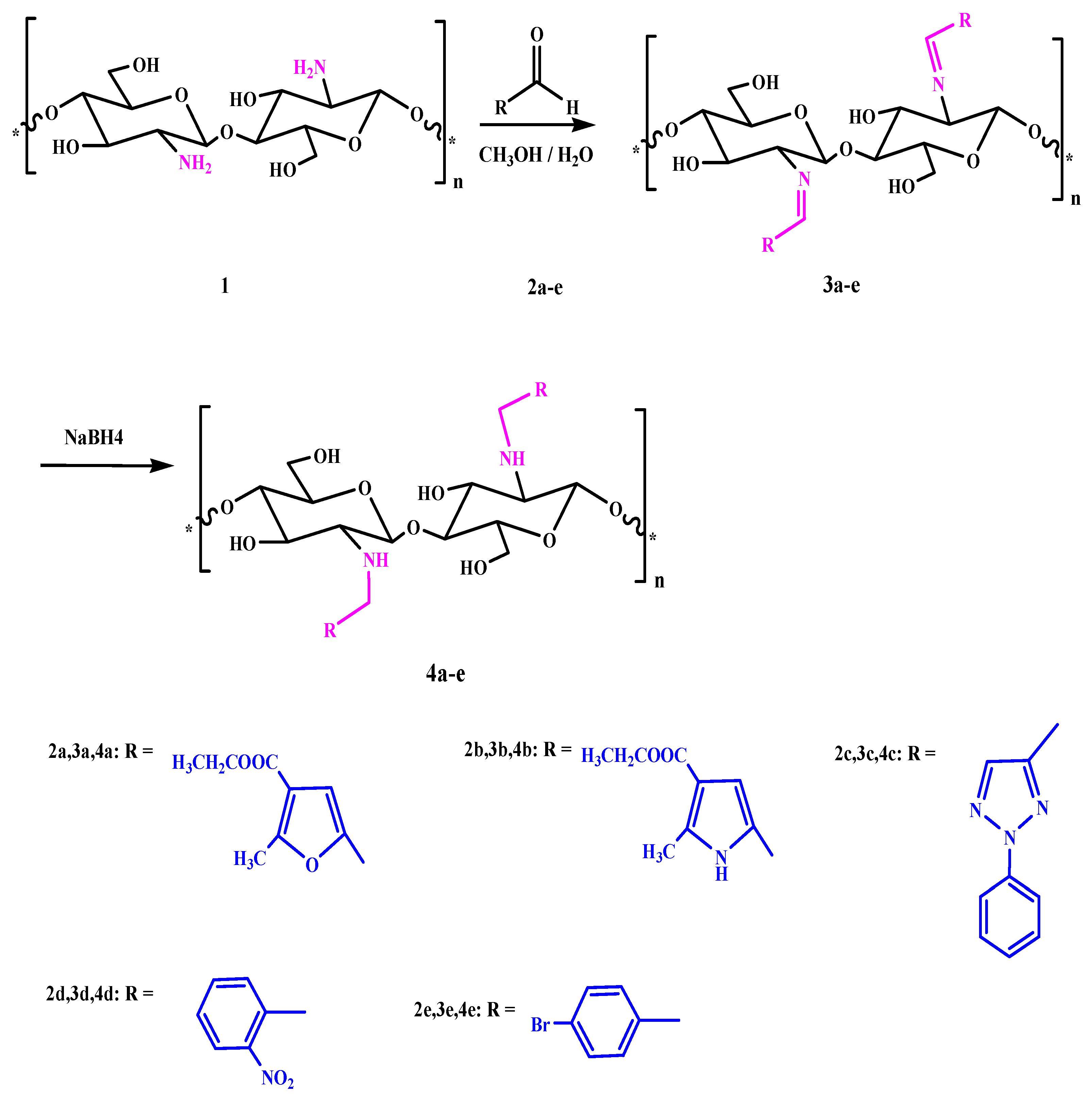
2.3. General Procedures for Chitosan Schiff-Base Synthesis
A solution of the aldehyde (20 mmol) in ethanol (20 mL) was added to chitosan (20 mmol) in 10% AcOH (50 mL). The mixture was stirred for 6–10 h at 70 °C, and then left overnight. After cooling, the homogenous hydrogels which formed were dried at 60 °C for dewatering to constant weight to give the product.
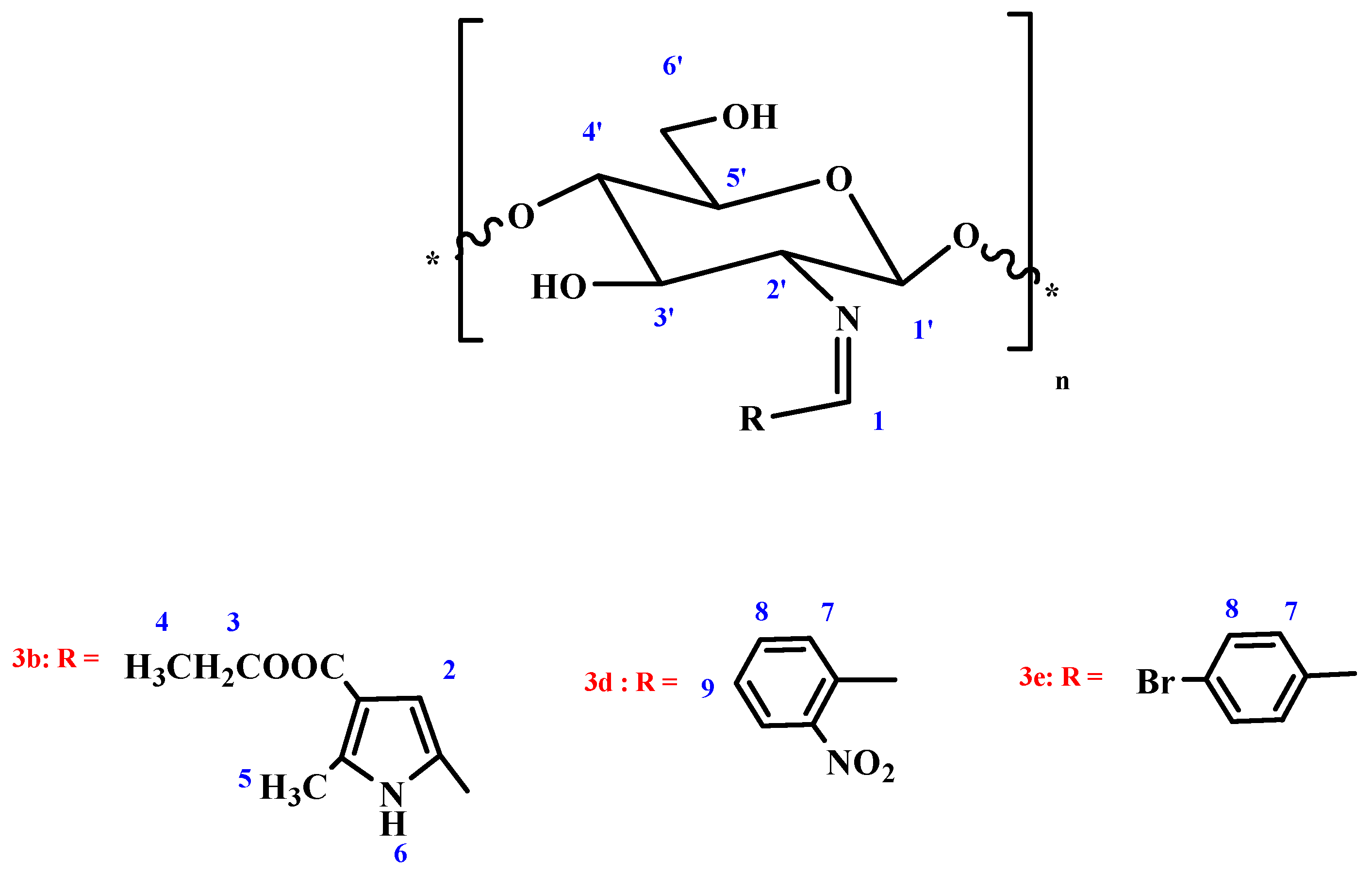
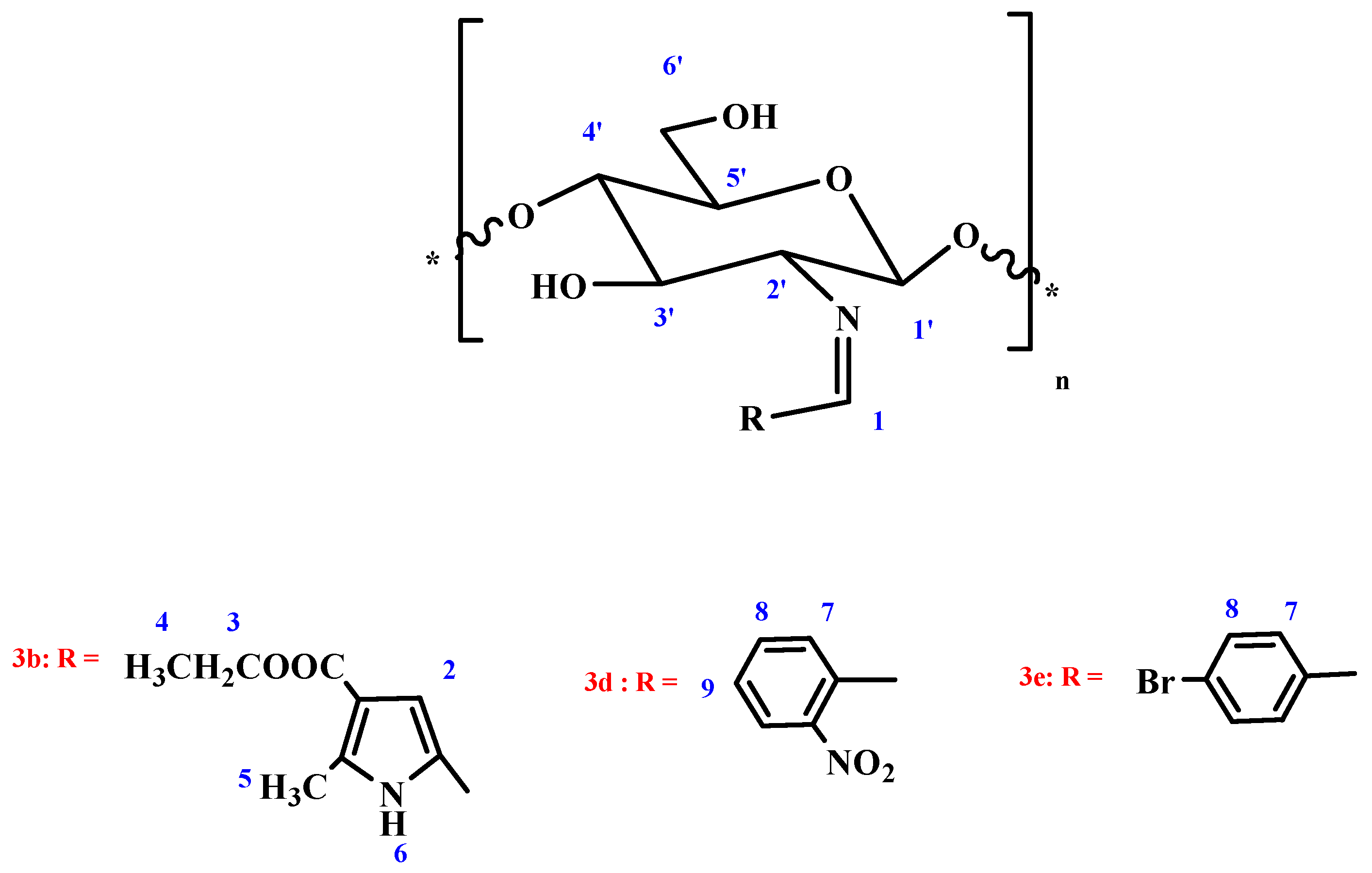
Chitosan furan Schiff-base (CFSB) 3a. Obtained from ethyl 5-formyl-2-methylfuran-3-carboxylate 2a in 91% yield as white powder; the mixture was stirred for 6 h; IR (KBr): 1642 (C=N), 1687 (COOEt), 3440 cm−1 (OH); Anal. Found: C, 52.18; H, 5.31; N, 4.13; O, 37.51.
Chitosan pyrrole Schiff-base (CPSB) 3b. Obtained from 5-formyl-2-methyl-1H-pyrrole-3-carboxylate 2b in 89.3% yield as white powder; The mixture was stirred for 8 h; IR (KBr): 1599 (C=N), 1655 (COOEt), 3322 cm−1 (OH); 1H NMR (300 MHz, DMSO): δ = 1.93 (q, 2H, CH2-ester; J1,2 = 2.3 Hz, J1,3 = 6.9 Hz), 2.38 (m, 1H, H-1′), 2.41(m, 2H, H-2′, H-3′), 2.61 (d, 2H, H-4′, H-5′; J1,2 = 2.3 Hz), 2.75 (m, 2H, H-6a′, H-6b′), 3.17 (t, 3H, CH3-ester; J1,2 = 2.3 Hz, J1,3 = 6.9 Hz), 3.25 (s, 3H, CH3-pyrrole), 4.53 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 6.51 (s, 1H, CH=N), 6.55 (s, 1H, H-pyrrole), 10.33 (bs, 1H, NH; exchangeable with D2O), Anal. Found: C, 52.84; H, 5.94; N, 8.43; O, 32.86.
Chitosan triazole Schiff-base (CTSB) 3c. Obtained from 2-phenyl-2H-1,2,3-triazole-4-carbaldehyde 2c in 95.1% yield as faint gray powder; The mixture was stirred for 10 h; IR (KBr): 1639 (C=N), 3455 cm−1 (OH); Anal. Found: C, 59.58; H, 5.17; N, 18.34; O, 15.82.
Chitosan nitrophenyl Schiff-base (CNPSB) 3d. Obtained from o-nitrobenzaldehyde 2d in 94.3% yield as yellow powder; The mixture was stirred for 6 h; IR (KBr): 1638 (C=N), 3480 cm−1 (OH); 1H NMR (300 MHz, DMSO): δ = 3.56 (m, 1H, H-1′), 3.68 (m, 1H, H-2′), 3.83 (d, 2H, H-3′, H-4′; J1,2 = 2.4 Hz), 3.90 (m, 1H, H-5′), 4.07 (m, 2H, H-6a′, H-6b′), 5.44 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 6.77 (s, 1H, CH=N), 6.98 (d, 2H, o-H; J1,2 = 2.9 Hz), 7.83 (d, 2H, m-H; J1,2 = 2.9 Hz), Anal. Found: C, 56.01; H, 5.11; N, 10.03; O, 28.65.
Chitosan bromophenyl Schiff-base (CBPSB) 3e. Obtained from p-bromobenzaldehyde 2e in 96.7% yield as white powder; The mixture was stirred for 7 h; IR (KBr): 1637 (C=N), 3466 cm−1 (OH); 1H NMR (300 MHz, DMSO): δ = 3.27 (m, 1H, H-1′), 3.38 (m, 1H, H-2′), 3.54 (d, 1H, H-3′; J1,2 = 1.8 Hz), 3.61 (m, 2H, H-4′, H-5′), 3.73 (m, 2H, H-6a’, H-6b′), 5.73 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 7.42 (s, 1H, CH=N), 7.53 (d, 1H, o-H; J1,2 = 2.8 Hz), 7.84 (d, 2H, m-H; J1,2 = 2.8 Hz), Anal. Found: C, 45.03; H, 4.26; N, 4.05; O, 23.14.
2.4. General Procedures for Reduction of Imine by NaBH4
A solution of imine (20 mmol) in methanol (20 mL) was added to 10% AcOH (20 mL). The mixture was stirred for 10 min, and then 0.1 g of sodium borohydride was slowly added to the mixture with continuous stirring in ice bath for 24 h. After that the product was dried.
Chitosan-furan amine derivative 4a. Obtained from imine 3a in 84% yield as white powder; IR (KBr): 1647(COOEt), 3254, 3322 cm−1 (NH), (OH); Anal. Found: C, 61.07; H, 7.08; N, 4.64; O, 27.18.
Chitosan-pyrrole amine derivative 4b. Obtained from imine 3b in 86.2% yield as white powder; IR (KBr): 1659 (COOEt), 3334, 3387 cm−1 (NH), (OH); 1H NMR (300 MHz, DMSO): δ = 1.83 (q, 2H, CH2-ester; J1,2 = 2.3 Hz, J1,3 = 6.9 Hz), 2.41 (m, 1H, H-1′), 2.41 (m, 2H, H-2′, H-3′), 2.61 (d, 2H, H-4′, H-5′; J1,2 = 2.3 Hz), 2.73 (m, 2H, H-6a′, H-6b′), 3.27 (t, 3H, CH3-ester; J1,2 = 2.3 Hz, J1,3 = 6.9 Hz), 3.29 (s, 3H, CH3-pyrrole), 4.61 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 6.51 (s, 2H, CH2), 6.53 (s, 1H, H-pyrrole), 9.53 (bs, 1H, NH; exchangeable with D2O), 10.35 (bs, 1H, NH; exchangeable with D2O), Anal. Found: C, 57.95; H, 7.05; N, 9.02; O, 25.68.
Chitosan-triazole derivative 4c. Obtained from imine 3c in 84% yield as faint green powder; IR (KBr): 3260, 3321 cm−1 (NH), (OH); Anal. Found: C, 58.79; H, 6.04; N, 18.33; O, 15.58.
Chitosan-nitrophenyl amine derivative 4d. Obtained from imine 3d in 91% yield as faint yellow powder; IR (KBr): 3331, 3340 cm−1 (NH), (OH); 1H NMR (300 MHz, DMSO): δ = 3.80 (d, 2H, H-1′, H-2′ J1,2 = 2.2 Hz), 3.90 (m, 2H, H-3′, H-4′), 4.14 (m, 3H, H-5′, H-6a′, H-6b′), 5.46 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 6.97 (s, 2H, CH2), 7.01 (d, 2H, o-H; J1,2 = 2.4 Hz), 7.83 (d, 2H, m-H; J1,2 = 2.4 Hz), 9.14 (bs, 1H, NH; exchangeable with D2O), Anal. Found: C, 55.61; H, 5.47; N, 9.89; O, 28.14.
Chitosan-bromophenyl amine derivative 4e. Obtained from imine 3e in 91% yield as white powder; IR (KBr): 3264, 3398 cm−1 (NH), (OH); 1H NMR (300 MHz, DMSO): δ = 3.37 (m, 2H, H-2′, H-1′), 3.48 (m, 3H, H-3′, H-4′, H-5′), 3.83 (m, 2H, H-6a′, H-6b′), 5.81 (bs, 2H, 3′-OH, 6′-OH; exchangeable with D2O), 7.41 (s, 2H, CH2), 7.53 (d, 2H, o-H; J1,2 = 2.0 Hz), 7.83 (d, 2H, m-H; J1,2 = 2.0 Hz), 9.31 (bs, 1H, NH; exchangeable with D2O), Anal. Found: C, 44.76; H, 5.25; N, 4.12; O, 22.78.
2.5. Antimicrobial Activity
The antimicrobial activity of CSB derivatives were evaluated against Staphylococcus aureus (RCMBA 2004) and Bacillissubtilis (RCMBA 6005) as Gram-positive bacteria and against Pseudomonas aeruginosa and Escherichia coli (RCMBA 5003) as Gram-negative bacteria and against Aspergillus fumigates (RCMBA 06002), Syncephalastrum racemosum (RCMB 05098), as fungi. Agar disk diffusion method was used for the determination of the antibacterial and antifungal activity, the well diameter was 6 mm (100 µL was tested), and the concentration of the tested sample was 5 mg/mL.
The susceptibility tests were performed according to the NCCLS recommendations (National Committee For Clinical Laboratory Standards, 1993). Screening tests regarding the inhibition zone were carried out by the well diffusion method [
22].
The inoculums suspension was prepared from colonies grown overnight on an agar plate, and inoculated into Mular Hinton broth (Merk, Darmstadt, Germany). A sterile swab was immersed in the bacterial suspension and used to inoculate Mueller-Hinton agar plates. Amphotericin B, Ampicillin and Gentamicin were used as references for anti-fungi, anti-Gram positive bacteria, and anti-Gram negative bacteria, respectively. The compounds were dissolved in dimethylsulfoxide (DMSO). The inhibition zone was measured around each well after 24 h incubation at 37 °C; controls using DMSO were adequately done.
MIC determinations were performed in the same way using agar disc diffusion method, but by using different concentrations from the testing compound.
2.6. Antiproliferative Activity Screening
Regarding cell line propagation, the cells were propagated in (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 1% l-glutamine, HEPES buffer and 50 μg/mL gentamycin. All cells were maintained at 37 °C in humidified atmosphere with 5% CO2 and were sub cultured two times a week. Cell toxicity was monitored by determining the effect of the examined compound on cell morphology and cell viability.
For cytotoxicity assay, the cells were seeded in 96-well plate at a cell concentration of 1 × 104 cell per well in 100 μL of growth medium. Serial two-fold dilutions of the tested chemical compound were added to confluent cell monolayers that were then dispensed into 96-well flat-bottomed microtiter plates (Falcon, NJ, USA) using a multichannel pipette. The microtiter plates were incubated at 37 °C in a humidified incubator with 5% CO2 for a period of 48 h. Three wells were used for each concentration of each tested sample. Control cells were incubated without test samples and with or without DMSO. After incubation of the cells for 24 h at 37 °C, various concentrations of each sample (50, 25, 12.5, 6.25, 3.125 and 1.56 μg) were added separately. Then the incubation was continued for 48 h.
The viable cells yield was determined colorimetrically using MTTB (3,4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide). The water insoluble tetrazolium salt is converted to purple formazon by the mitochondrial dehydrogenase of viable cells. After the end of incubation period, media were aspirated, and the crystal violet solution (1%) was added to each well for at least 30 min. The stain was removed and plates were rinsed using tap water until all excess stain was removed. Glacial acetic acid (30%) was then added to all wells and mixed thoroughly, then the absorbance of the plates were measured after gently being shaken on Micro plate Reader (TECAN, Inc., Olympus Europa Holding GmbH, Männedorf, Switzerland), at 490 nm. All results were corrected for background absorbance detected in wells without added stain. Treated samples were compared with the cell control in the absence of the tested compound. All experiments were carried out in the triplicate. The cell cytotoxicity effect of the tested compound was calculated [
16,
17].



{kind=link}
{kind=link}
{kind=link}
{kind=link}