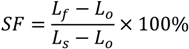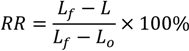3.1.1. Glass Transition and Melting
The value of soft segment
Tg is needed to determine a suitable value of
Ttrans, such that
Ttrans >
Tg.
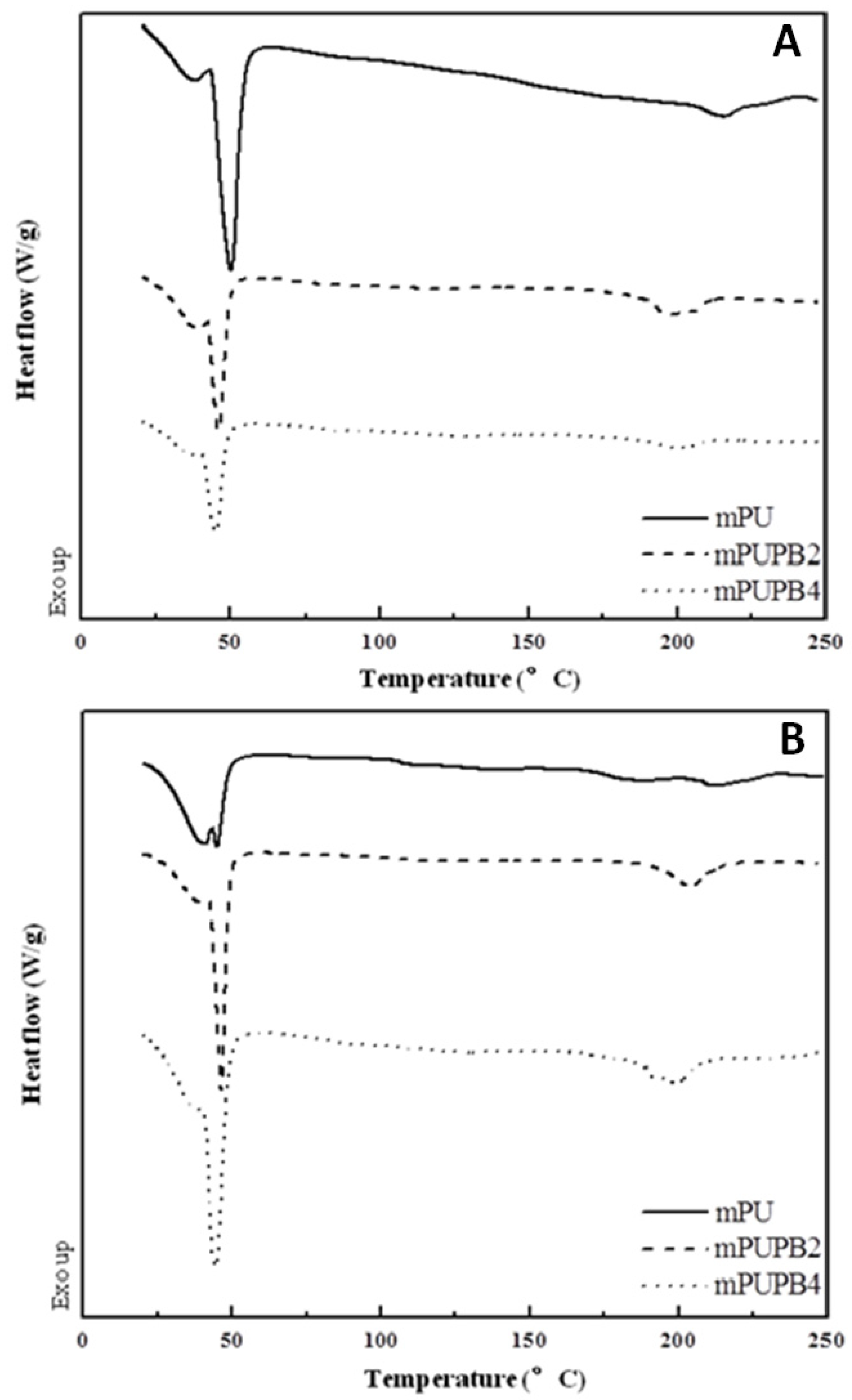
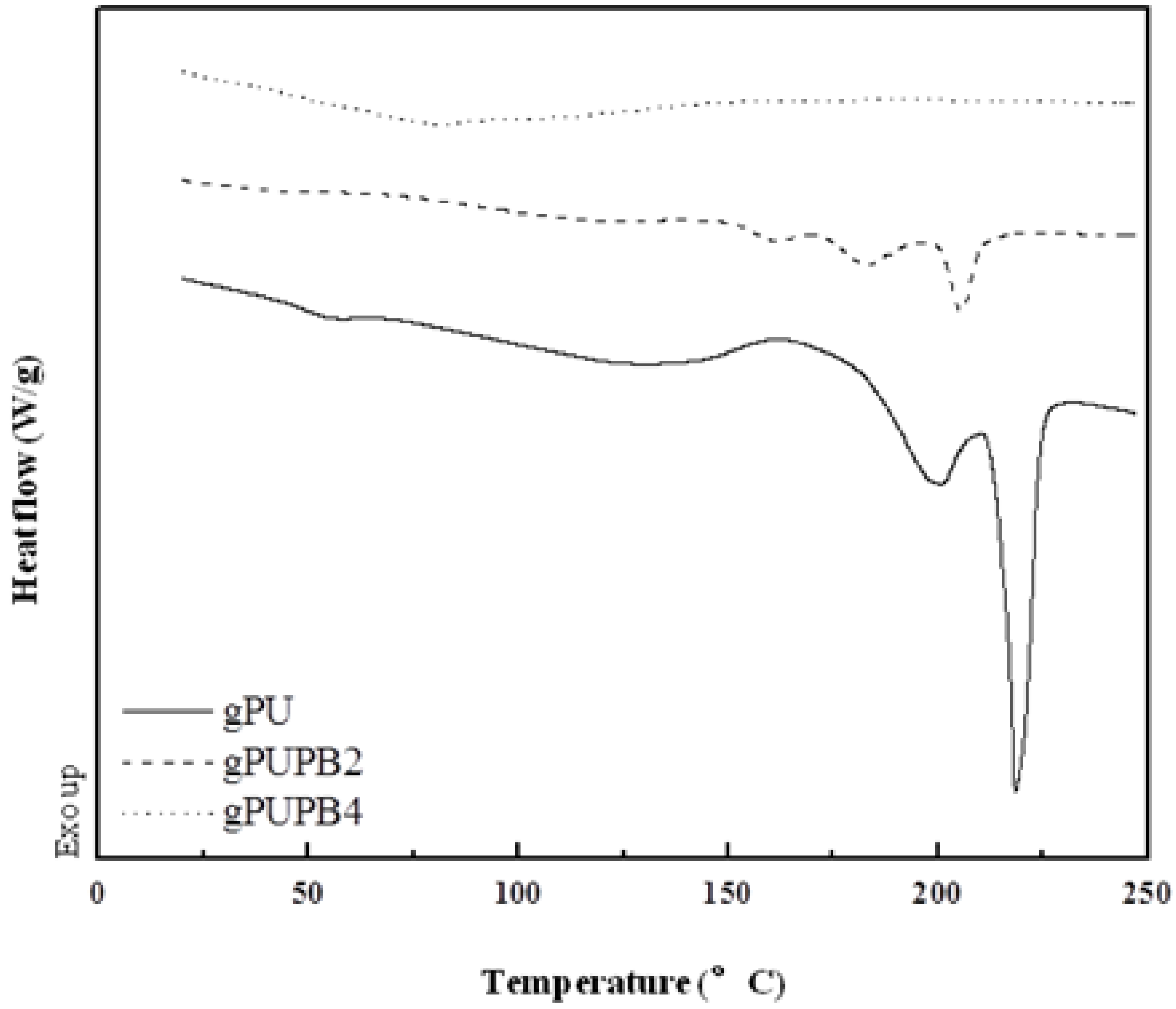
Figure 2 shows the DSC traces of three specimens. These samples were heated from room temperature to 250 °C with a heating rate of 10 °C min
−1. The curve for gPU (
Table 1) shows two weak zones of glass transition with
Tg at around 49 °C and 95 °C, attributed respectively to soft- and hard-segment rich domains. However, these two transitions do not appear in DSC traces of compounds gPUPB2 and gPUPB4, indicating that the PB domains probably promoted mixing between the hard and soft-segment phases as was seen in another study [
47]. Note that the
Tg of PB is around 170 °C [
42]. Thus, phase mixing of PB with the soft and hard domains should increase their respective
Tg values. The DSC traces of PU-PB compounds (gPUPB2 and gPUPB4) in
Figure 2 show heat flow curves with small slope, implying that accurate values of
Tg of PU-PB compounds cannot be computed from these DSC traces.
Figure 2.
DSC traces of PU with amorphous soft-segment and its compounds with PB. The compounds were cured at 180 °C. Scan rate was 10 °C min−1.
Figure 2.
DSC traces of PU with amorphous soft-segment and its compounds with PB. The compounds were cured at 180 °C. Scan rate was 10 °C min−1.
The DSC traces in
Figure 2, however, show melting transitions of the crystalline domains. Sample gPU exhibits two melting peaks with a total heat of fusion of 26.88 J g
−1, with the peak temperature respectively 200 °C and 218 °C. The melting peak due to crystalline hard segment also appeared in the DSC trace of gPUPB2 compound with peak temperatures of 161 °C, 183 °C, and 205 °C and the total heat of melting of 13.36 J g
−1. The DSC trace of gPUPBZ4 compound, however, does not show the melting peak due to hard segment. The reduction of heat of melting observed in gPUPB2 compound and the absence of melting peaks in gPUPBZ4 compound together indicate that hard segment crystallization was hindered due to the presence of PB in these compounds.
3.1.2. Phase Mixing and PB Domain Size
Figure 3 shows the phase images of SMPU from atomic force microscopy scans. In
Figure 3a, the dark and bright areas represent respectively the soft and hard domains of SMPU.
Figure 3b,c also shows some extremely bright spots; these can be identified as the aggregates of PB. The typical size of PB domains inferred from
Figure 3b is 150 nm, which grew to a size of 250 nm in
Figure 3c due to an increase of the PB content from 6–12 wt%.
In view of the phase mixing seen in
Figure 3, one can envisage that the PB domains are connected to PU chains by hydrogen bonding and covalent linkages. The hydrogen bonds form between the phenolic ‒OH groups on PB chains and the soft and hard segments of PU. The covalent bonds link the PB and PU chains via chemical reactions between the ‒NCO groups of PU and the phenolic ‒OH groups of PB chains. A comparison of the
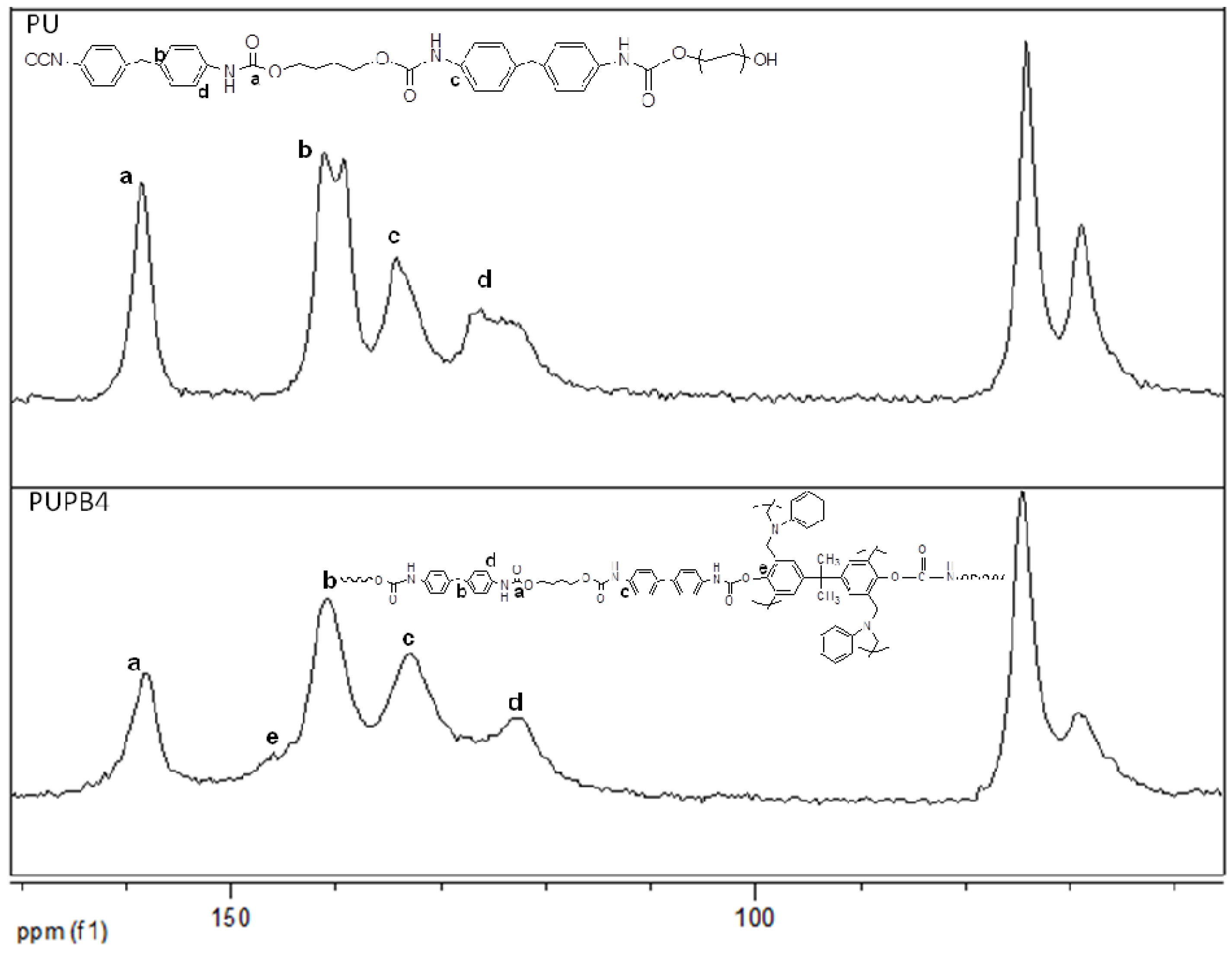
13C NMR spectra of gPU and gPUPB4 presented in
Figure 4 confirms the existence of covalent bonds between PU and PB chains. A new peak at around 146 ppm appearing in the NMR spectra of gPUPB4 matches with the
13C NMR signature of the carbon on the benzene ring of PB and adjacent to the oxygen connected with PU chains. The unique carbon atoms are labeled in the chemical structure of PU-PB chains and presented in
Figure 4. However, the peak at 146 ppm is relatively weak, apparently due to small concentration of PB chains in the system. This is in good agreement with the earlier reports [
45,
46,
47,
51]. The covalent bond formation between PU chains and PB and the AFM phase morphology in
Figure 3 can now explain the absence of soft-segment glass transition in DSC traces and the reduction of heat of fusion of the hard segments in PU-PB compounds.
Figure 3.
AFM phase images of samples (a) gPU. (b) gPUPB2. (c) gPUPB4. The contrast covers phase variations in 0°–30° range.
Figure 3.
AFM phase images of samples (a) gPU. (b) gPUPB2. (c) gPUPB4. The contrast covers phase variations in 0°–30° range.
Figure 4.
13C NMR spectra of gPU and gPUPB4.
Figure 4.
13C NMR spectra of gPU and gPUPB4.
Further evidence of the reduction of microphase separation was obtained from FT-IR data. The values of hydrogen bonding index (α) were calculated from Equation (3) and the values are listed in
Table 4.
The areas under the peaks at 1730 cm
−1 due to free CO (A
FCO), at 1710 cm
−1 due to hydrogen-bonded CO (A
aHCO) in amorphous hard domains and at 1703 cm
−1 due to hydrogen-bonded CO (A
cHCO) in crystalline hard domains were used to calculate the hydrogen bonding index, α. Two conclusions can be drawn from the data in
Table 4: (1) the extent of phase separation in SMPU reduced due to the incorporation of PB and (2) the reduction of phase separation is due to loss of crystalline urethane hard segment domains.
Table 4.
Hydrogen bonding index (α) of SMPU with amorphous soft segment.
Table 4.
Hydrogen bonding index (α) of SMPU with amorphous soft segment.
| Sample | AcHCO | AaHCO | AFCO | α |
|---|
| gPU | 5.2 | N/A | 3.0 | 0.63 |
| gPUPB2 | 0.8 | 2.2 | 7.0 | 0.30 |
| gPUPB4 | 0.7 | 2.0 | 6.5 | 0.29 |
3.1.3. Dynamic Mechanical Properties
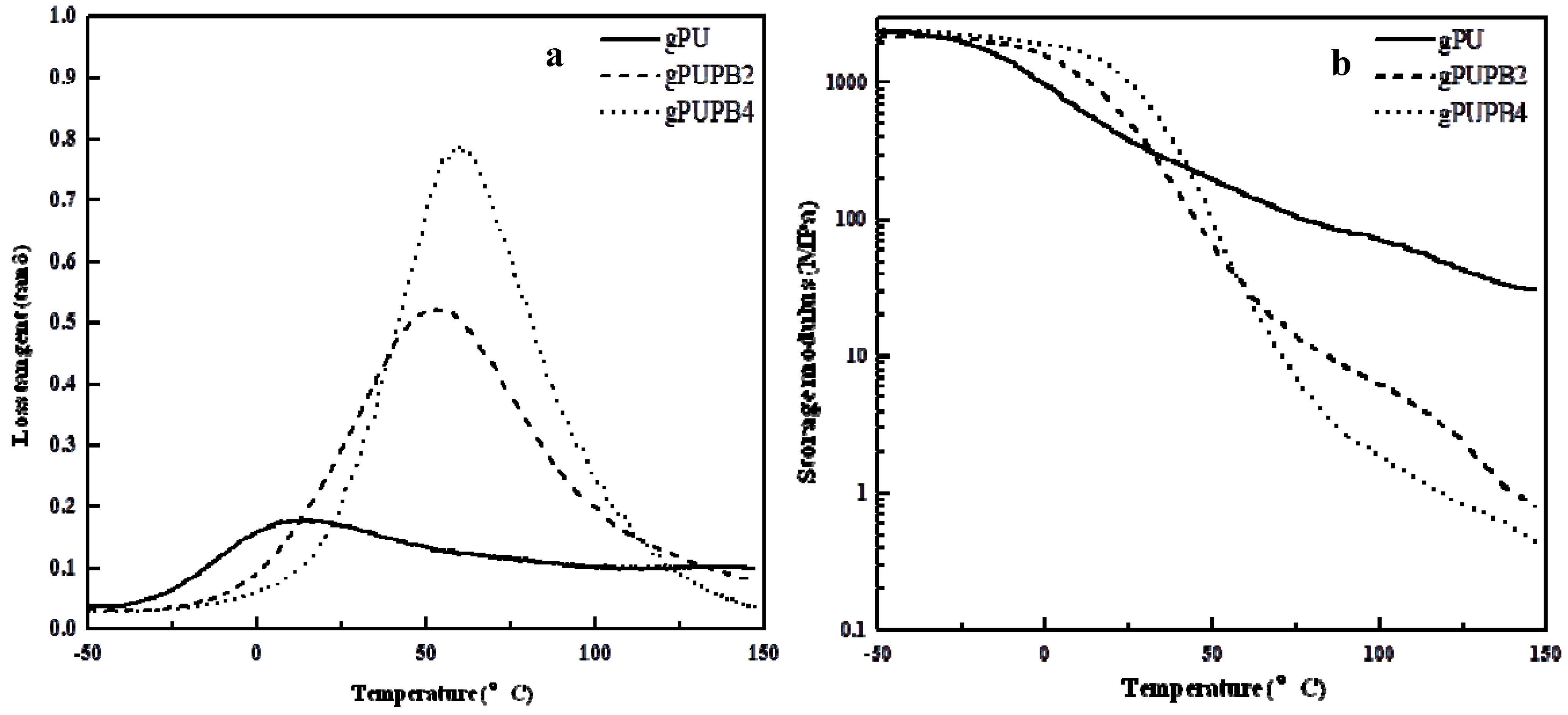
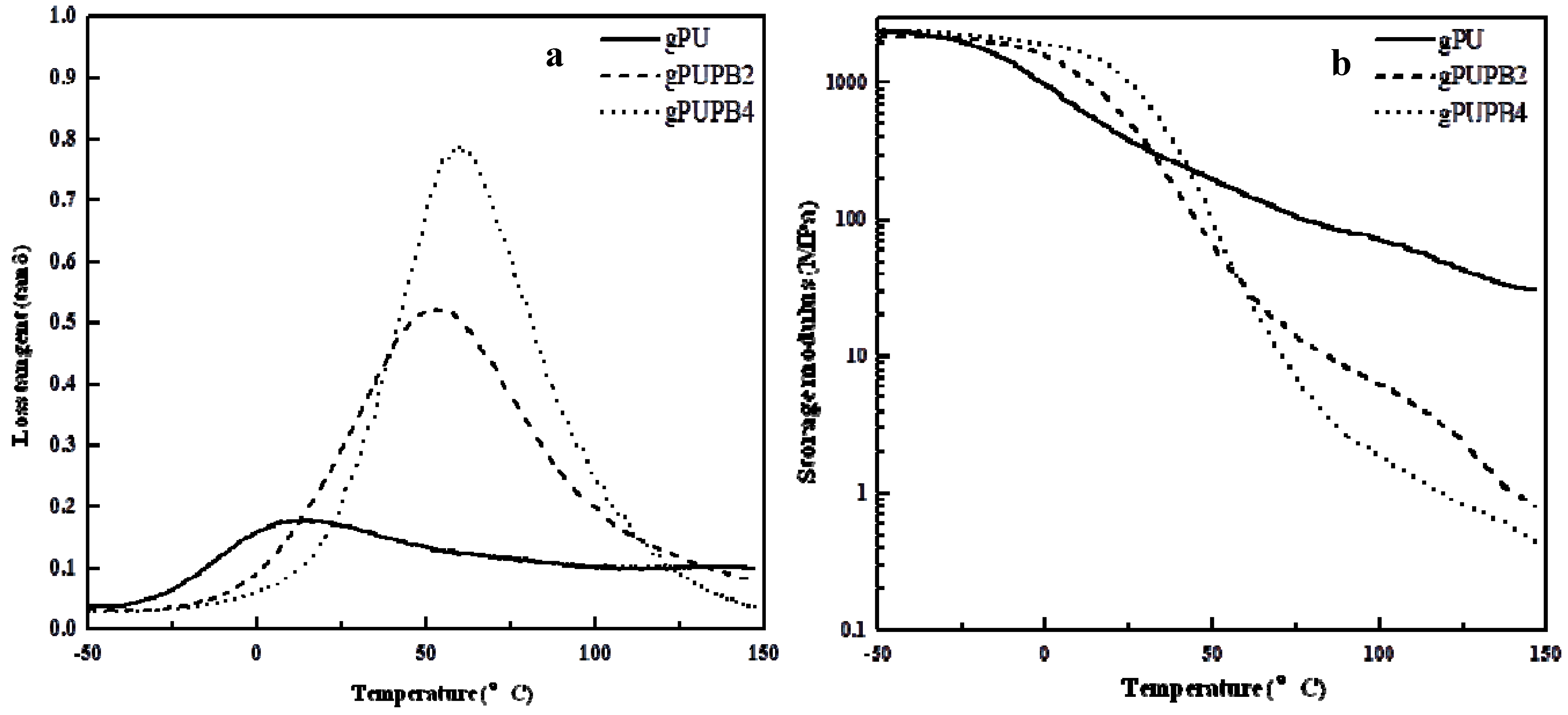
The values of glass transition temperature can be inferred from tan δ
vs. temperature curves, as listed in
Table 3. The tan δ
vs. temperature traces in
Figure 5a reveal that all materials exhibit a single peak of tan δ, indicating the
Tg of soft domains of gPU, gPUPB2, and gPUPB4, respectively, at 14°, 52°, and 60 °C. One can attribute such increases in
Tg to mixing of PB domains.
Figure 5b shows the values of storage modulus as a function of temperature. The larger storage modulus of gPU than gPUPB2 and gPUPB4 above the glass transition temperature of the soft segment is attributed to greater quantity of crystalline PU hard segment. The values of storage modulus (
E') at (
Tg − 20 °C) and (
Tg + 20 °C) and the ratio of
E'(
Tg − 20 °C) and
E'(
Tg + 20 °C) are listed in
Table 5. The modulus ratios were 4 of gPU, 102 of gPUPB2, and 156 of gPUPB4. It was earlier proposed that a large difference of storage modulus above and below the glass transition temperature and a sharp glass to rubber transition are prerequisites for efficient shape recovery from the deformed state [
52]. Generally, the ratio of modulus in glassy and rubbery states of 20 indicates good shape memory performance. In view of this, the values of
E'(
Tg − 20 °C)/
E'(
Tg + 20 °C) reported in
Table 5 imply the following: (1) The unmodified PU with amorphous soft-segment considered in this work cannot exhibit shape recovery as it cannot hold the temporary shape due to
Tg value lower than the room temperature. (2) The two PU-PB compounds prepared in this work should show excellent shape-memory properties, as will be seen below.
Figure 5.
(A) Loss tangent (tan δ). (B) Storage modulus (E') as a function of temperature. Heating rate, 4 °C min−1; frequency, 1 Hz.; scan between −50 °C and 150 °C.
Figure 5.
(A) Loss tangent (tan δ). (B) Storage modulus (E') as a function of temperature. Heating rate, 4 °C min−1; frequency, 1 Hz.; scan between −50 °C and 150 °C.
Table 5.
The values of storage modulus at Tg – 20 °C and Tg + 20 °C.
Table 5.
The values of storage modulus at Tg – 20 °C and Tg + 20 °C.
| Sample | E'(Tg – 20 °C) (MPa) | E'(Tg + 20 °C) (MPa) | E'(Tg – 20 °C)/E'(Tg + 20 °C) |
|---|
| gPU | 1100 | 292 | 4 |
| gPUPB2 | 818 | 8 | 102 |
| gPUPB4 | 313 | 2 | 156 |
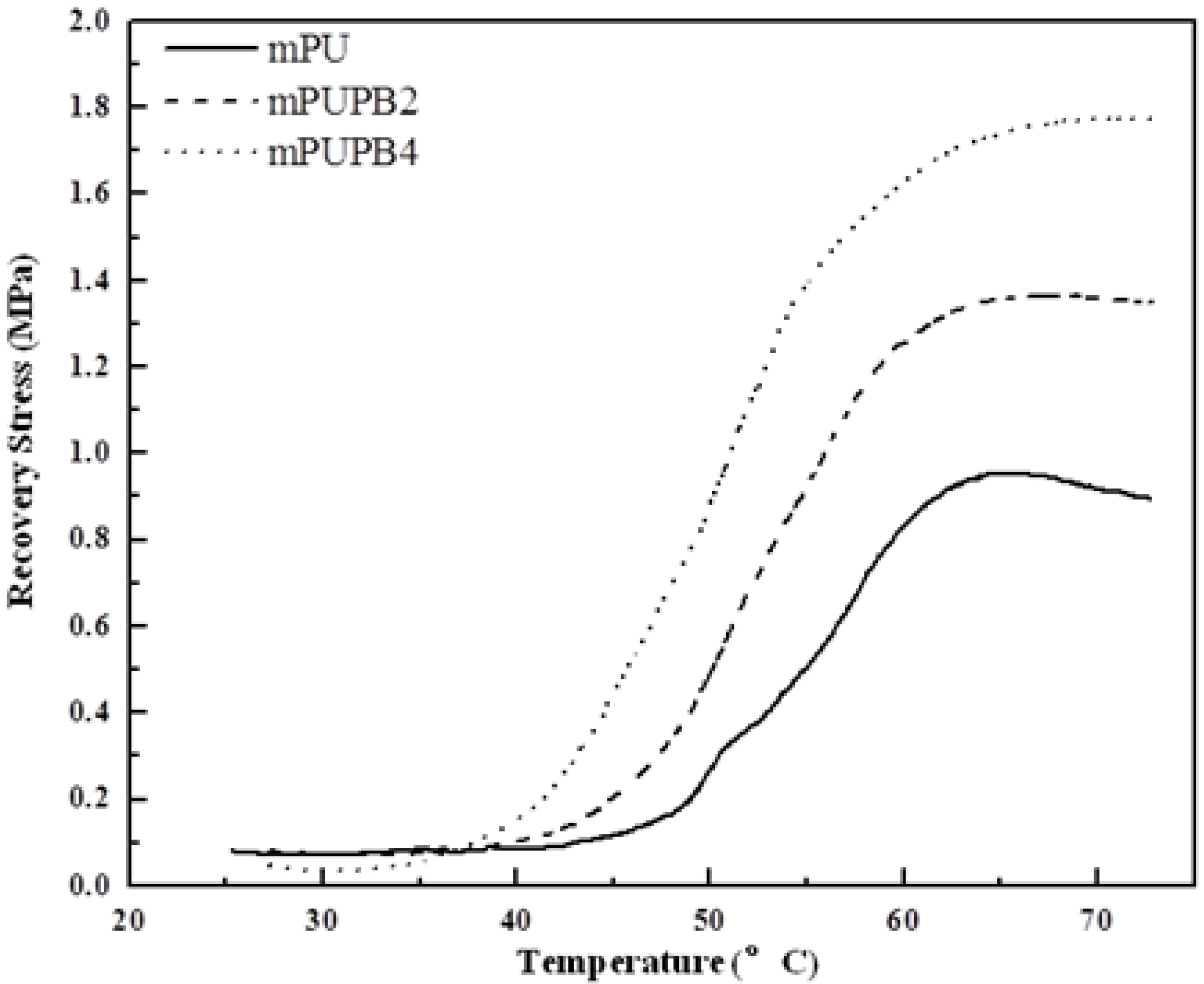
3.1.4. Shape-Memory Properties
Shape fixity. A rectangular specimen was first stretched at Ttrans to 30% its original length and then cooled down to Tg − 40 °C at a cooling rate of 5 °C min−1 to obtain the temporary shape. The specimen dimension was measured at (Tg − 40) °C. The values of shape fixity are as follows: 97.2% for gPU, 95.3% for gPUPB2, and 92.8% for gPUPB4. Shape fixity refers to the unconstrained recovery of SMPs after the stretching load was removed. A value less than 100% indicates that an instantaneous shrinkage of the stretched polymer chains cannot preserve the imposed strain. Recall that the values of Tg of samples obtained from DMA were 14, 57 and 70 °C, respectively, for gPU, gPUPB2 and gPUPB4. Accordingly, the temporary shape was fixed at −26, 17 and 30 °C, respectively, for gPU, gPUPB2, and gPUPB4. Of the three materials, the compounds gPUPB2 and gPUPB4 were able to retain the deformed shapes at room temperature.
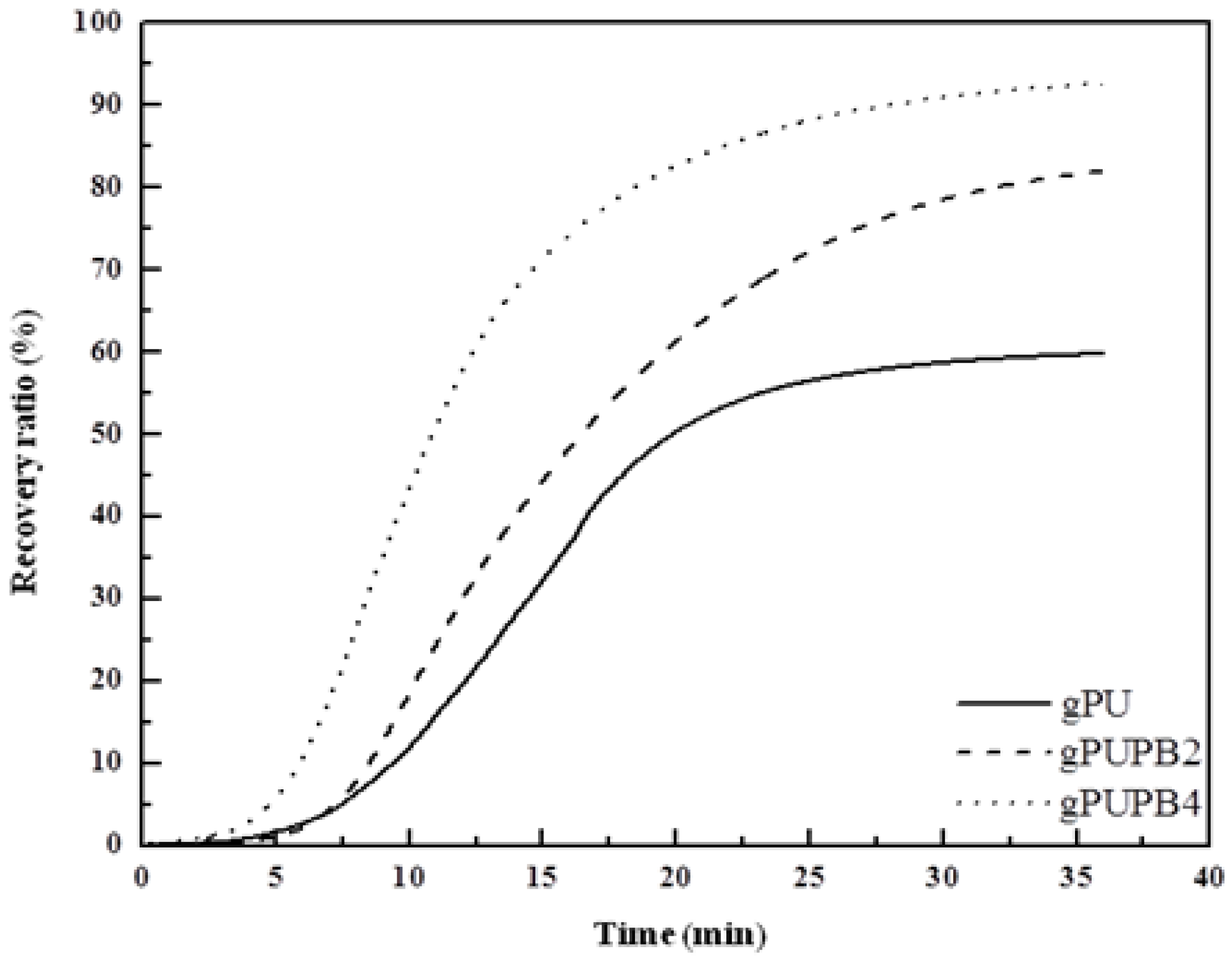
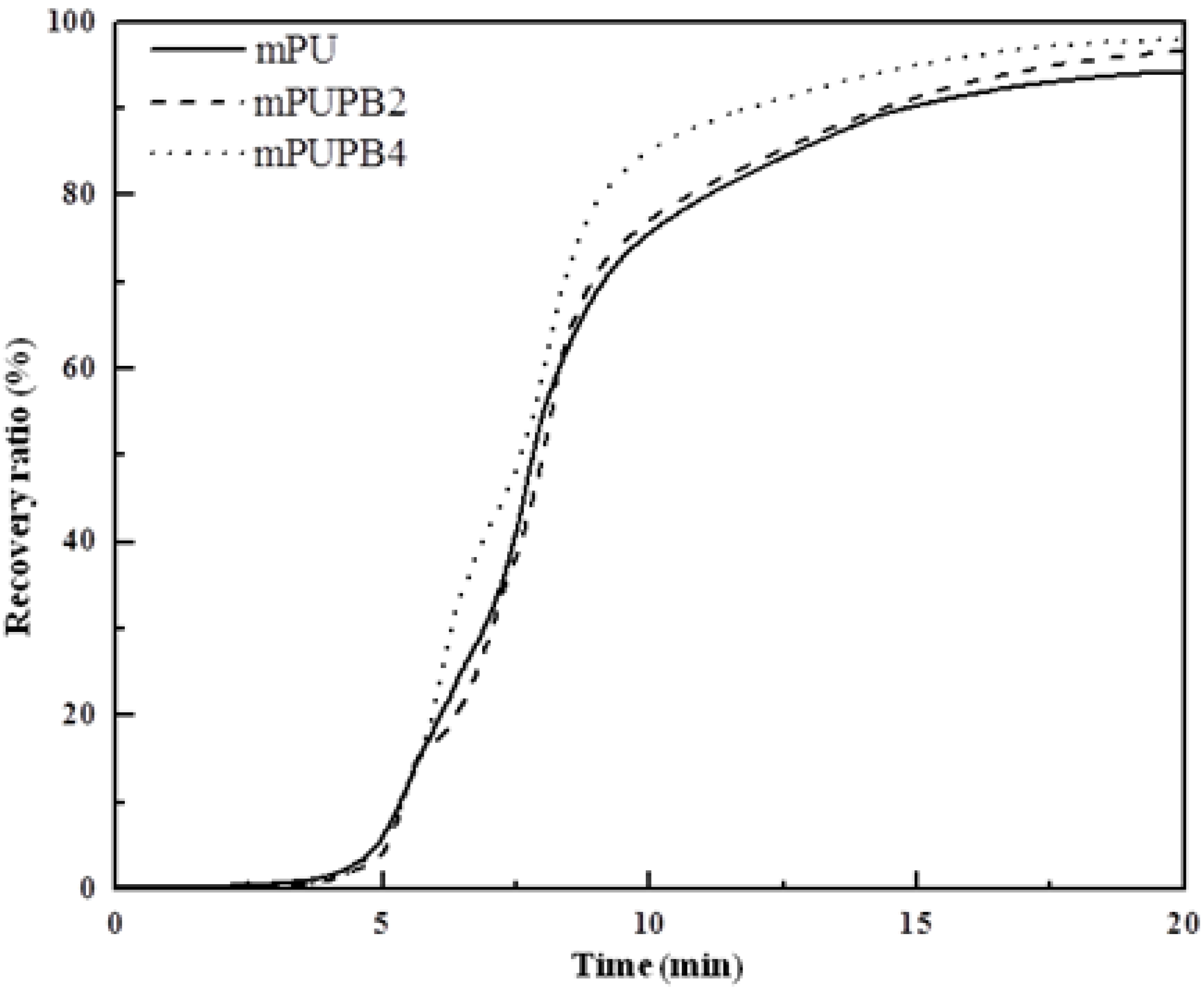
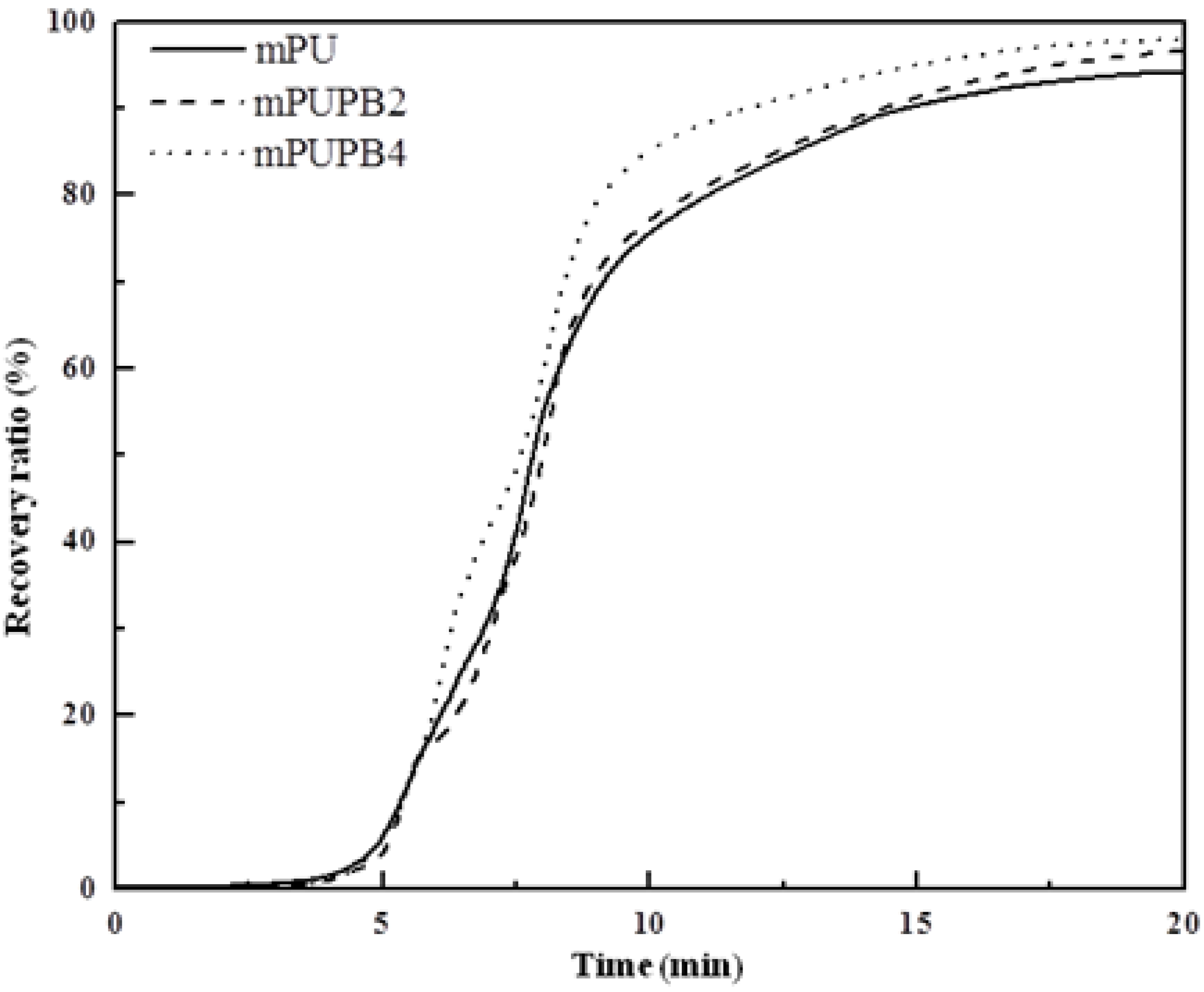
Recovery ratio.
Figure 6 presents the shape recovery values of samples as a function of time. The test began with the material kept at the fixing temperature, e.g., at −26, 17 and 30 °C, respectively, for gPU, gPUPB2, and gPUPB4 and continued to
Ttrans of each sample with a heating rate of 5 °C min
−1 and kept isothermal for a period of 20 min. The maximum values of recovery ratio of samples are respectively 60% for gPU, 82% for gPUPB2, and 93% for gPUPB4. It is evident that incorporation of PB led to obvious enhancement in shape recovery ratio. The PB domains acted as the hard phase in the compounds with PU and strengthened the polymer networks, keeping the permanent shape of specimen via physical and chemical interactions with PU chains. The rate of recovery can also be inferred from
Figure 6. The higher the values of modulus ratio
E'(
Tg − 20 °C)/
E'(
Tg + 20 °C), the higher is the recovery rate as is apparent from the greater slope seen in
Figure 6. The specimen gPUPB4 shows the fastest recovery rate among three samples considered in this work.
Figure 6.
Shape recovery ratio as a function of time. Samples were heated from fixing temperature to respective Ttrans at a heating rate of 5 °C min−1 and kept isothermal for 20 min.
Figure 6.
Shape recovery ratio as a function of time. Samples were heated from fixing temperature to respective Ttrans at a heating rate of 5 °C min−1 and kept isothermal for 20 min.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}