Ring-Opening Polymerization—An Introductory Review

1
Wacker-Lehrstuhl für Makromolekulare Chemie, Technische Universität München Lichtenbergstr.4, Garching D-85747, Germany
2
SteDaPa Consulting, Im Gerstenfeld 44, Dormagen D-41542, Germany
*
Author to whom correspondence should be addressed.
Polymers 2013, 5(2), 361-403; https://doi.org/10.3390/polym5020361
Submission received: 21 March 2013
/
Revised: 9 April 2013
/
Accepted: 10 April 2013
/
Published: 25 April 2013
(This article belongs to the Special Issue Ring-Opening Polymerization)
Abstract
:This short, introductory review covers the still rapidly growing and industrially important field of ring opening polymerization (ROP). The review is organized according to mechanism (radical ROP (RROP), cationic ROP (CROP), anionic ROP (AROP) and ring-opening metathesis polymerization (ROMP)) rather than monomer classes. Nevertheless, the different groups of cyclic monomers are considered (olefins, ethers, thioethers, amines, lactones, thiolactones, lactams, disulfides, anhydrides, carbonates, silicones, phosphazenes and phosphonites) and the mechanisms by which they can be polymerized involving a ring-opening polymerization. Literature up to 2012 has been considered but the citations selected refer to detailed reviews and key papers, describing not only the latest developments but also the evolution of the current state of the art.
1. Introduction
Ring-opening polymerization (ROP) is, together with chain (radical and ionic) polymerization and condensation polymerization, one of the three paths to the polymers that are so important to life in the 21st century. Some ROP can be considered as a chain polymerization (addition of monomer to a growing chain end) but many reactions are more complicated and involve, e.g., activated monomers. Particularly ROP has proved to be a useful synthetic route to technologically interesting polymers with very specific, and controllable properties (e.g., refractive index), for preparing synthetic variants to naturally occurring polymers (e.g., chitin, see Section 3.1) or to optimize biodegradable polymers for agricultural, medicinal and pharmaceutical applications (see Section 2.4).
Since this review is intended more as an introduction to the field of ROP, no attempt will be made to describe the particularities of the thermodynamics of these polymerizations. It should however be noted that rather than the driving force for the polymerization being the conversion of a multiple to a single bond, for most ROP the driving force is the ring strain and associated steric considerations. Additionally, there is the consideration of the ring chain equilibrium, which leads to the considerable influence of the initial monomer concentration and temperature on the microstructure of the products. The interested reader is referred to the excellent recent summary by Duda and Kowalski [1].
With some exceptions, such as the ROP of Leuchs’ anhydrides (see Section 4.2) or the radical ROP of 2,2-diphenyl-4-methylene-1,3-dioxolane (see Section 2.2), is not accompanied by the elimination of small molecules. Additionally, ROP is not aided by the enthalpy difference between a single and double C–C bond, which counteracts the loss of entropy occurring as a consequence of polymerization via chain polymerization. ROP have in common that the monomers are rings, irrespective of ring size, but the reason why polymerization takes place can vary. Rings composed of 3–8 atoms may polymerize due to the loss of enthalpy associated with the loss of ring strain. Thus, the ring strain for oxiranes [2] is 116 kJ mol−1 and even for the 7 and 8 membered lactones and lactams [3] the polymerization is driven by an enthalpic contribution even though, in these cases the ring strain is only ca. 6 J mol−1. Strain-free six-membered rings do not generally polymerize. For rings containing disulfide, silicon or carbonate moieties an additional effect is the increased rotational freedom of these groups in the resulting linear chains which leads to an increase in entropy to give the driving force for polymerization [4].
Many polymers of industrial importance are produced via ROP, e.g., olycyclooctene, polynorbornene, polyethylene oxide, polysiloxane and polyphosphazene. A large family of commercially available polymers called polyethylenimines (many of which are highly branched), are produced via a CROP of aziridine or oxazoline monomers. Polyoxymethylene (POM), derived from trioxane and most of the diols (produced, e.g., via CROP of THF) used in polyurethanes as well as Nylon 6 (poly ε-caprolactam) are perhaps, in terms of volume, the most significant. More recently, polyglycolides and polylactides (see Section 4.1) [4] are becoming important as packaging materials due to the bio-compatibility and natural decomposition of these materials.
Another important development is the use of metathesis catalysts to open rings with exo functional groups to yield novel polymer structures (Section 5).
Table 1 lists the major groups of ring structures and the mechanism by which they are usually polymerized.
This review is organized according to mechanism rather than molecular types and is designed to demonstrate the usefulness of this type of polymerization to yield novel polymers, particularly for those new to the subject. It is hoped that it will provide a useful start for entering the field rather than being an exhaustive review of all the literature to date. It is recommended that the interested reader refer to, e.g., the chapters of [5].

Table 1.
Typical cyclic monomers and their usual polymerization mechanism [4].
| Name | Structure | Ring size | Mechanism |
|---|---|---|---|
| Olefin |  | 4,5,8 | Metathesis |
| Ether |  | 3–5,7 | Cationic, anionic |
| Thioether |  | 3,4 | Cationic, anionic |
| Amine |  | 3,4,7 | Cationic |
| Lactone |  | 4,6–8 | Anionic, cationic |
| Thiolactone |  | 4–8 | Anionic, cationic |
| Lactam |  | ≥4 | Anionic, cationic |
| Disulfide |  | ≥8 | Radical |
| Anhydride |  | 5 and ≥7 | Anionic |
| Carbonate |  | 6–8 and ≥20 | Anionic |
| Silicone |  | 6,8 and ≥10 | Anionic, cationic |
| Phosphazene |  | 6 | Cationic |
| Phosphonite |  | 3,5–7 | Anionic |
2. Radical Ring-Opening Polymerization
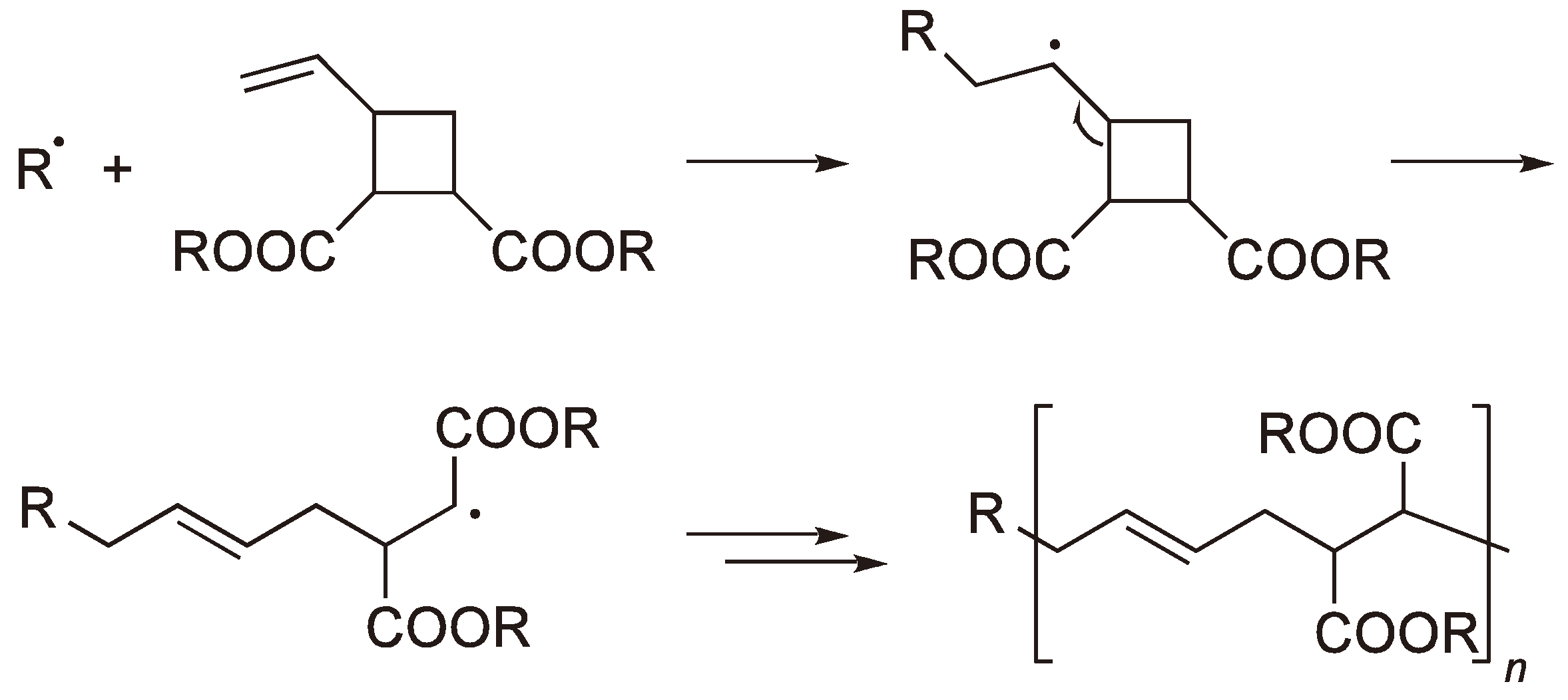
Via a ring-opening polymerization, especially radical ROP (RROP), it is possible to produce polymers with the same or lower density than the monomers. This is interesting for applications where it is desirable to maintain a constant volume during the polymerization such as tooth fillings, coatings and accurate molding of electrical and electronic components. RROP offers novel routes to polyesters and polyketones particularly using vinyl substituted cyclic monomers (see Section 2.2) [6]. A less common group of monomers which undergo RROP are the bicyclobutanes, whereby the bridge across the four-membered ring can be viewed as a concealed double bond (see Figure 1).
Figure 1.
The radical ring opening polymerization (RROP) of a bicyclobutane [7].
Figure 1.
The radical ring opening polymerization (RROP) of a bicyclobutane [7].

2.1. Vinyl Substituted Cyclic Monomers
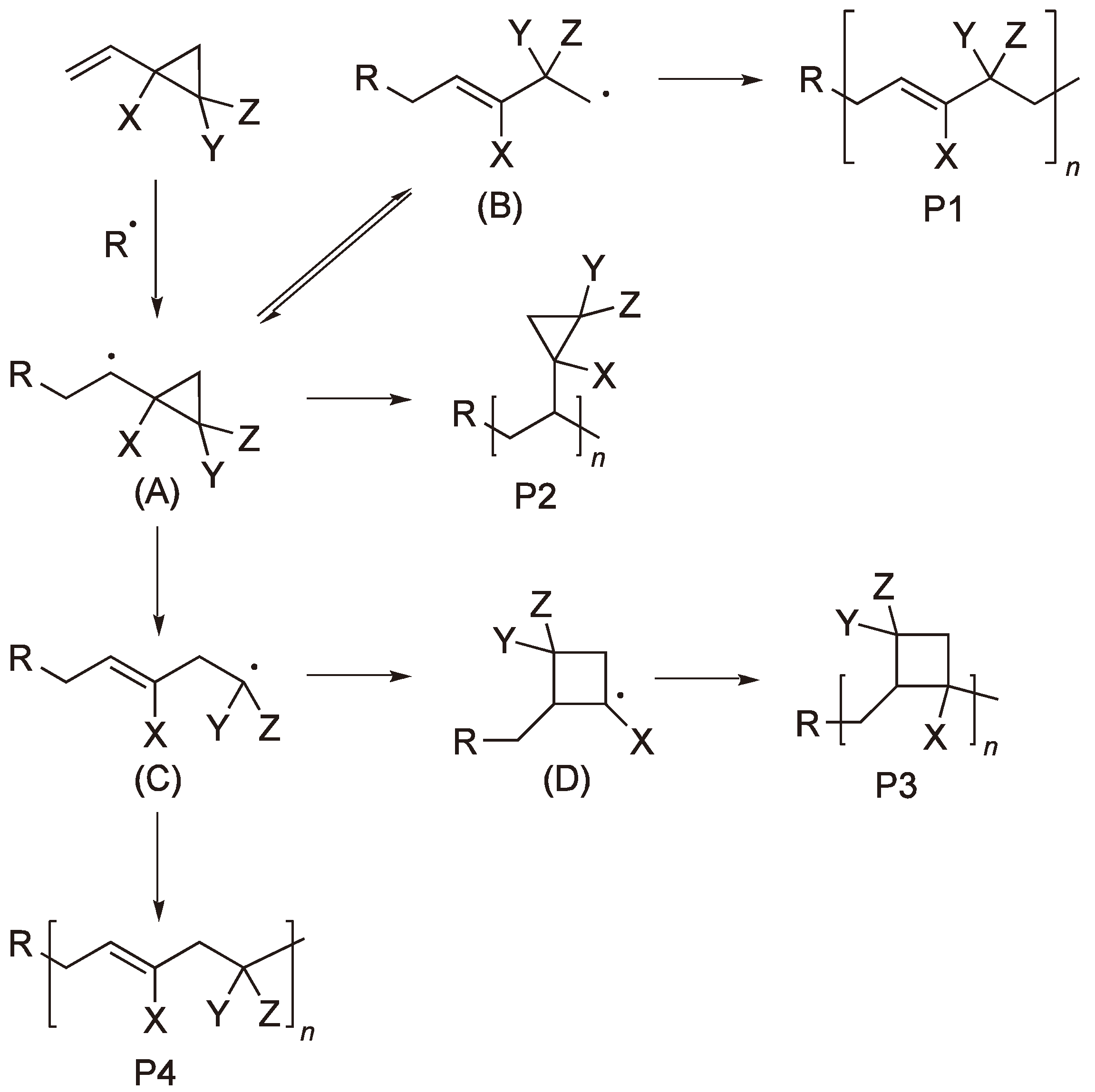
Vinyl cyclopropane and its derivatives have been studied by several groups [8,9,10,11]. The three membered ring has considerable ring strain (115 kJ mol−1) [2] and the simple RROP thus competes with the vinyl chain polymerization (see Figure 2).
Figure 2.
The RROP of vinyl cyclopropane showing the competing reaction pathways.
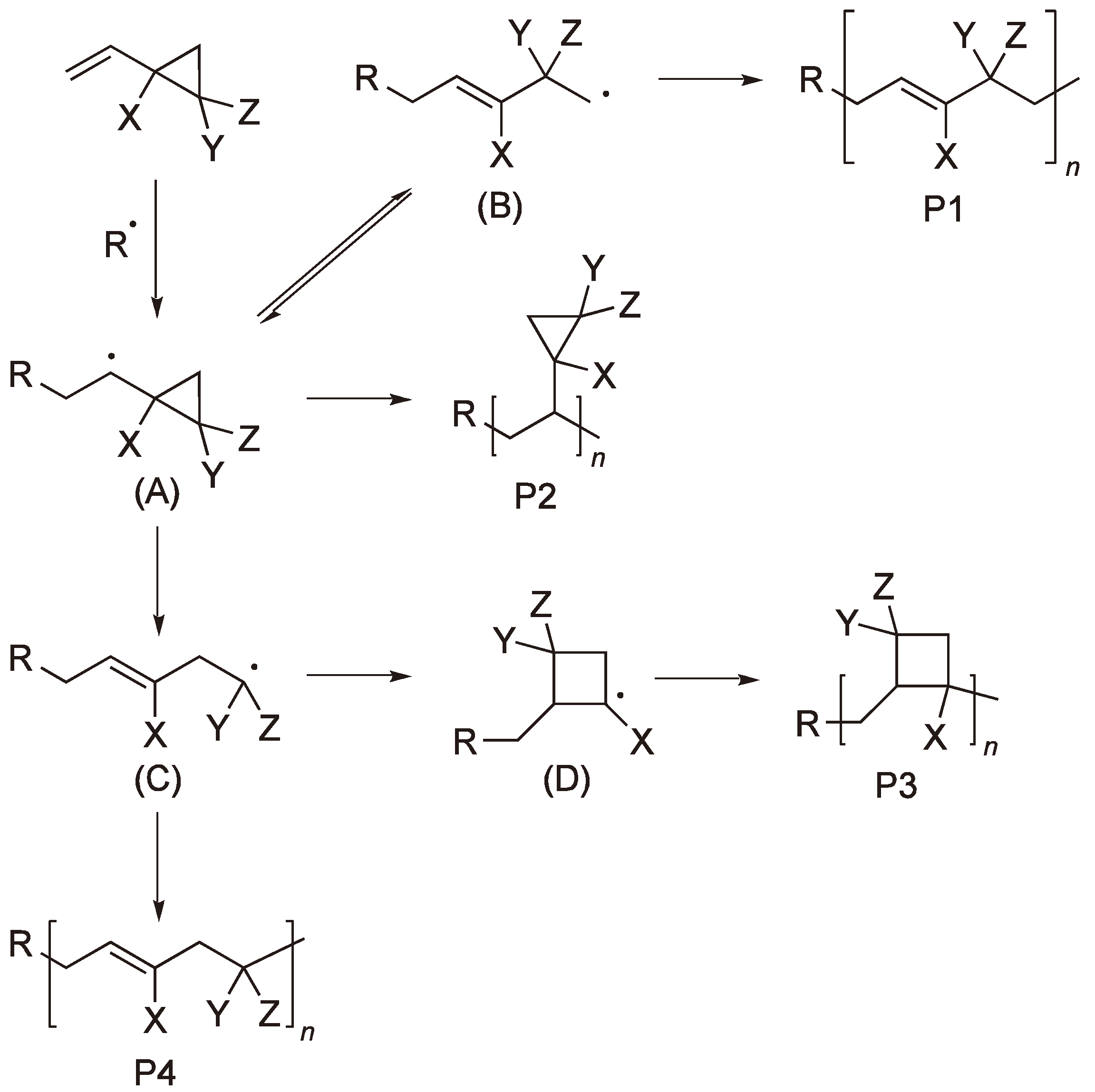
Which of the simple ROP products P1 and P4 predominates depends on the substituents Y and Z and the preference of the ring to open at the CX–CH2 or the CX–CY bond. The simple vinyl chain polymerization P2 and the intermediate formation of a four-membered ring to yield P3 are additional possibilities.
Figure 3.
The RROP of phenyl (Ph) vinyl oxirane.
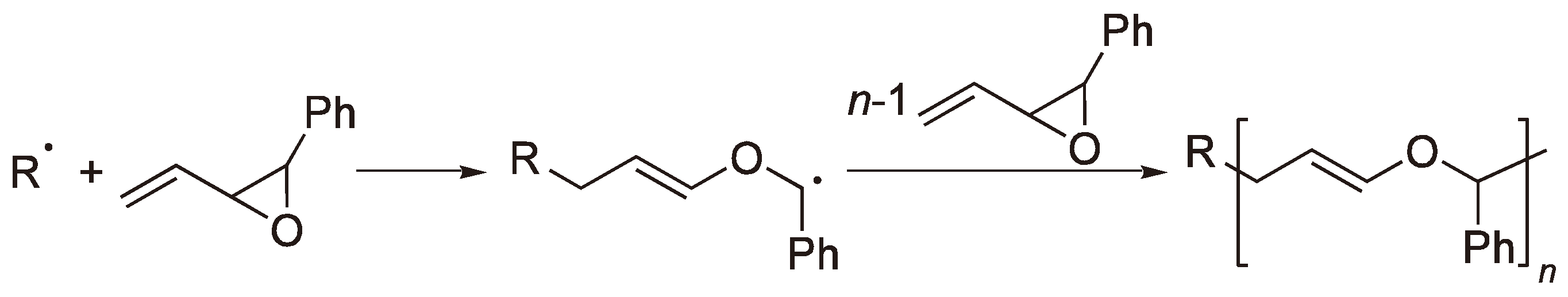
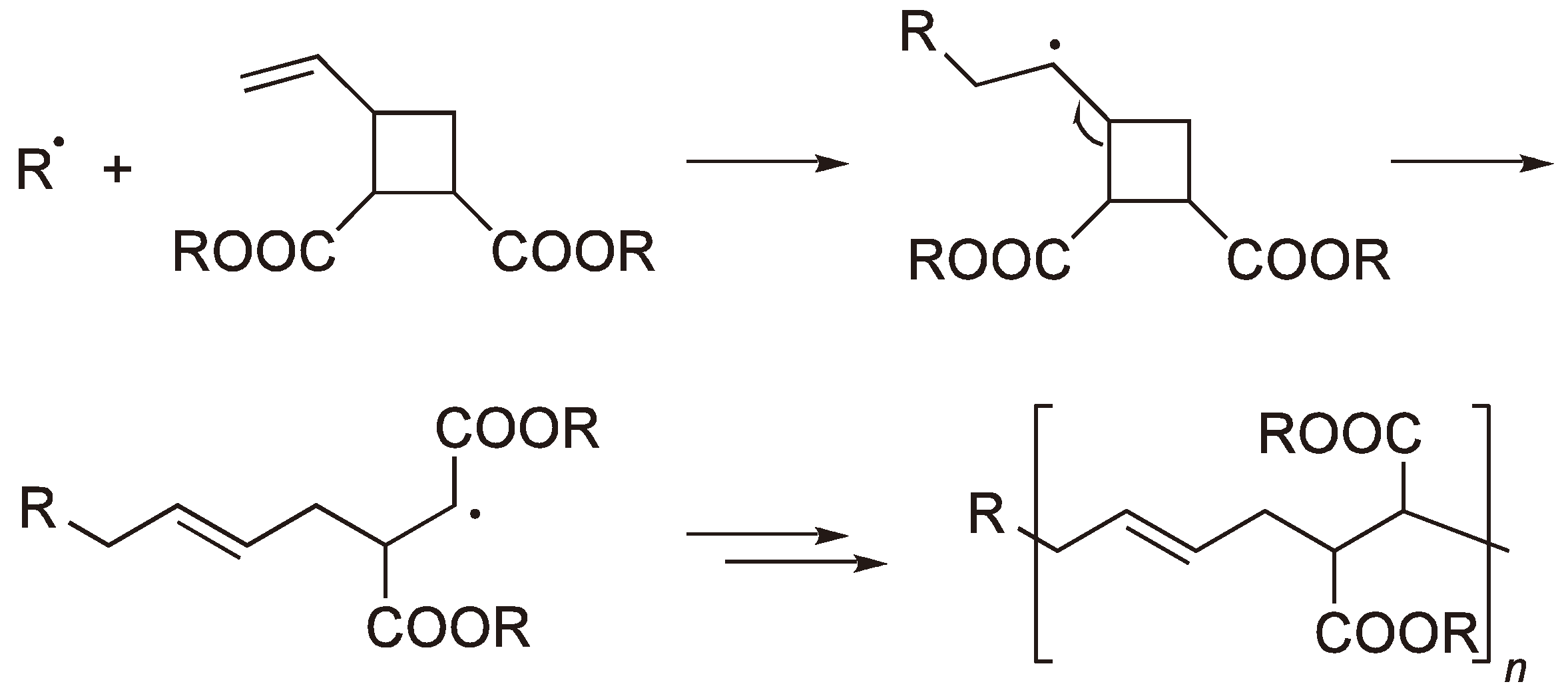
Figure 4.
The RROP of a substituted vinyl cyclobutane.
2.2. Methylene Substituted Cyclic Monomers
The ring-opening of ketene acetals has proved to be a novel route to useful polyesters [16] (see Figure 5) although the sensitivity of the monomers to acid hydrolysis makes their handling difficult.
Figure 5.
The RROP of ketene acetals.

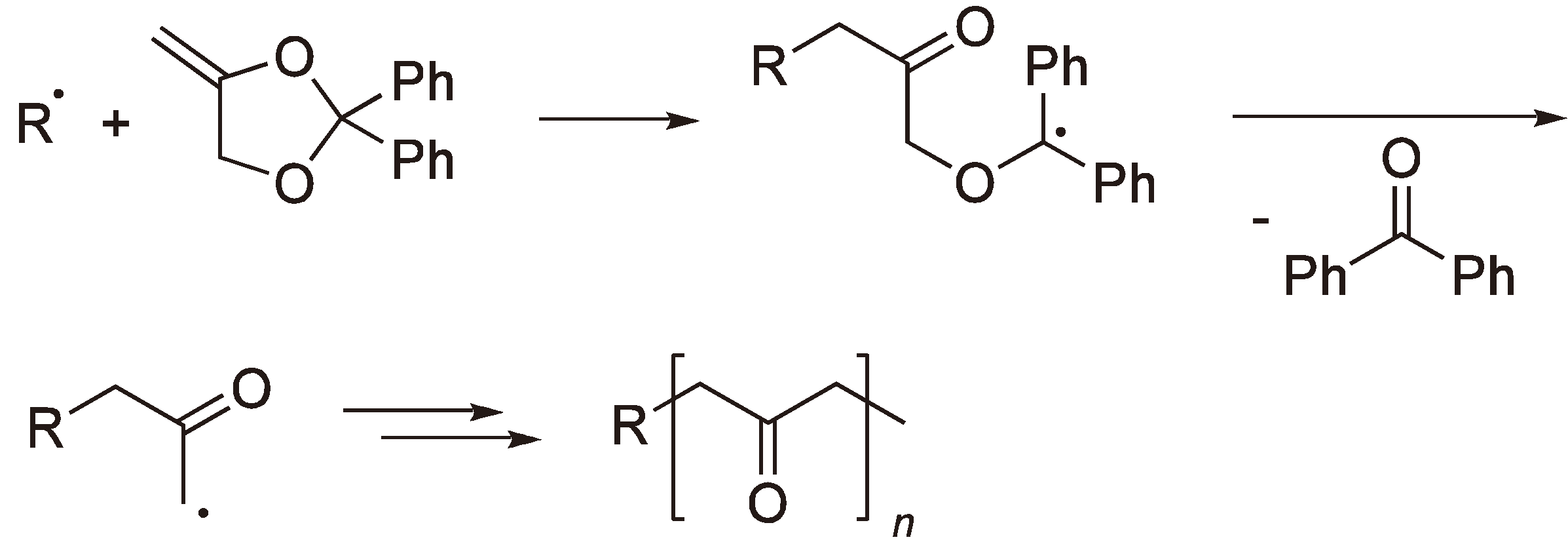
The polymerization of 2,2-diphenyl-4-methylene-1,3-dioxolane [17] yields a polyketone rather than a polyester since benzophenone is eliminated by each ring-opening step (see Figure 6).
Figure 6.
The radical polymerization of 2,2-diphenyl-4-methylene-1,3-dioxolane to yield a polyketone.
Figure 6.
The radical polymerization of 2,2-diphenyl-4-methylene-1,3-dioxolane to yield a polyketone.
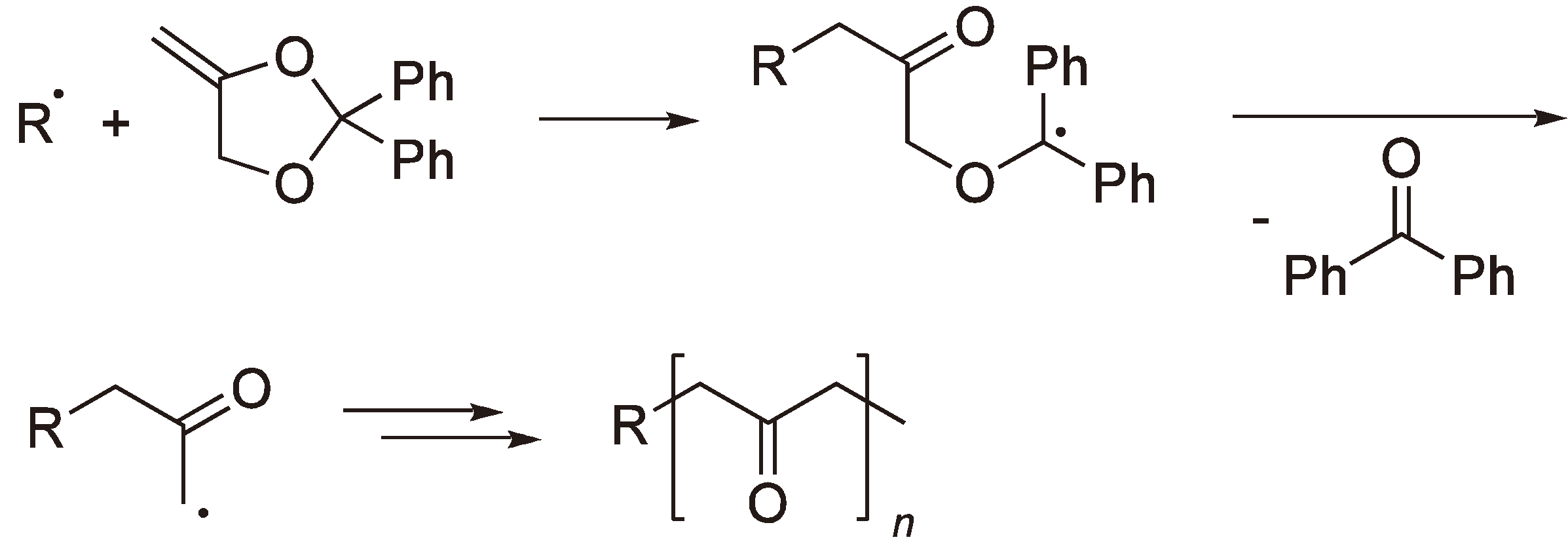
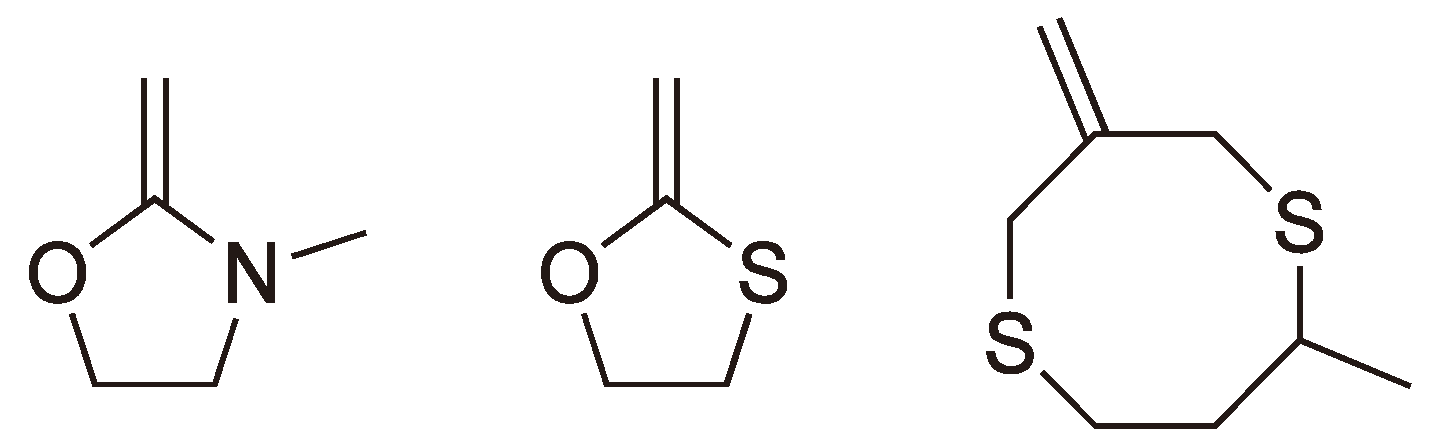
Heterocyclic monomers with nitrogen or sulfur heteroatoms have also been shown to undergo radical ROP (see Figure 7).
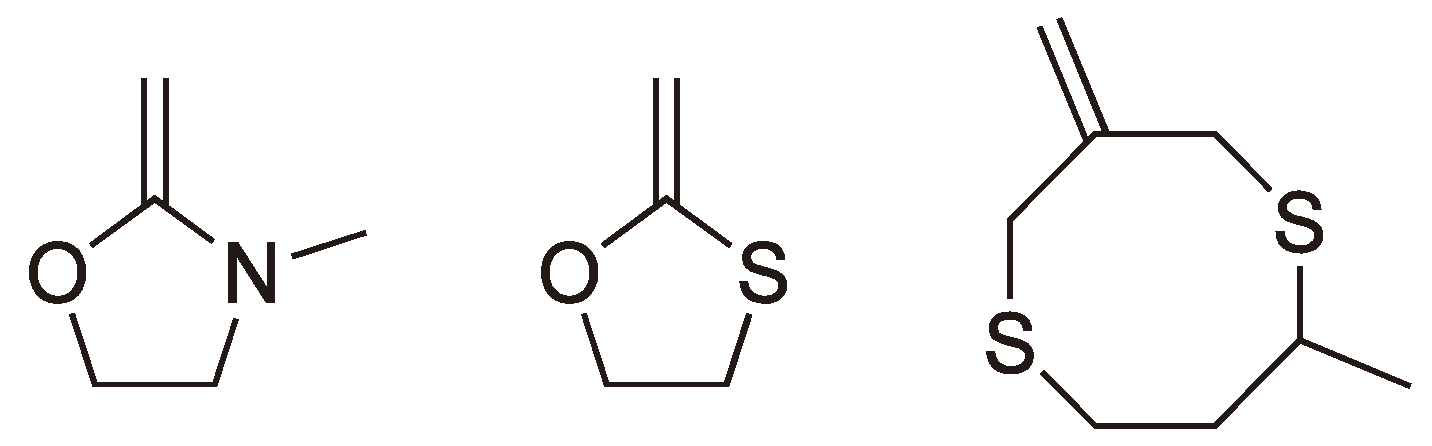
2.3. Double Ring-Opening
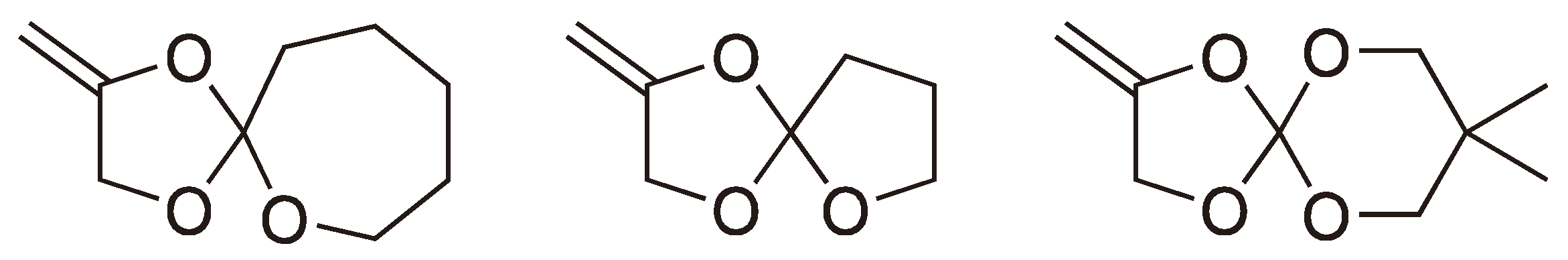
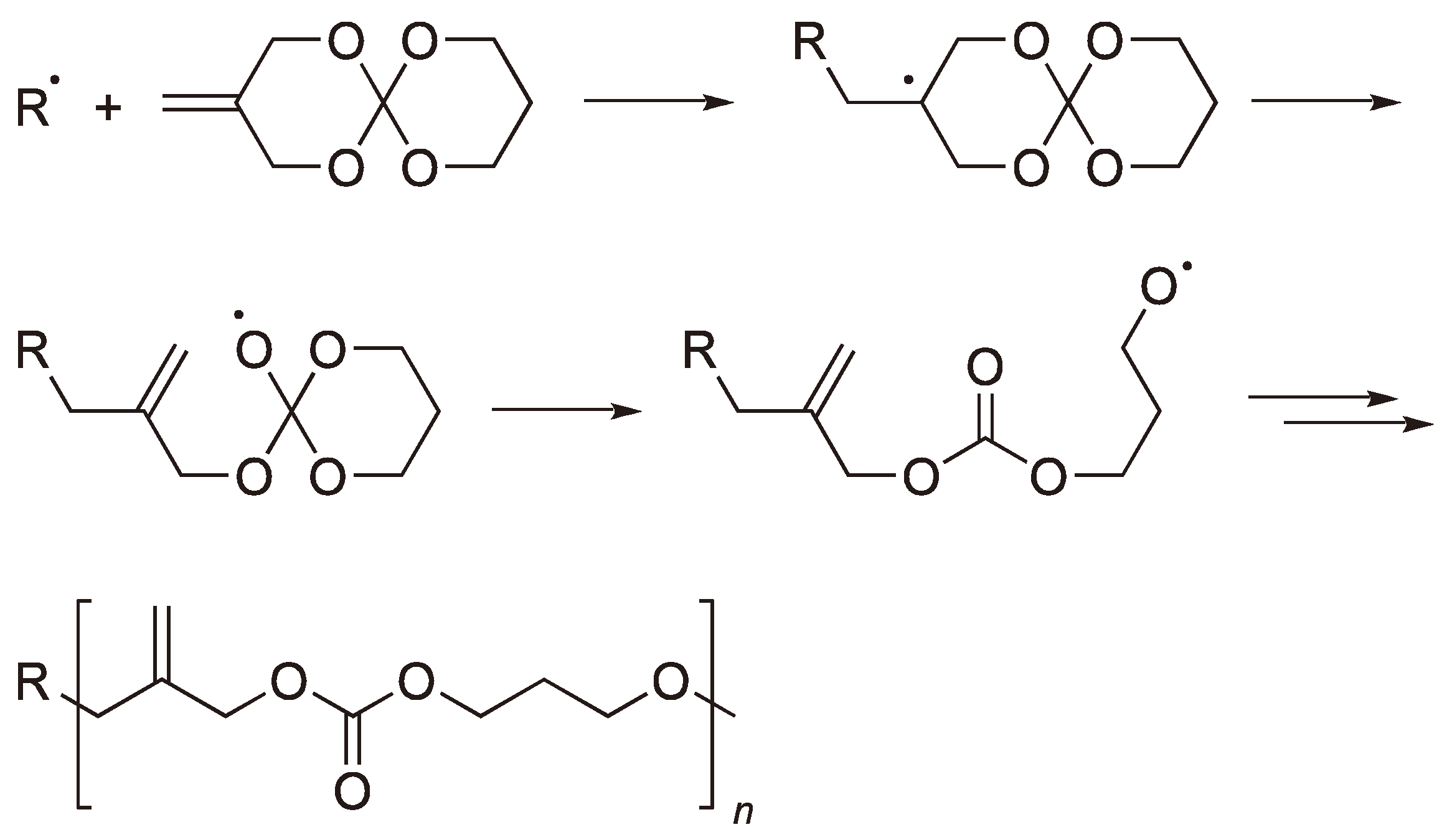
Spiro ortho-carbonates and their analogues have been the subject of considerable interest due to the volume expansion which occurs when the crystalline monomers are polymerized (If the monomers are first melted then a volume contraction takes place) [20]. A typical reaction scheme is shown in Figure 8 [16].
Figure 8.
The radical ROP of a spiro carbonate.

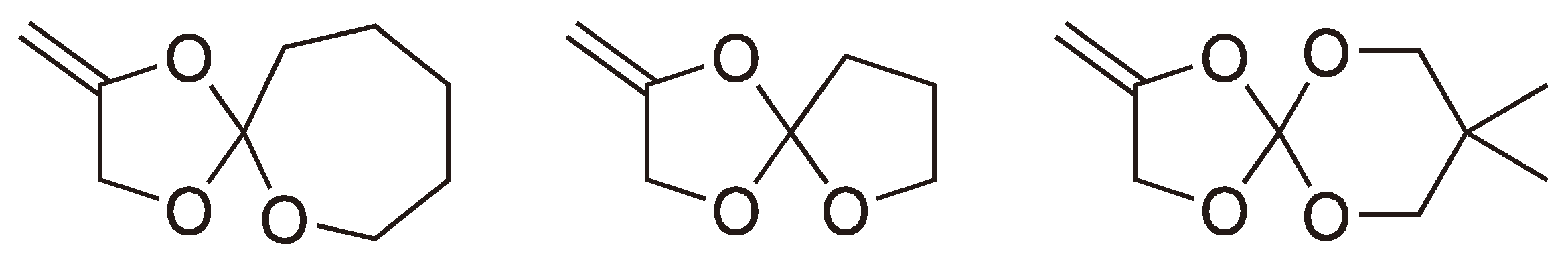
The spiro ortho esters shown in Figure 9 have also been shown to polymerize, essentially quantitatively, via a radical ROP [21,22,23,24].
Figure 9.
Some other examples for the radical ROP spiro monomers.
2.4. Degradable Polyesters via Radical Ring-Opening Homo- and Copolymerization
The polyester synthesis described above under 2.2 (Figure 5) is not a typical CROP, AROP or RROP in that a rearrangement takes place as each ring opens. Despite the apparently promising work of Bailey [16] the synthesis of polyesters via a RROP of ketene acetals initially received little interest. However, with the increasing interest in biodegradable polyesters and the versatility of RROP with respect to the structures accessible via very different monomers [25] and their copolymerization with conventional vinyl monomers [26,27,28,29,30,31,32,33,34] has changed the situation significantly.
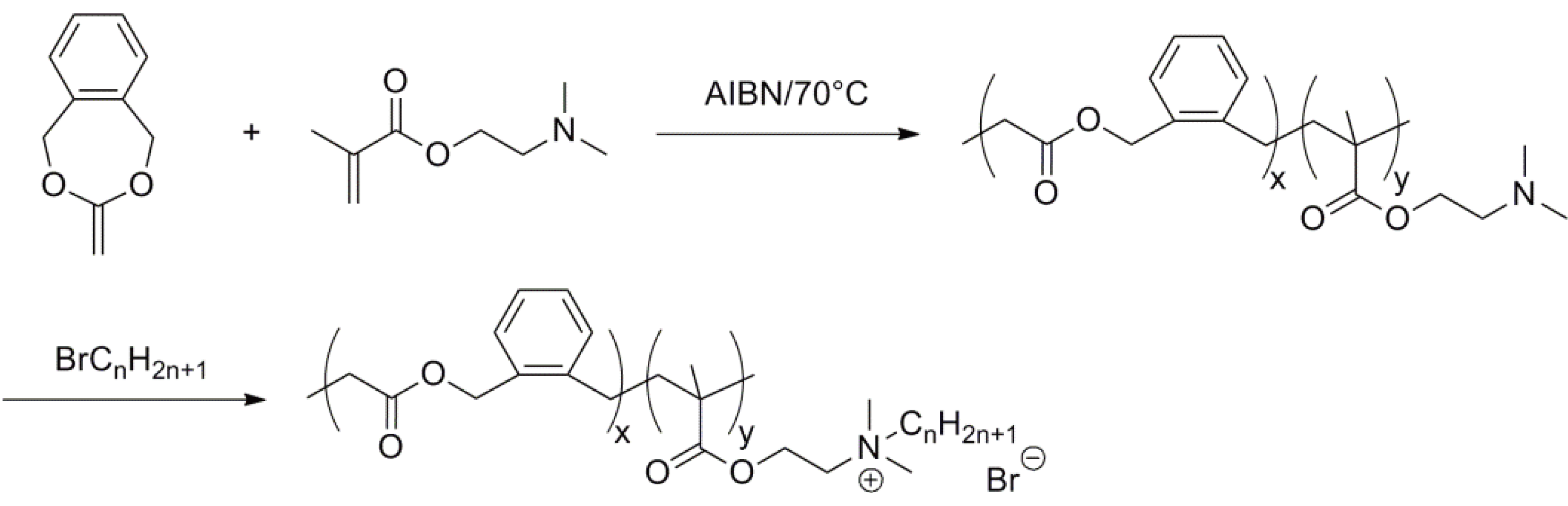
Figure 10 shows an example of such a copolymerization. It is clear that by varying the monomers or their proportions, or indeed by varying the mode of initiation, e.g., NMP (nitroxide mediated polymerization) [35], RAFT (reversible addition fragmentation transfer polymerization) [36] or ATRP (atom transfer radical polymerization) [37] many and varied polymers can be obtained, optimized for their use in agricultural, medicinal or pharmaceutical fields.
Figure 10.
An example of a radical ring opening copolymerization of a ketene acetal.

3. Cationic Ring-Opening Polymerization
Ring-opening polymerizations involving a positively charged intermediate (Cationic ROP or CROP) provide several important industrial polymers. These include polyacetals, copolymers of 1,3,5-trioxane and oxirane or 1,3,5-trioxane and 1,3-dioxolane, polytetrahydrofurans, copolymers of tetrahydrofuran and oxirane, poly (3,3-bis(chloro-methyl)oxetanes), polysiloxanes, polymers of ethyleneimine and polyphosphazenes [38,39].
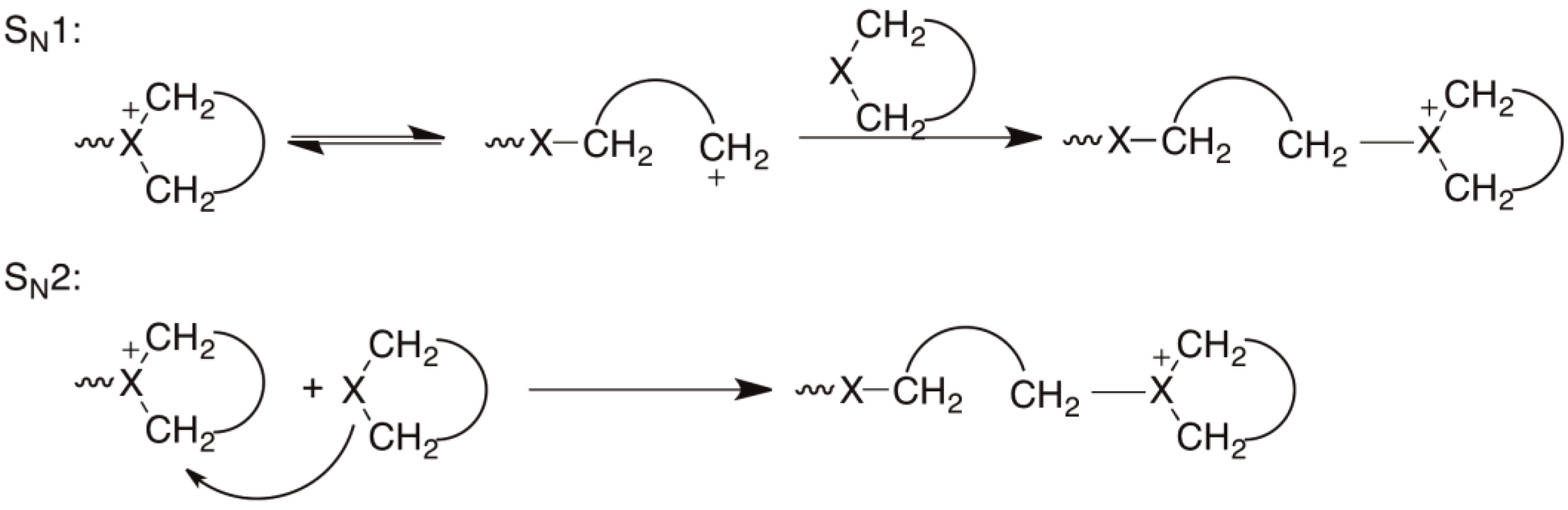
There are essentially two mechanisms that are discussed in the literature [38] for CROP: The first involves a growing chain with a cationic center at the chain end which adds to a monomer molecule via a SN1 or SN2 mechanism (Figure 11), whereby X represents an oxygen, a nitrogen or a sulfur atom.
Figure 11.
CROP involving active chain ends; SN1 and SN2 mechanisms.
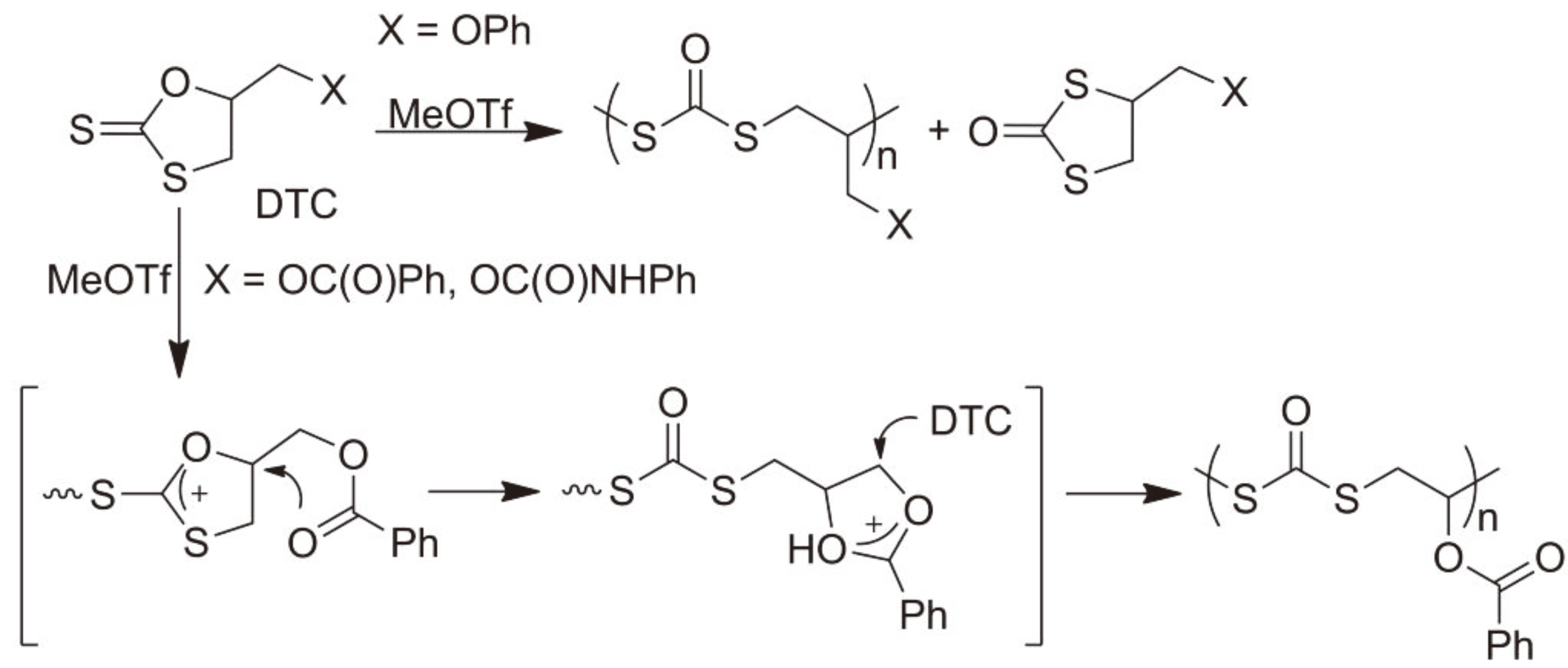
The polymer structure can be further controlled by the use of appropriate side groups. Thus, the exchange of an ether side group for an ester or carbamate (So-called NGP: Neighboring Group Participation) results in a different, well defined, microstructure during the CROP of cyclic dithiocarbonates (Figure 12) with methyl triflate initiator due to effective suppression of the isomerization of the monomer and the stabilization of the growing chain via the ester (carbamate) moiety [40].
Figure 12.
Controlling the reaction path of a CROP of dithiocarbonates using Neighboring Group Participation (NGP).
Figure 12.
Controlling the reaction path of a CROP of dithiocarbonates using Neighboring Group Participation (NGP).
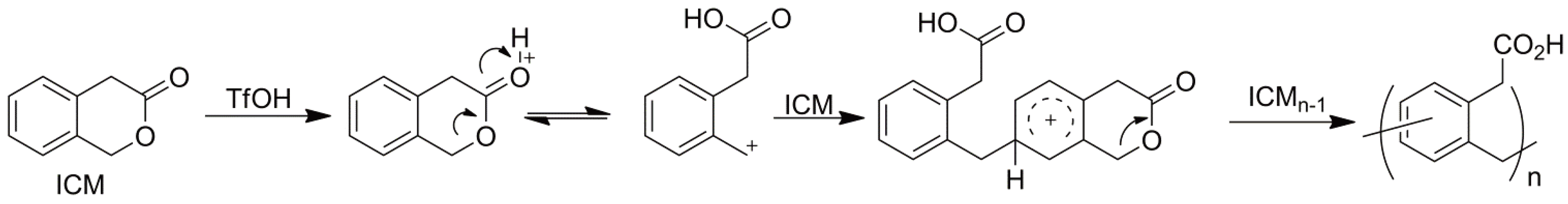
An extreme example of NPG is reported by Suzuki et al. [41] TfOH who describe the polymerization of an aromatic lactone in which the lactone is part of a fused ring system (3-Isochromanone). Using a variety of spectroscopic techniques and an elegant analysis, the authors suggest that the resulting polymer, which could be prepared in excellent yield via a bulk polymerization (2 h at 120 °C, 5 mol % triflic acid (TfOH) as catalyst), is formed via an intermediate Friedel-Crafts alkylation step (Figure 13) as a result of the stabilization of the growing chain end in the form of a benzyl cation.
Figure 13.
The CROP of 3-isochromanone.

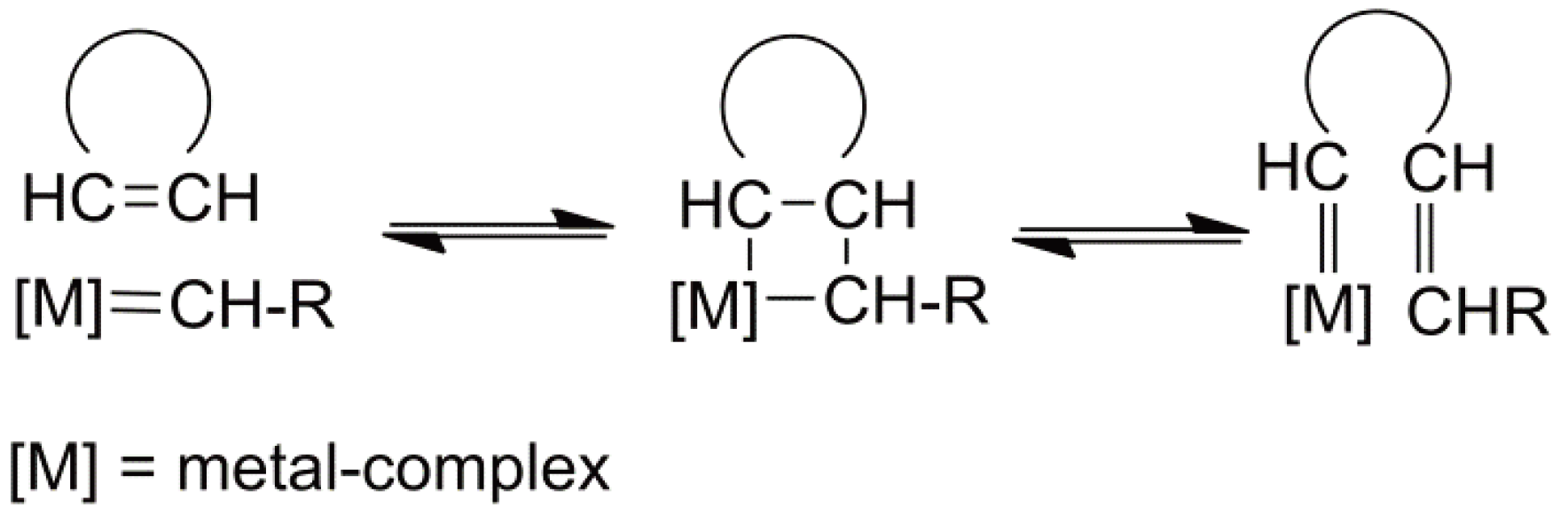
A treatment of the propagation of CROP would not be complete without reference to a long discussion, which can be considered as resolved (vide infra), specifically concerning, e.g., 1,3-dioxolanes, as to whether the propagating species are linear tertiary or secondary ions and thus, the mechanism is one of ring expansion or of linear growth. In terms of the microstructure of the polymers produced this is not trivial. In a very detailed analysis Plesch et al. [42] showed that under conditions where the concentration of water was less than 10−4 mol L−1 and at relatively low monomer concentration, the secondary oxonium ion predominates and the ring expansion mechanism prevails. In contrast to the suggestions of Plesch, Jaacks [43] and later Penczek [44] postulated, based on extensive experimental results that propagation takes place essentially via open chains. Finally, from even more detailed studies it was found that ring-expansion is favored at low monomer concentrations and the open chain mechanism dominates the propagation at higher monomer concentration [45] (see also [5]). This discussion is worthy of detailed study since it demonstrates the need to carefully examine the experimental techniques and the prevailing conditions before making conclusions about reaction mechanisms, especially where cationic systems are involved.
An alternative, third, mechanism proposed for the propagation of CROP is that the monomer is activated and carries the cationic center; the growth reaction is an electrophilic attack of the activated monomer on the chain end (Figure 14).
Figure 14.
CROP via an activated monomer mechanism.

For some systems it has been shown that both mechanisms operate simultaneously [46].
There are at least three types of reactions [38], which compete with the propagation reaction during a CROP as shown in Figure 15.
Figure 15.
Three principal side reactions during a CROP.

3.1. Initiation
CROP can be initiated by Brönsted acids, carbenium ions, onium ions, photoiniators and covalent initiators:
- Brönsted acids:
Protons can initiate the polymerization of numerous heterocyclic monomers, e.g., oxiranes, 1,3-dioxolanes, thiiranes, aziridines, tetrahydrofurans, lactones, 1,2 oxazolines and cyclic siloxanes as shown in Figure 16 assuming an activated chain end mechanism, in analogy to Figure 12 (SN2). “Dry” acids such as HCl, H2SO4, as well as HClO4 and HOSO2CF3 have frequently been used [40,41,42].
Figure 16.
Initiation of a cationic ROP by a Brönsted acid.
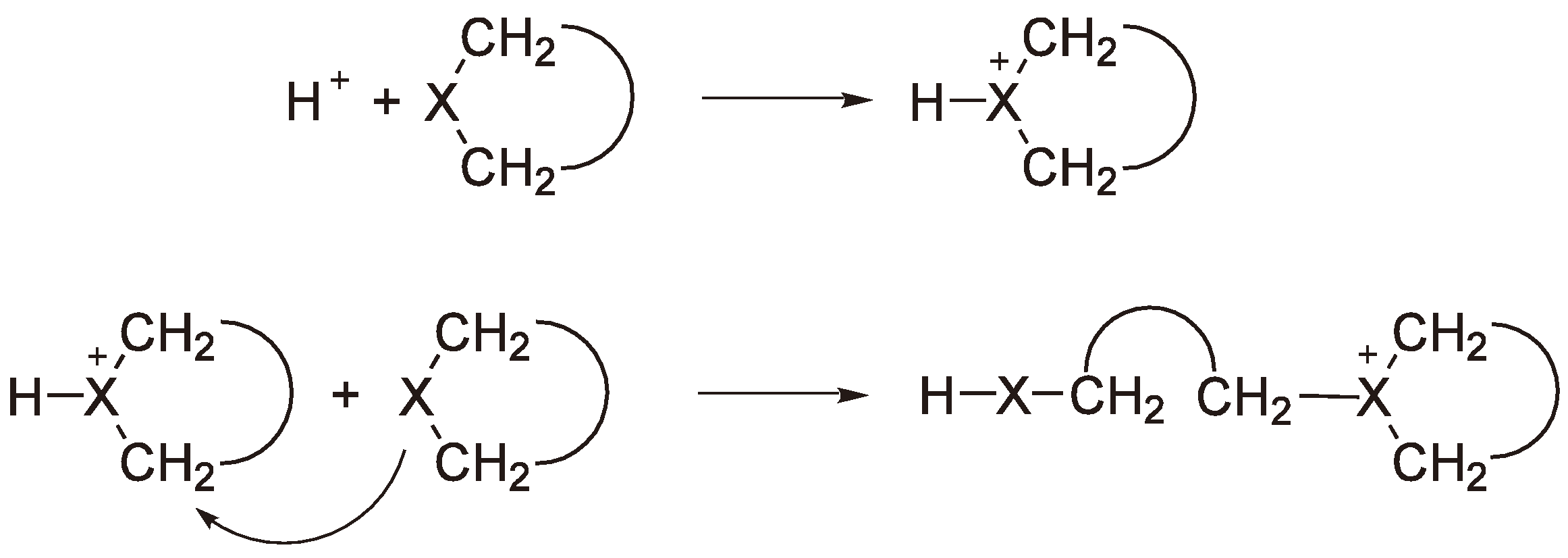
Alternatively, the heteroatom poly-acids of the general structure shown in Figure 17 can be employed as initiators [47].
Figure 17.
General structure of heteroatom poly-acids.
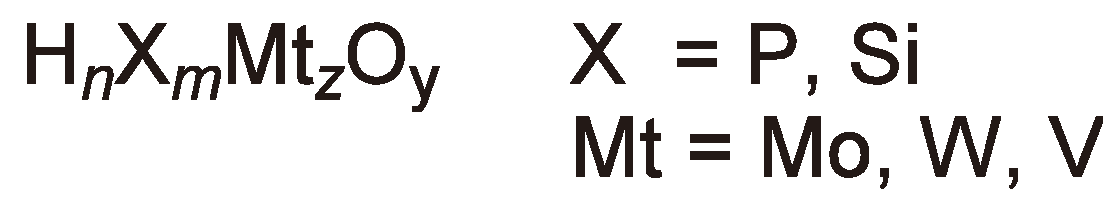
The acids with X = P can be relatively easily prepared from phosphoric acid and the required metal oxide; they are stable in air and are soluble in a variety of organic solvents. Two examples are well documented in the literature: H3PMo12O40 and H3PW12O40 [47].
The CROP of epoxides and oxetanes via Brönsted acids are often characterized by an induction period and, although not completely explained, the more recent studies of Crivello’s group [48] modifying the kinetics of the photo initiated CROP by the addition of crown ethers (which are similar in structure to the oligomers formed in the early stages of polymerization) or 2,6 di-tert-butyl pyridine do offer a plausible explanation for some of the observed effects. Thus, control of an auto accelerated thermal polymerization is improved by such additives.
- Carbenium Ions:
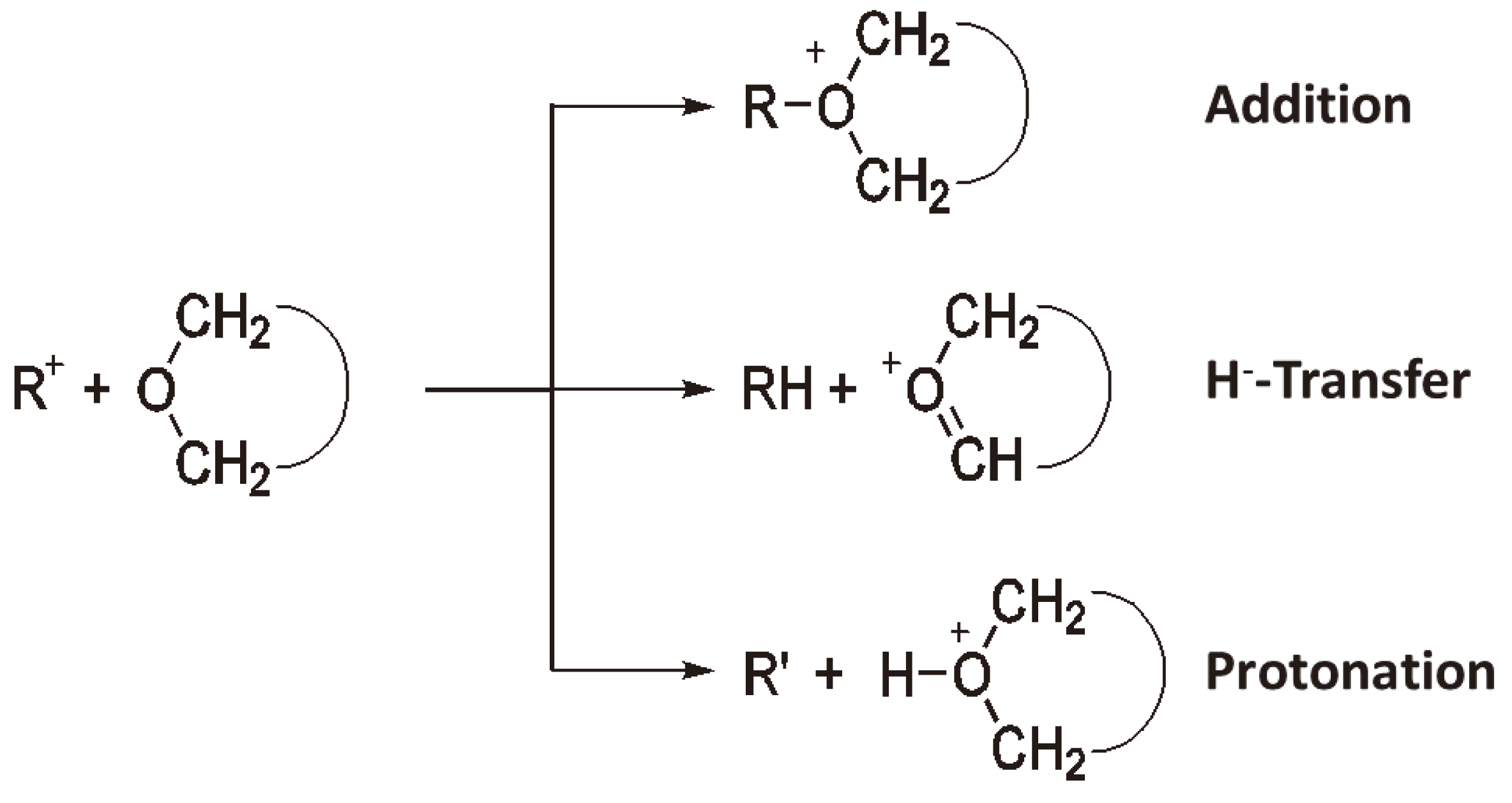
Initiation of CROP by various carbenium ions has been frequently studied, the general reaction scheme is shown in Figure 18 (R+ = (C6H5)3C+ [49], (C6H5)2CH+ [50], (C6H5)CH2+, H2C = CH–CH2+ and (CH3)3C+ [38]). The carbenium ions can react with rings of various structures and having various hetero atoms:
Figure 18.
Initiation of CROP by carbenium ions (R = R+(–H+)).
- Onium-Ions:
Onium-Ions can be prepared in a separate reaction or in situ [51]. Initiation by onium ions follows, in a general sense, the scheme shown in Figure 19; typically the reaction involves the transfer of a carbenium ion and can thus be seen as an extension of the initiation by carbenium ions (vide supra).
Typical examples are the reactions of trialkyl oxonium ions with cyclic acetals [38], ethers [52], or sulfides [53] as well as with amines [54].
A variation on this type of initiation is reported by Li and Yu [55] who have reported the use of glycosyl ortho-alkynyl benzoates and a gold triflate salt (PPh3AuNTf2) to prepare poly-THF with glycosyl terminal groups.
Figure 19.
Initiation by onium-ions (Z and X may be identical, e.g., oxygen).

- Covalent Initiators:
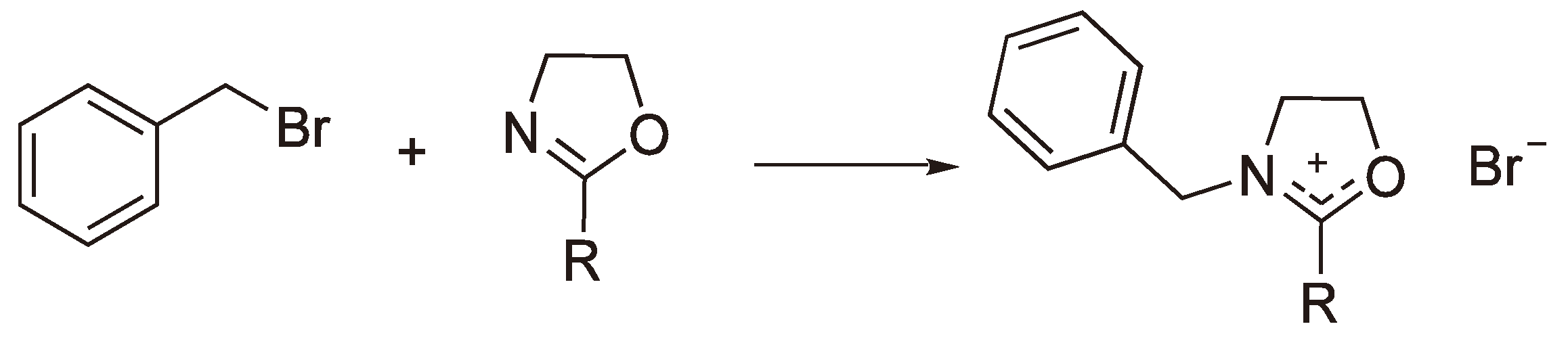
The polymerization of 2-Alkyl-2-oxazolines can be initiated by classical alkylating reagents, such as benzyl bromide [56] (see Figure 20).
Figure 20.
Initiation by covalent reagents.
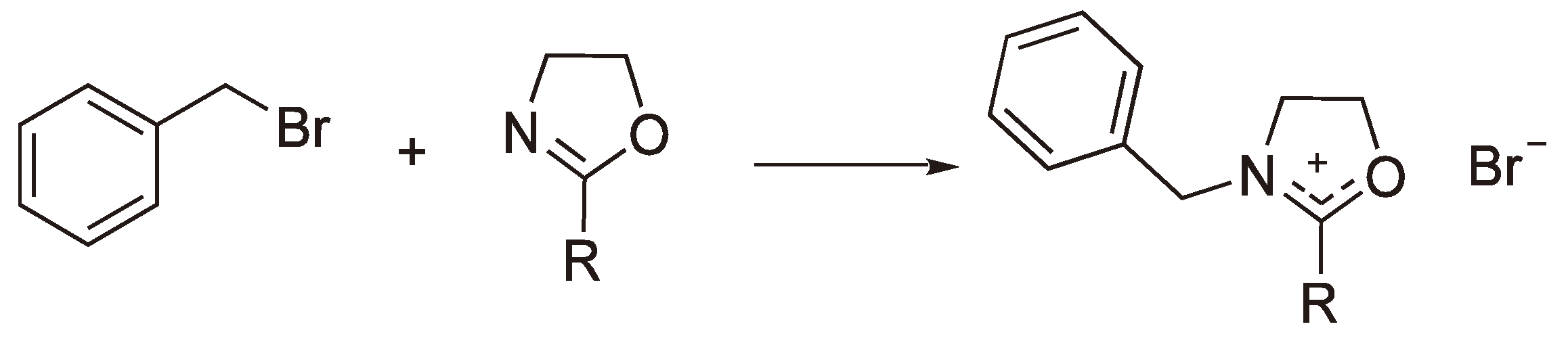
Esters of Brönsted acids (e.g., ROSO2CF3 and ROSO2F) can also be used as initiators [57,58,59], whereby the covalent initiators act as precursors for a carbenium ion initiation (vide supra). In particular, the use of triflic acid anhydride [57] is worth noting as a route to growing polymers of THF with two active ends.
- Lewis acids:
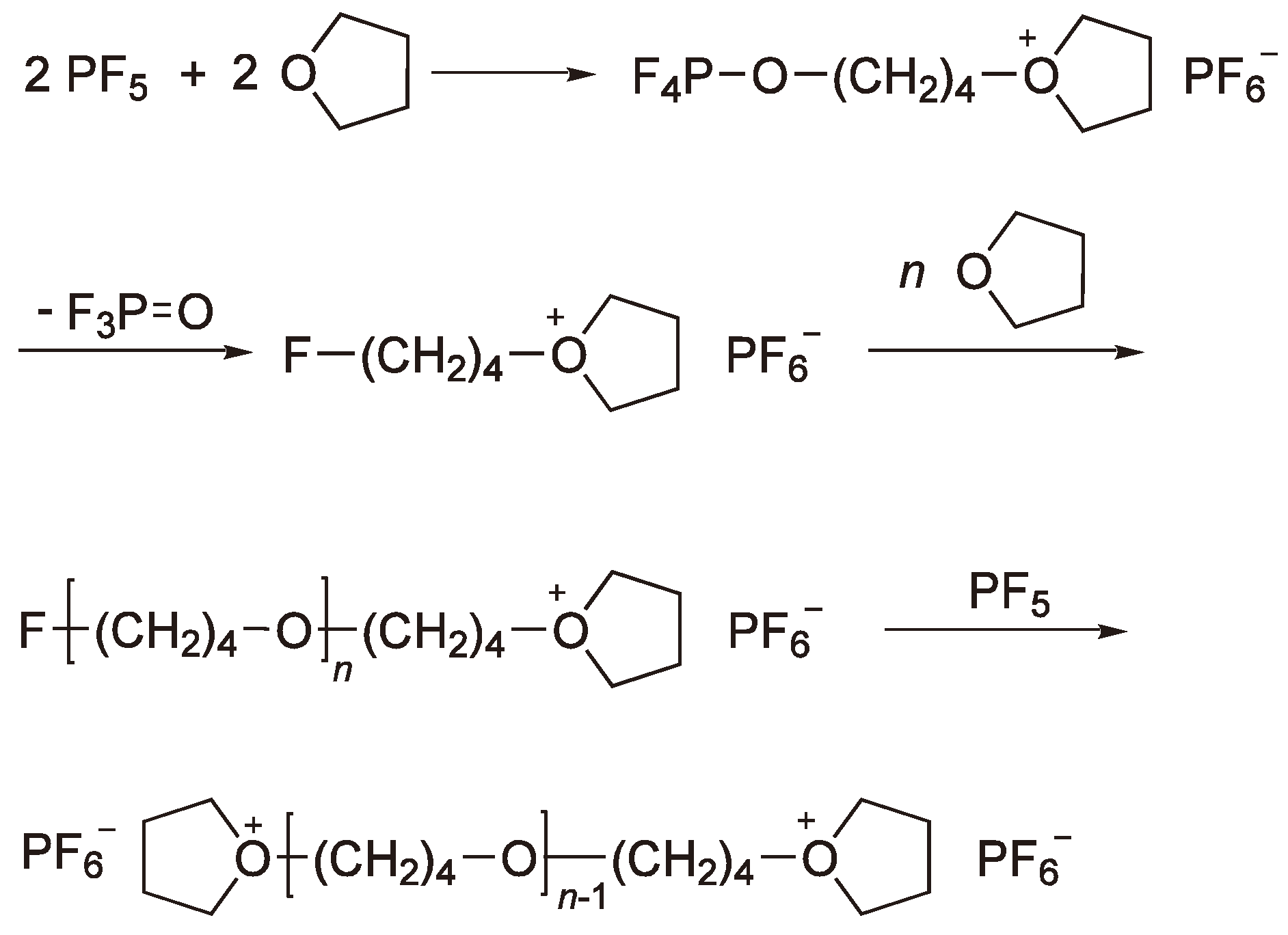
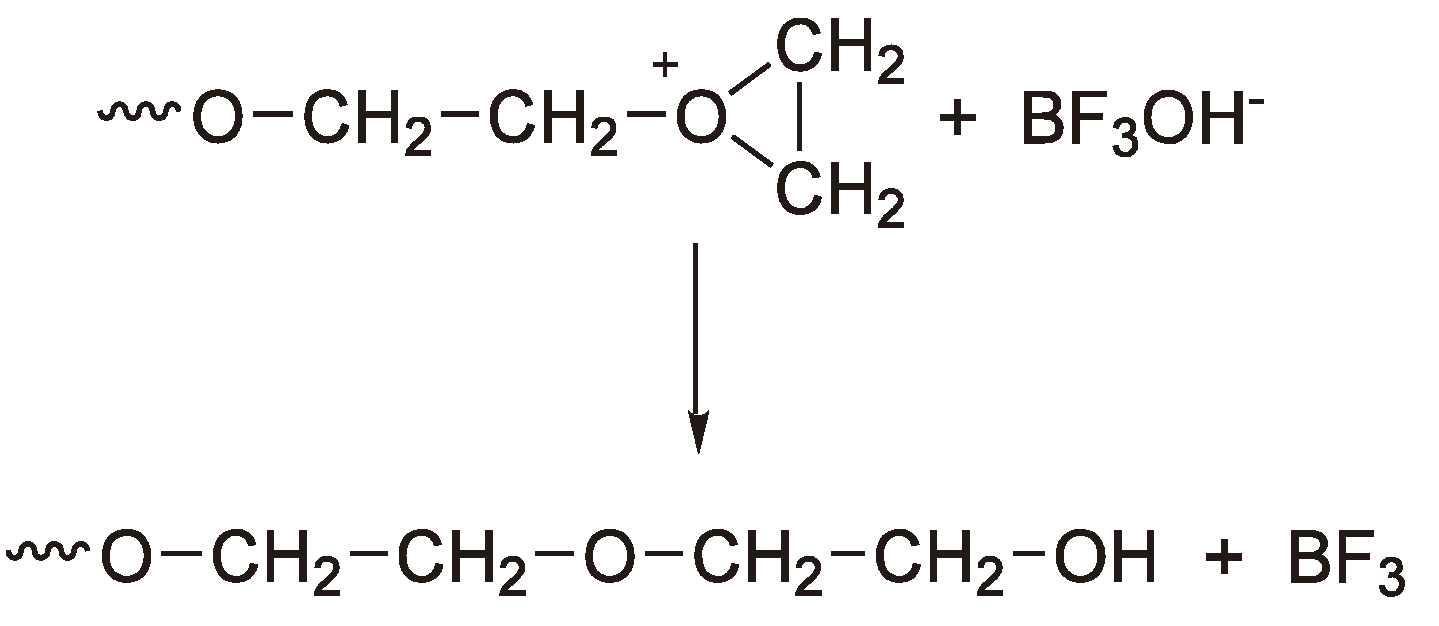
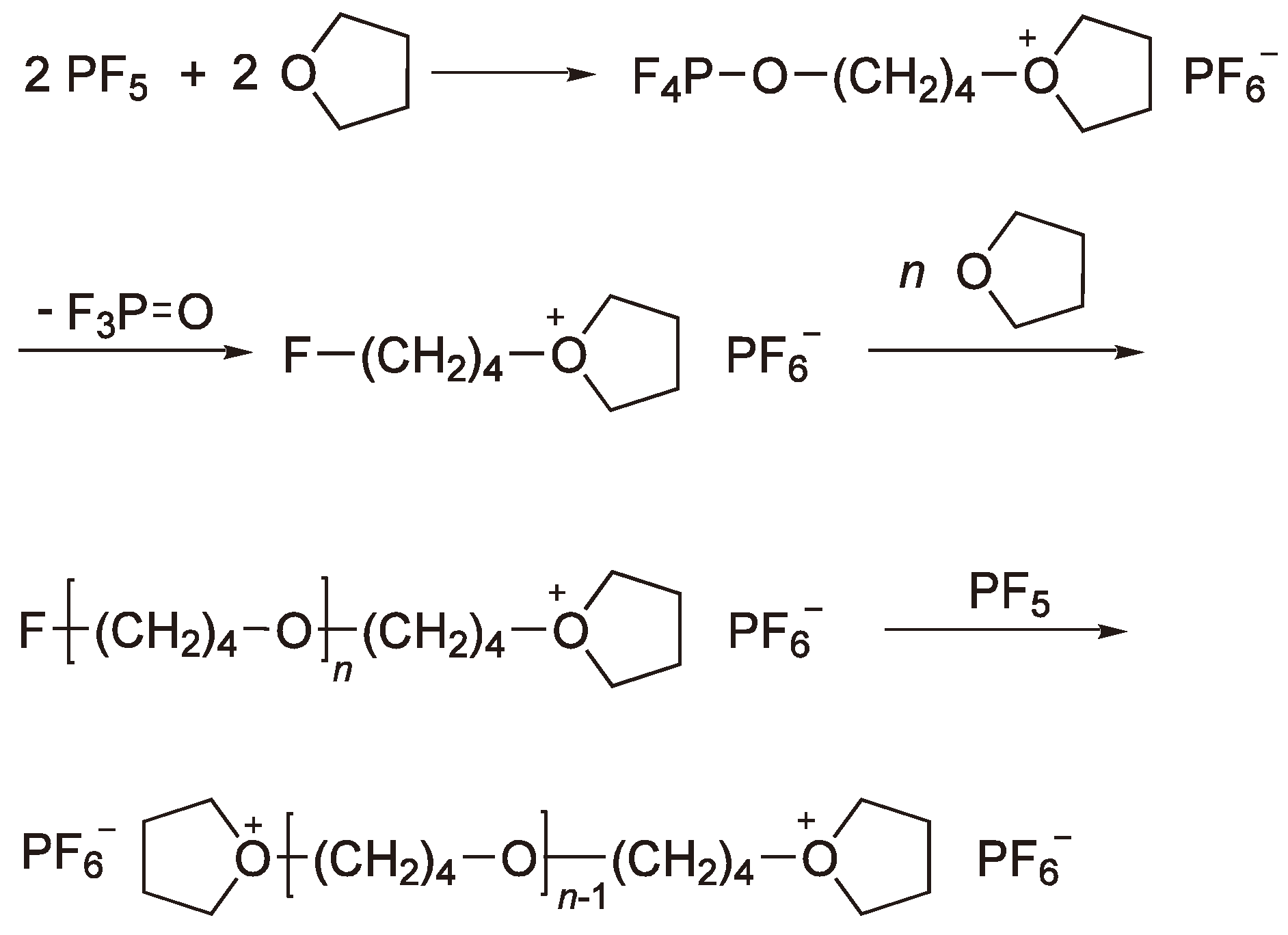
These compounds require a coinitiator (activator), e.g., an alkyl halide or water, in order to polymerize heterocyclic monomers [2,38,60]. The most frequently employed Lewis acid for the cationic polymerization is BF3 and its precursor BF3OEt2. However, in order to apply them for the ROP of THF the addition of cationogenic compounds such as acid halides is required. Detailed studies have also been made using PF5, e.g., for the polymerization of THF for which a mechanism supported by 19F and 31P NMR spectroscopy has been proposed (see Figure 21) [61].
Figure 21.
Initiation of the CROP of THF by PF5. Chain growth from both chain ends.
- Photoinitiators
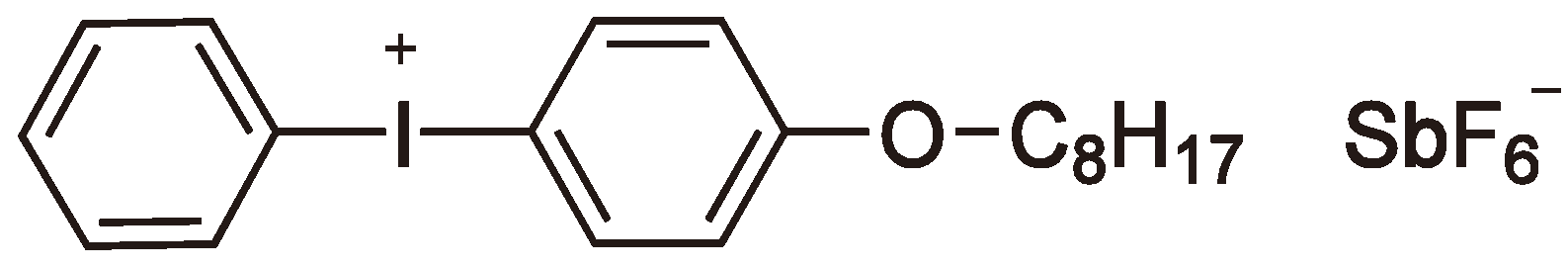
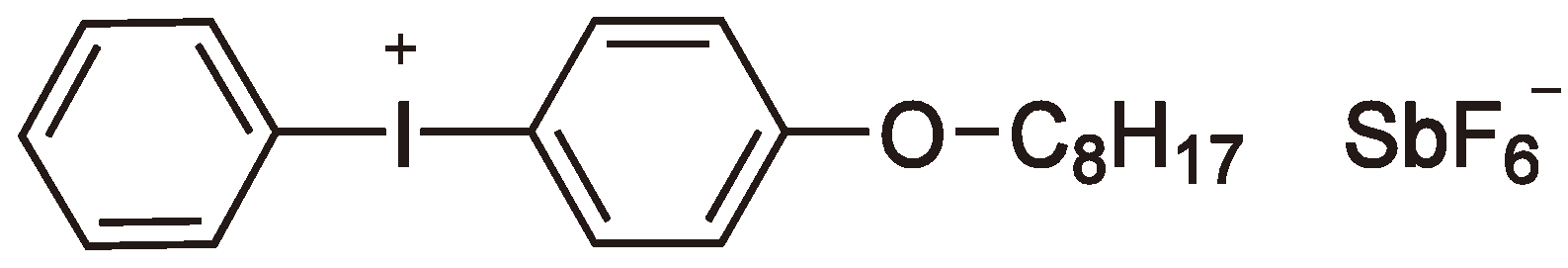
Sulfonium- and iodonium-salts have achieved industrial importance as cationic photo initiators [39]. Both heterolytic and homolytic reaction mechanisms are possible. Taking sulfonium salts as an example, via a heterolytic process the sulfonium salt is raised to a singlet-state, which then relaxes to give Ph2S and Ph+ (from Ph3S+). The Ph+ moiety can then react with e.g., an alkyl group (this may be the solvent, polymer or monomer [48,62,63,64,65]) to yield a proton and PhR (see Figure 22).
Figure 22.
Formation of an initiator by heterolytic or homolytic splitting of a photoinitiator (*: Singlet-state).
Figure 22.
Formation of an initiator by heterolytic or homolytic splitting of a photoinitiator (*: Singlet-state).
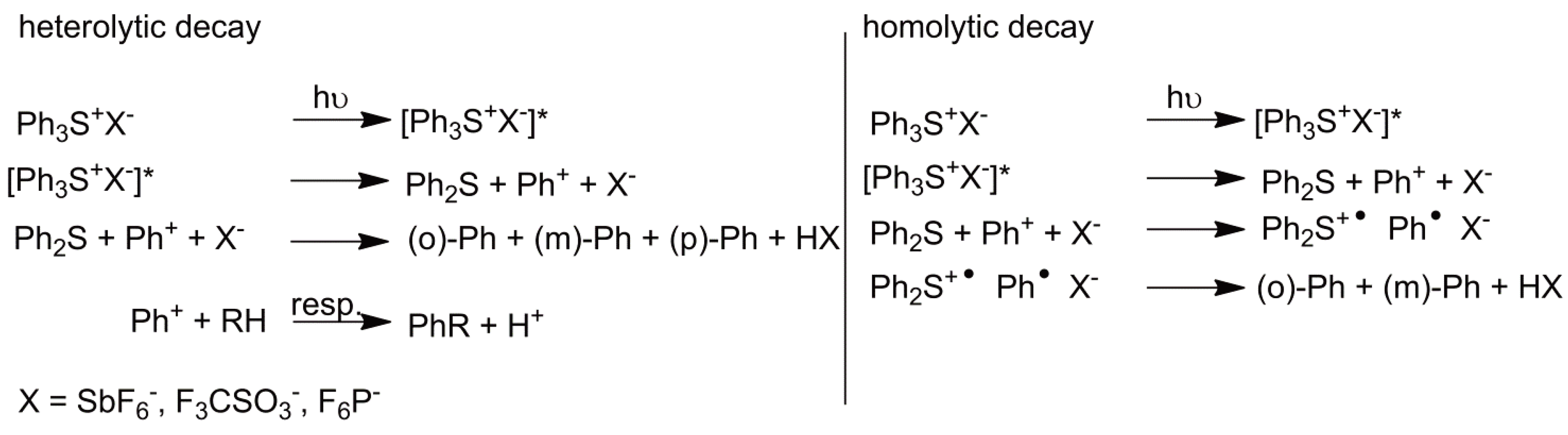
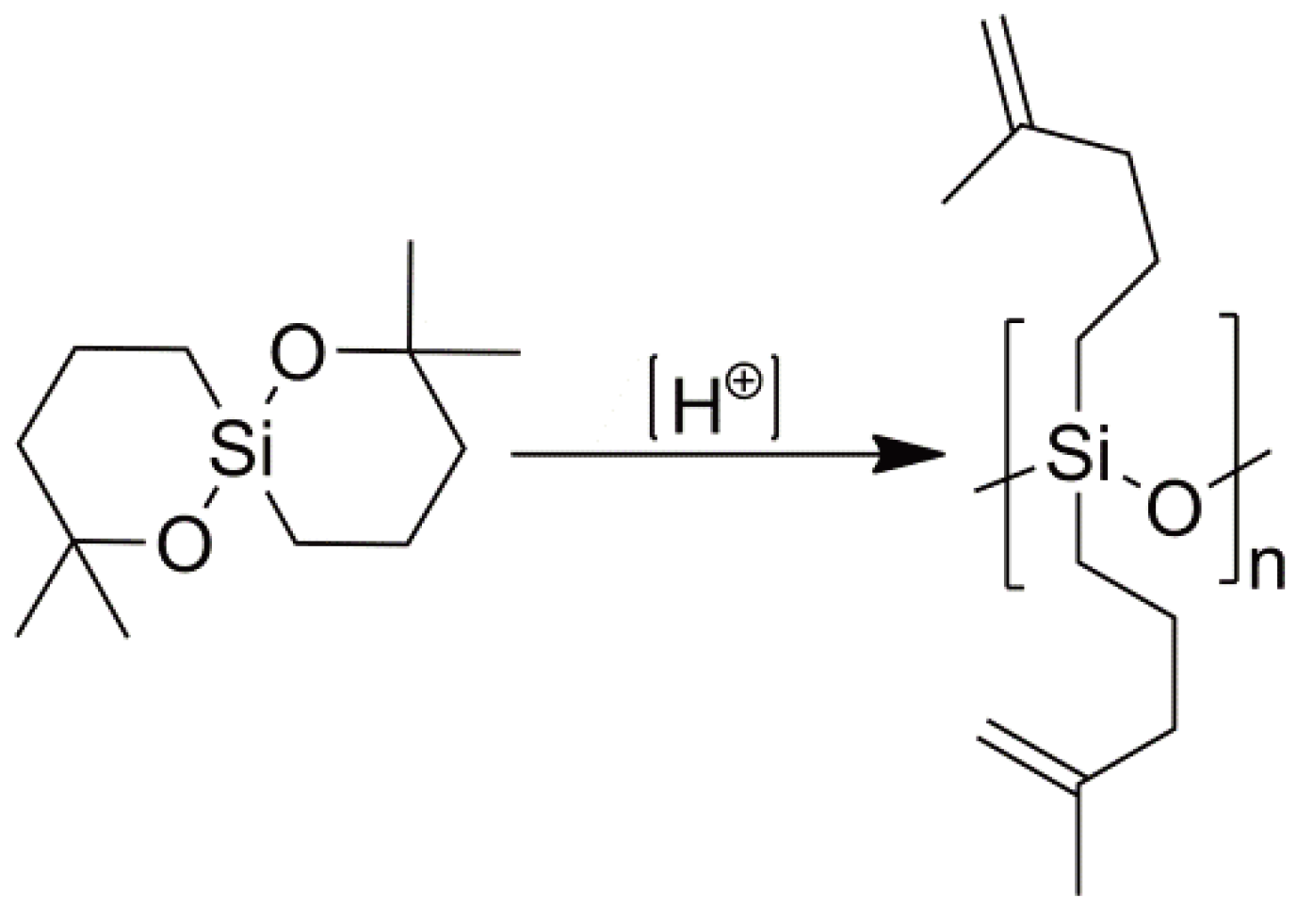
Via the heterolytic splitting of the initiator can be considered as an acid generator or photoacid generator (PAG). Typical examples are triphenylsulfonium triflate and diphenyliodonium triflate. The latter has been studied recently [66] to selectively cleave the carbon-oxygen bonds in a vinyl functionalized oxasilaspirocycle to form both homo- and copolymer polysiloxanes via a double ring-opening CROP (cf. Section 2.3, see Figure 23).
Figure 23.
The formation of a polysiloxane via a double ring-opening cationic polymerization.

The homolytic splitting of triphenylsulphonium cations leads to a phenyl radical, which then undergoes a Fries photo-rearrangement together with the diphenylsulphonium cation radical to yield by-products such as p- und o-(Phenylthio)biphenylenes [67]. That some m-substituted products are observed can be explained by a heterolytic decomposition followed by an electrolytic aromatic substitution of the diphenylsulphonium cation radical [68]. Photo initiators have proved useful as crosslinking agents for hole (defect) conductors [69,70] (e.g., aromatic amine polymers which can give up an electron to yield a radical cation, an electronic defect) with oxetane side groups, which are employed in the manufacture of OLED. The reaction schemes are shown in Figure 24, Figure 25.
Figure 24.
CROP of oxetanes (R = Polymer chain with oxetane side groups (for the source of H+, see e.g., Figure 22)).
Figure 24.
CROP of oxetanes (R = Polymer chain with oxetane side groups (for the source of H+, see e.g., Figure 22)).
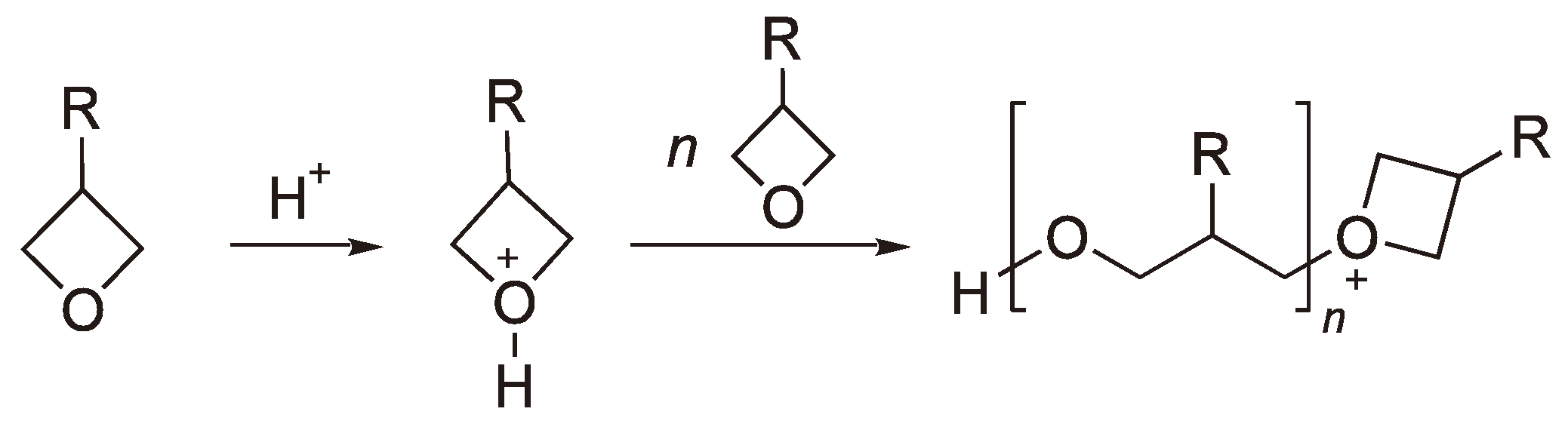
Figure 25.
The cationic crosslinking of a hole defect polyarylamine.
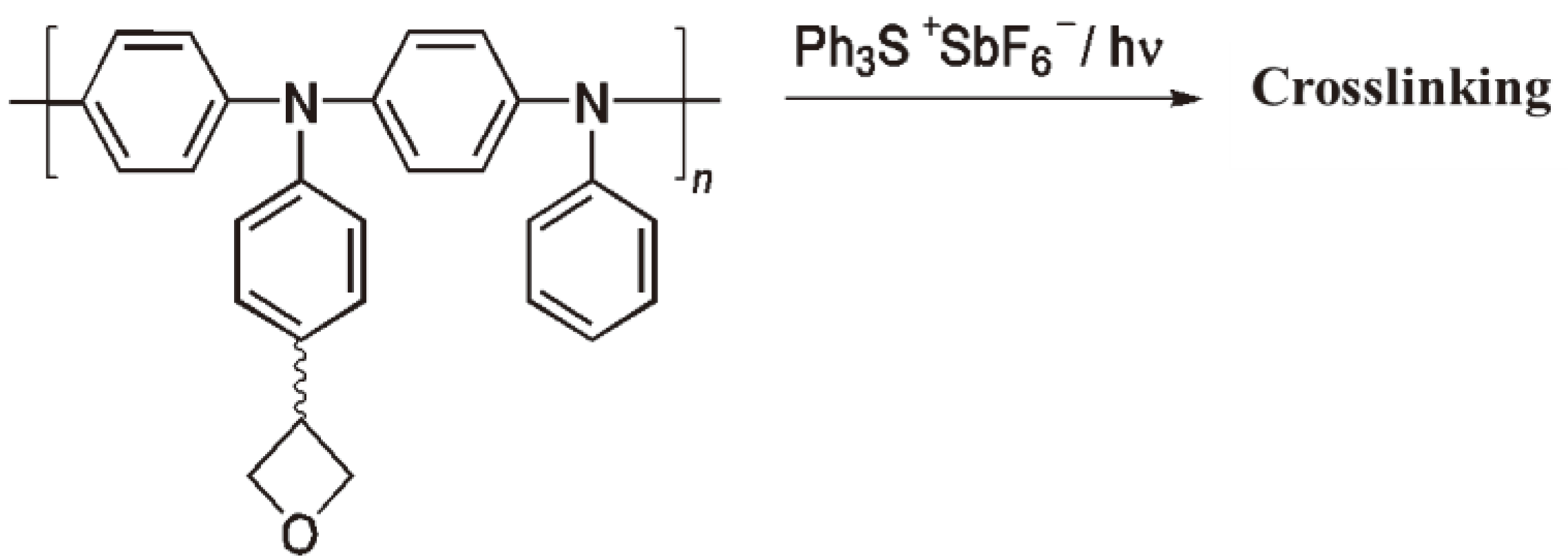
If the hole defect polymer has numerous oxetane side groups, the ROP leads to crosslinking and the polymer becomes insoluble. The soluble polymer can be coated (e.g., by spin-coating), dried and then photochemically crosslinked so that further coating is possible without dissolving the deposited film [71,72].
The solubility of the photo initiator can be improved by increasing the length of the side chain (e.g., Figure 26).
Enzymes have also been shown to initiate the ring-opening polymerization of oxazolines (EROP). By employing different sugar molecules having a methyl oxazoline side group a variety of natural and synthetic polysaccharides could be produced, in particular some of the most complicated synthetic polymeric structures and synthetic chitin. The fundamental mechanism, although the enzyme interacts with the intermediates and increases the reaction rate considerably, remains a CROP [73]. It is worth noting that polyoxazolines have been shown to be biocompatible and potentially useful as carriers of pharmaceutically active agents [74]. An additional group of initiators for the CROP of oxazolines are the activated metallocene complexes described by Kourti et al. [75]. In this study, zirconium and hafnium metallocenes were activated with pentafloroborates or their anilinium salts to initiate a CROP of 2-methyl and 2-phenyl oxazolines. That the polymerization is indeed a CROP was corroborated by appropriate blank experiments. However, since the metallocene does not take part in the propagation step the steric control associated with metallocene polymerizations cannot be expected for these systems and the polymerizations appear to show no special kinetic or mechanistic features. However, the authors do note an increase in the MWD and Mn at close to complete conversion, which may be explained by backbiting reactions (vide infra, Section 3.3).
3.2. Chain Growth
The active species in CROP are oxonium, sulfonium, ammonium or phosphonium ions (see Figure 27).
Figure 27.
The structure of the growing chain ends in CROP.
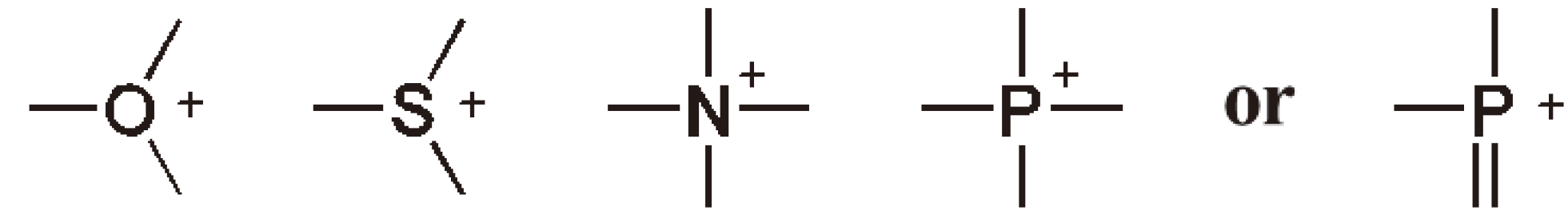
The active species can be identified by NMR (see examples in Table 2) and can be quantified e.g., by reacting with e.g., sodium phenolate [76,77] to yield a UV active end group (see Figure 28).
| Monomer | Structure of the Growing Chain End [51,52,53,56] |
|---|---|
 | |
Figure 28.
Termination of CROP by phenolate to allow quantitative determination of the growing chains.
Figure 28.
Termination of CROP by phenolate to allow quantitative determination of the growing chains.

An alternative method for the quantitative determination of the active chain ends is to react these with, e.g., triphenyl phosphine and to analyze the solution by 31P NMR (see Figure 29) [78]. It is worth noting that these polymers are not suitable for further analysis, as they tend to degrade although the solutions are stable, for days at least, provided they are kept under an inert atmosphere.
Figure 29.
Reaction of the CROP growing chain ends with a phosphine to allow quantitative analysis.
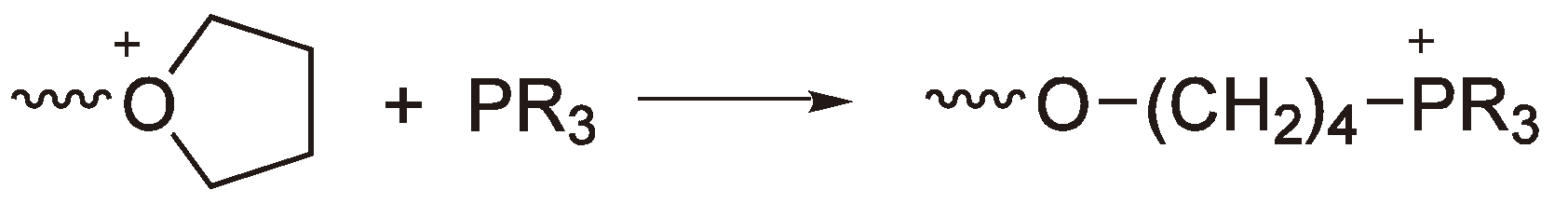
Figure 30.
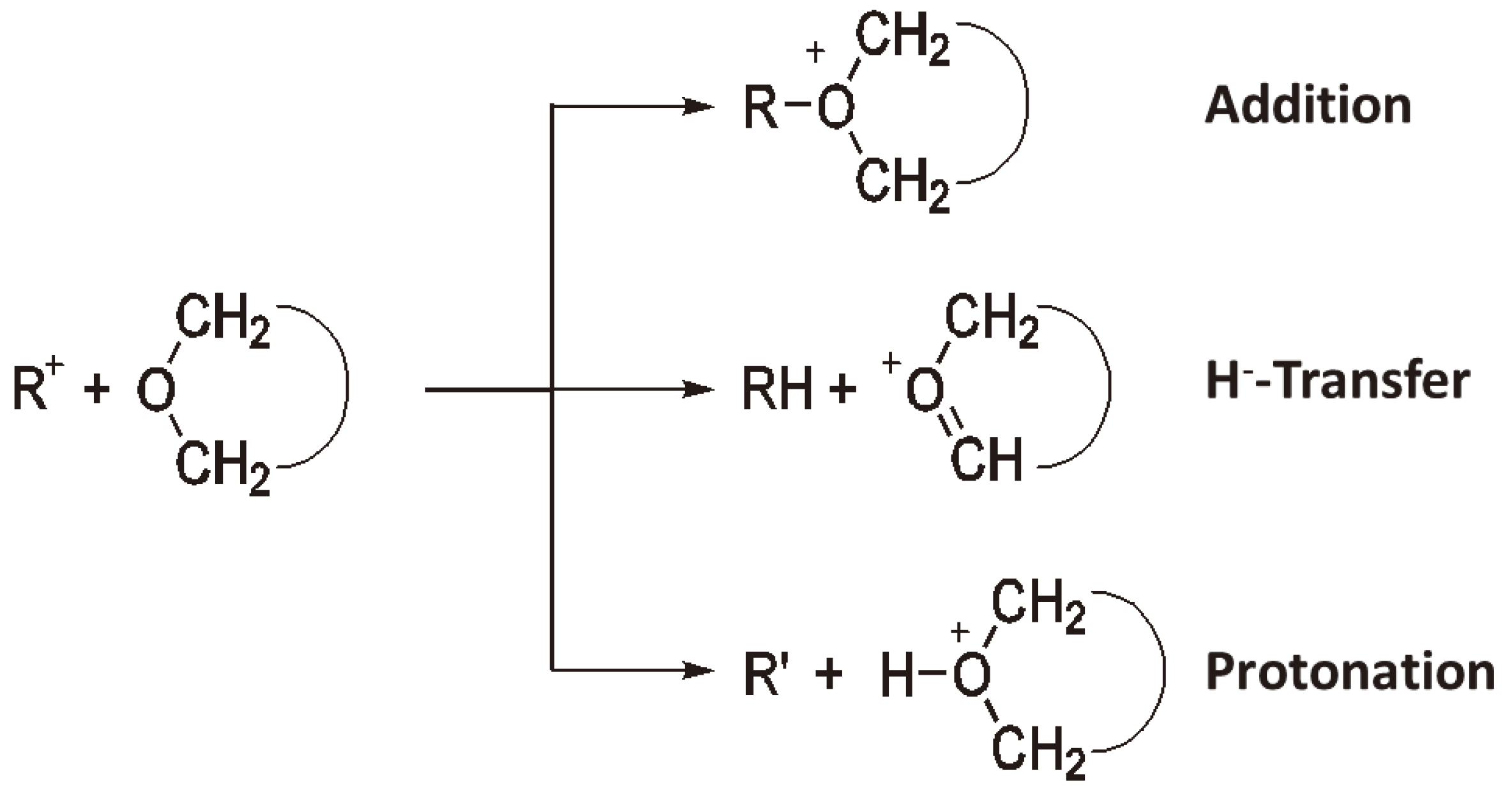
Chain growth via an activated monomer.

With asymmetric monomers chain growth via an activated monomer rather than an activated chain end can yield different polymer microstructures as shown in Figure 31.
Figure 31.
The microstructure of poly-glycidol: (a) Growth via an active chain end and (b) via an activated monomer mechanism.
Figure 31.
The microstructure of poly-glycidol: (a) Growth via an active chain end and (b) via an activated monomer mechanism.
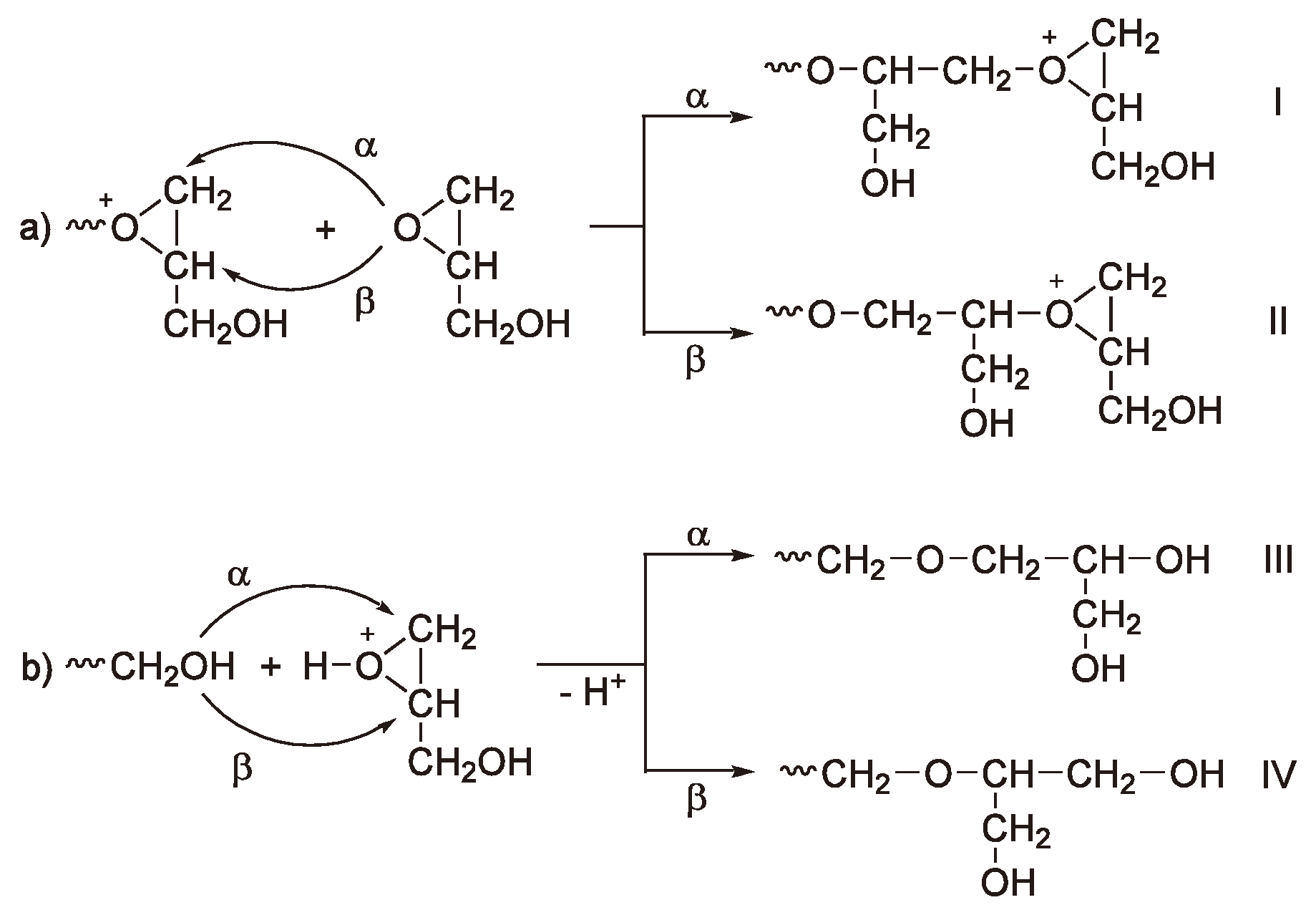
The experimental results suggest that the predominant structure is III [38,79,80] (Figure 31) so that it can be assumed that for this monomer the activated monomer mechanism predominates.
The CROP of hexamethylene cyclotrisiloxane (D3) is an important industrial process generally catalyzed by trifluoromethane sulphonic acid or metal salts of this acid [81]. For the CROP of cyclic siloxanes two propagation reactions have been proposed [82,83] (see Figure 32).
Figure 32.
Two alternative propagation routes for the ROP of cyclic siloxanes: (a) Step-growth and (b) Chain-growth.
Figure 32.
Two alternative propagation routes for the ROP of cyclic siloxanes: (a) Step-growth and (b) Chain-growth.
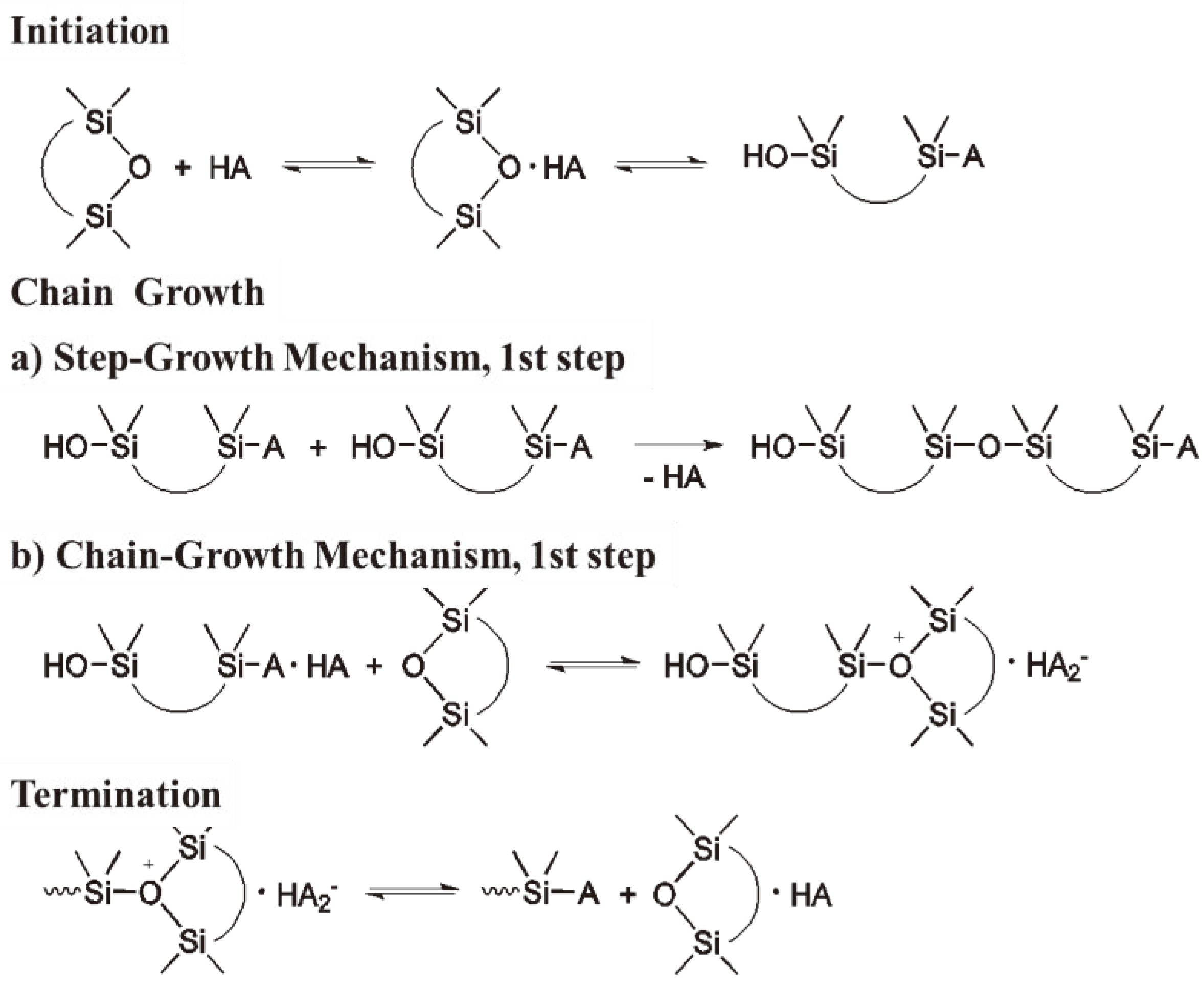
In contrast to the chain growth polymerization of olefins, where the termination leads to an active species being destroyed, the termination of cyclic monomers containing a heteroatom (such as the siloxane shown in Figure 32) leads in the first instance to the chain growth being stopped but the resulting monomer unit can start a new chain as long as monomer is available. Thus, the distinction between the terms transfer and termination becomes gradual. Since the reaction intermediates are extremely reactive, the mechanism of the CROP of cyclic siloxanes is still a matter for discussion [2,38].
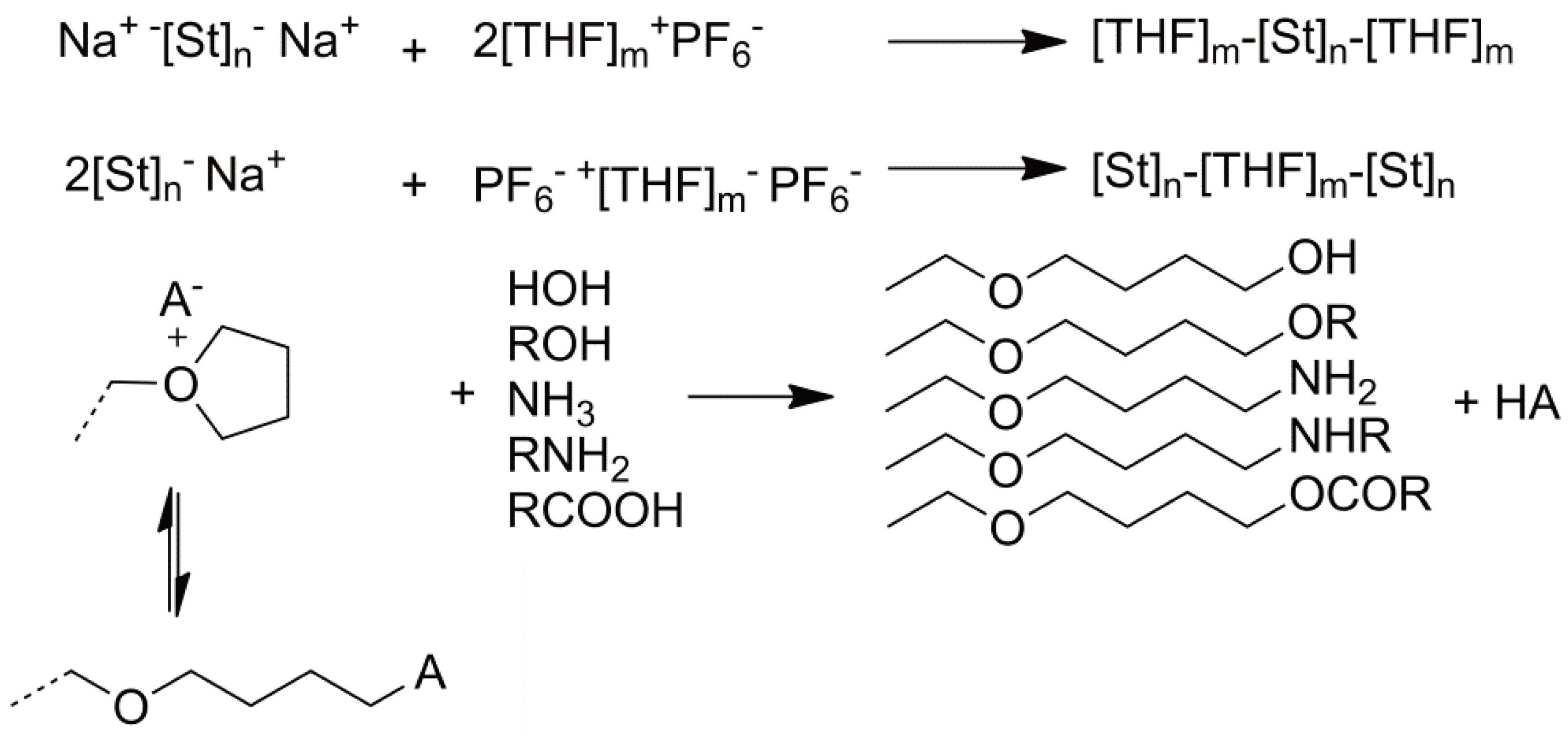
3.3. Termination
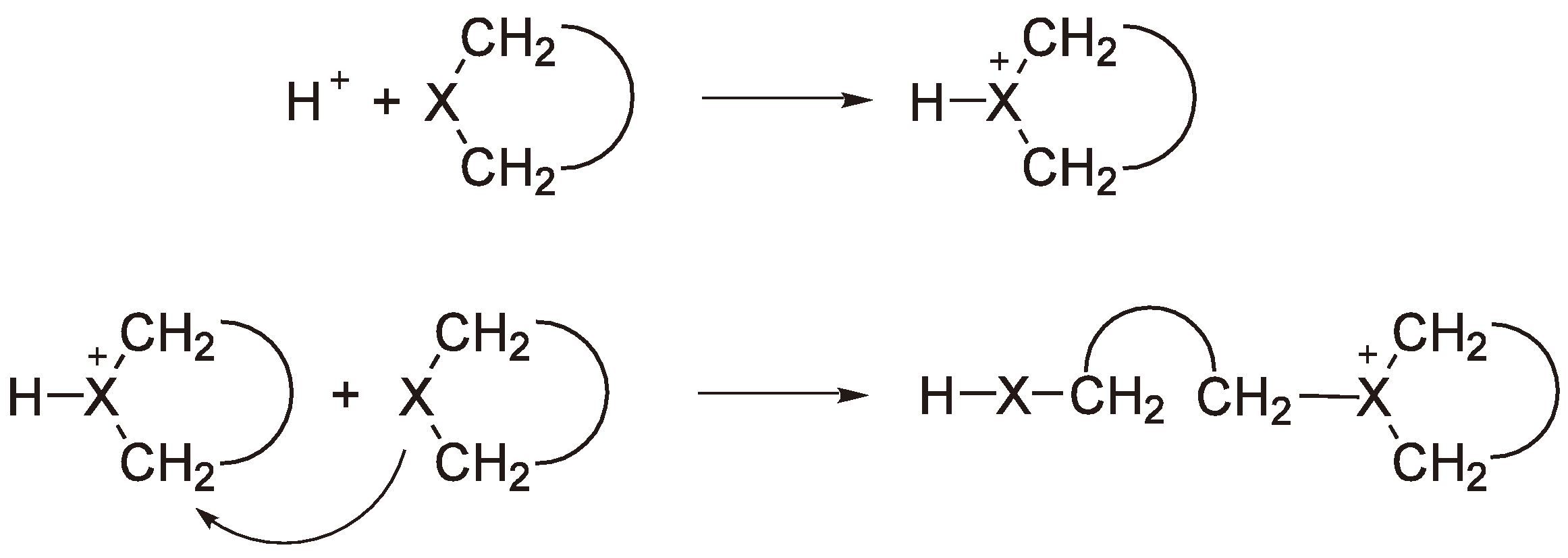
The CROP of some heterocycles, especially cyclic ethers such as THF and oxetane can, at appropriate temperatures and monomer concentrations, be considered as “living” (i.e., without termination). Intentionally adding termination reagents may e.g., be done to enable the end groups to be (quantitatively or qualitatively) analyzed. Examples, already discussed above, are the addition of phenoxy anions [76] or phosphines [78] but polyanions can also be used to yield block copolymers [84]. These reactions are summarized in Figure 33.
Figure 33.
Intentional termination of CROP.

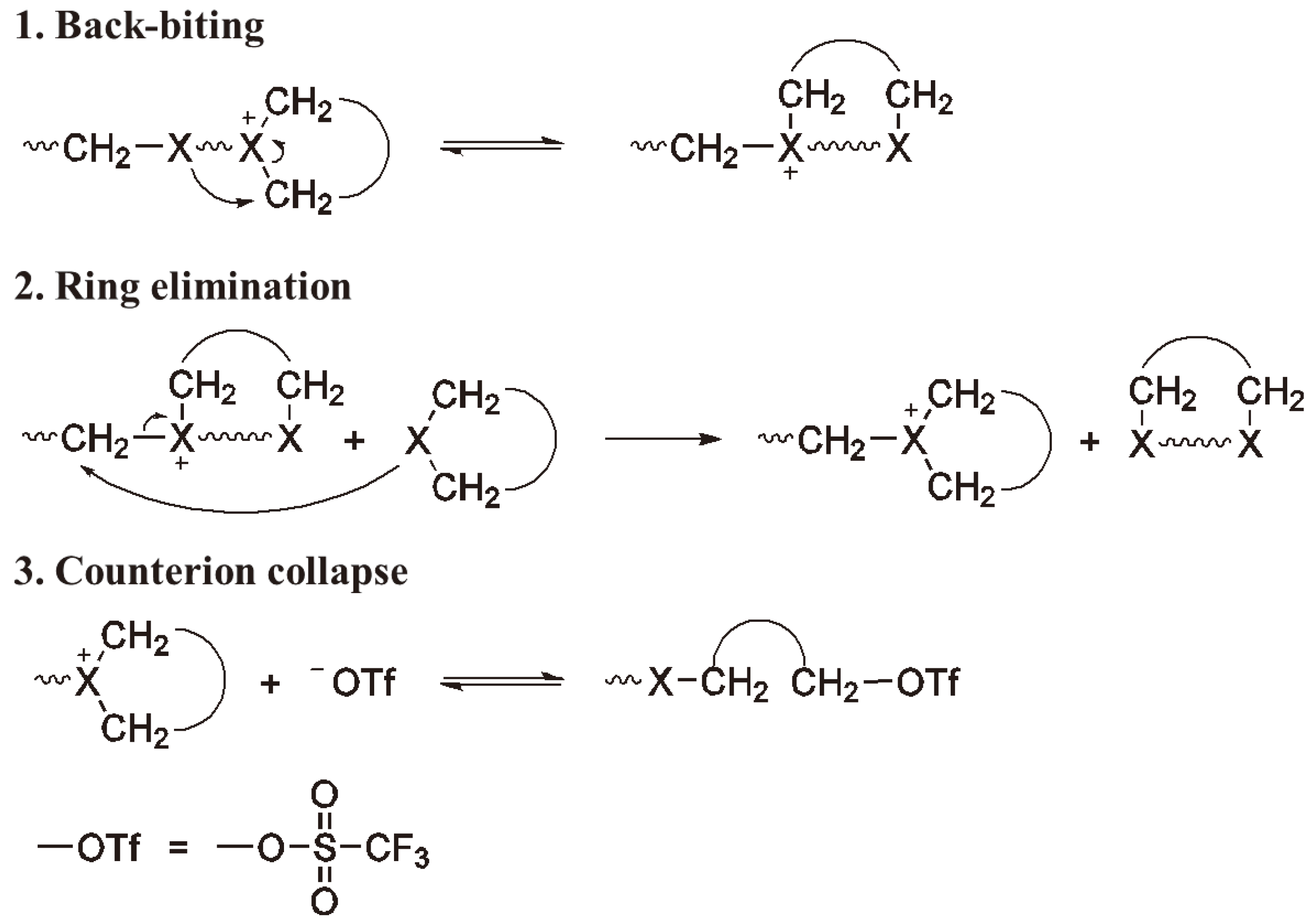
However, backbiting and/or intermolecular alkylation is intrinsic to such systems (see Figure 34).
Figure 34.
Transfer (Termination) via an alkyl transfer mechanism.
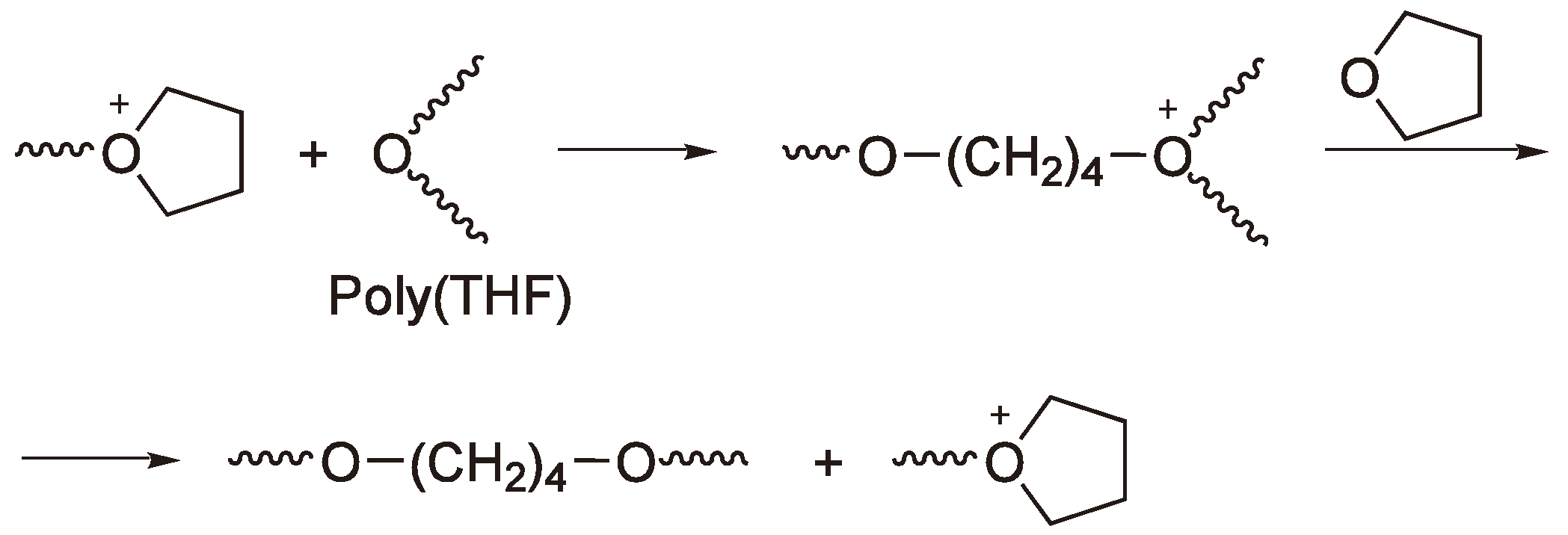
Since these reactions usually only become predominant when the monomer becomes depleted, only then do they represent chain termination rather than transfer; the charged species cease to grow due to lack of monomer and the second stage of the scheme in Figure 34 does not occur.
In some systems, backbiting seems to be negligible and the systems appear to be living [85]. In this work the authors report the CROP of 3-benzyl-1,3-oxazolidine-2-thione (BOT) to yield a polythiourethane (Figure 35) using BF3OEt2 in nitrobenzene solvent and although initiator efficiencies are low (ca. 0.1 based on the amount of BF3OEt2 employed), the Mw/Mn measured was ca. 1.0 for all initiator/monomer ratios studied and both di- and triblock copolymers could be produced with 3-benzyl tetrahydro-1,3-oxazolidine-2-thione (BTOT) as a second monomer. This work is especially worth citing for its use of an unusual end-capping agent (Et2NCS−Et2NH2+), which can then be analyzed by NMR, the use of a variety of analytical techniques to confirm the structures as well as for the excellent yields of polymers with improved thermal stability and perhaps interesting optical properties via a CROP. The kinetic analysis also corroborates the idea (vide supra) that the ROP is driven by the ring strain; thus, the rate of propagation for the 5 membered BOT ring is twice that of the 6 membered ring in BTOT, although the authors ascribe the difference to the different steric hindrance around the thiocarbonyl groups of the two monomers.
Figure 35.
The CROP of 3-benzyl-1,3-oxazolidine-2-thione initiated with BF3OEt2.
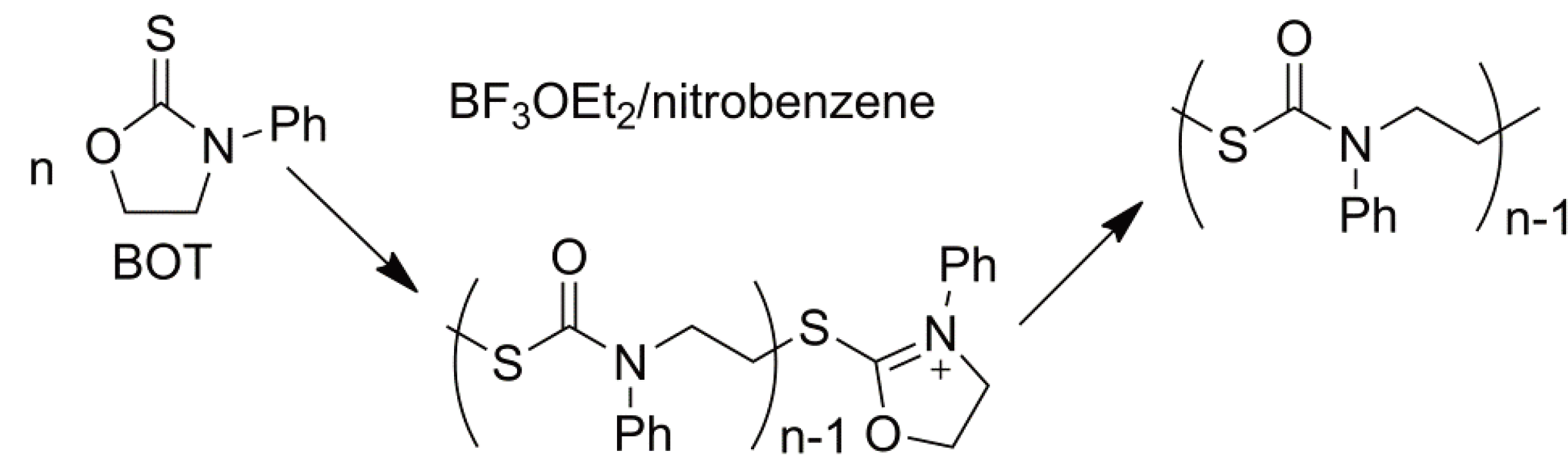
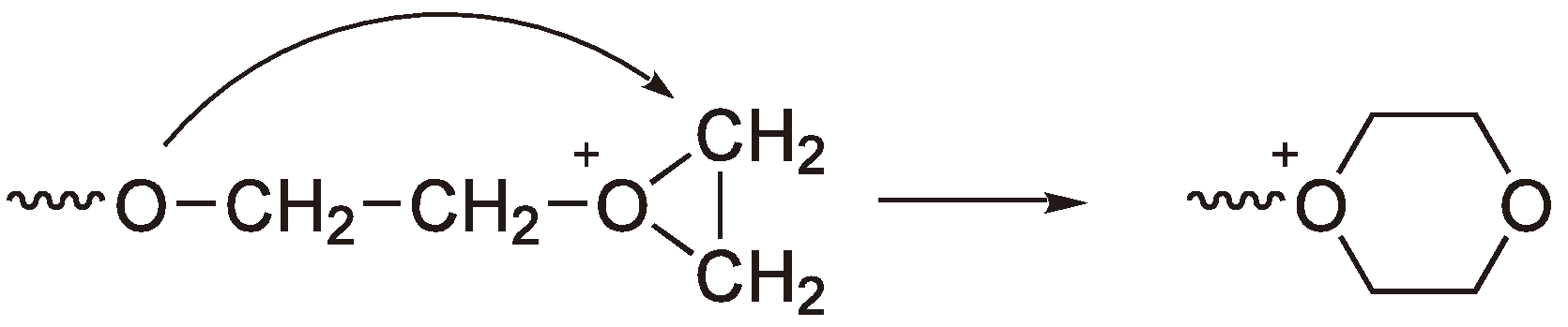
Figure 36 shows this reaction for the ROP of oxirane. The resulting dioxonium ring, in this case, is inert to further chain growth [38,79].
Figure 36.
Termination via back-biting; ring expansion.

The collapse or decomposition of the counter ion also leads to a termination of chain growth, whereby this reaction (Figure 37) may be an equilibrium reaction and thus, the chain ends considered as dormant rather than terminated.
Figure 37.
Termination of chain growth due to collapse of the counter ion.
The living nature of CROP has been utilized to form block copolymers from 2-methyl-2-oxazoline, 2-ethyl-2-oxazoline, 2-n-nonyl-2-oxazoline and, e.g., styrene or acrylates. The CROP of the oxazoline was terminated by a carboxylate functionalized chain transfer agent and the second block formed by a reversible addition-fragmentation chain transfer (RAFT) polymerization [86].
CROP reactions are often rather slow and reaction times even at higher temperatures are often of the order of hours. The reaction rate can be increased considerably (up to 400 fold) by the use of microwave rather than simple heating, although it has been shown that the effect is a simple thermal effect [87]. An interesting comparison of continuous micro reactor configurations and a batch reactor for the microwave assisted CROP of 2-ethyloxazoline is reported by the same group [88] who discuss a major effect on the MWD of the products resulting from the residence time distribution of the individual reactors whereby differences in conversion are conveniently assigned to probable inaccuracies in the temperature measurements. Poly (2-oxazolines) are becoming increasingly interesting for biomedical applications [89] and as micellar catalysts [90,91,92].
4. Anionic Ring-Opening Polymerization
Anionic ROP (AROP) can be described as the nucleophilic attack of the growing chain end on a heterocyclic monomer molecule (see Figure 38). The AROP of ε-caprolactam to produce Nylon 6 is an important industrial process.
Figure 38.
The AROP as a nucleophilic addition reaction [93].
Figure 38.
The AROP as a nucleophilic addition reaction [93].
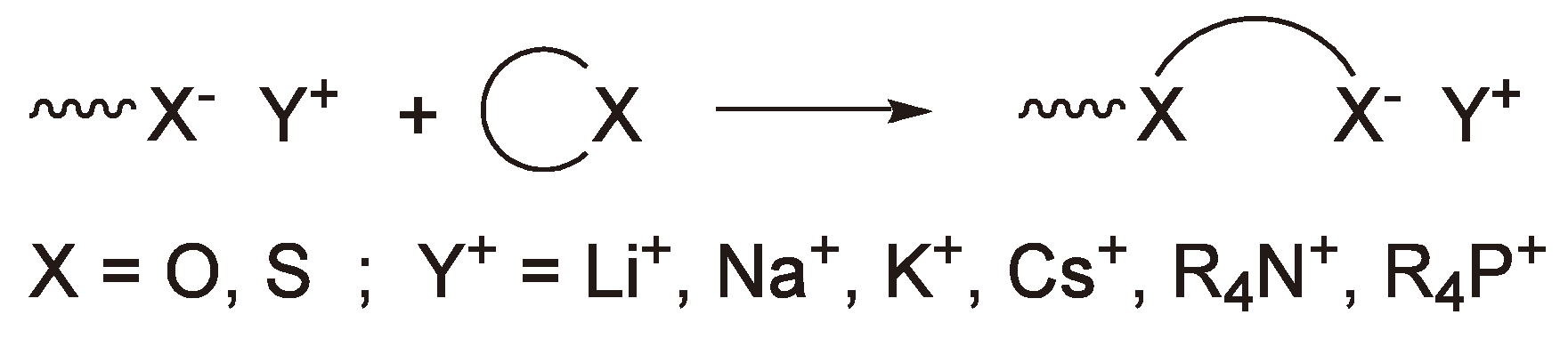
Some of the heterocycles that can be polymerized anionically are listed in Table 3 with their corresponding active chain ends.
| Monomer | Structure of the Growing Chain | Reference | ||
|---|---|---|---|---|
 |  | [94] | ||
 |  | [95] | ||
 |  | [96,99] | ||
 |  | [97] | ||
 |  | [98] | ||
4.1. Initiation
Some of the more important initiators for AROP are listed in Table 4.
| Description | Structure | Monomer | Reference |
|---|---|---|---|
| Radical anion |  | Ethylene oxide | [100] |
| Propylene sulfide | [101] | ||
| Carbanion | C2H5–Li+ | Thietane | [99] |
| n-C4H9–Li+ | Propylene sulfide Hexamethylcyclo-trisiloxane | [102] [102 ] | |
 | Ethylene oxide | [103] | |
| Alcoholate | CH3O−K+ | Styrene oxide | [104] |
| β-Propiolactone | |||
| Silanolate | (CH3)3SiO−K+ | β-Propiolactone | [105] |
| ε-Caprolactone | [106] | ||
| Carboxylate | CH3COO−K+ | β-Propiolactone | [106] |
| Ethylene oxide | [106] | ||
| Propylene oxide | [107] | ||
| Thiolate | C2H5S−K+ | Propylene sulfide | [107] |
| Lactam anion |  | ε-Caprolactam | [108] |
| Amine | (C2H5)3N | Propylene sulfide | [95] |
| Al-trialkoxide | Al(O-t-C3H7)3 | ε-Caprolactone | [109,110] |
| Al-Dialkyl monoalkoxide | (C2H5)2AlOCH3 | Lactide ε-Caprolactone | [111,112] |
4.2. Propagation
For unsymmetrically substituted rings there are formally two ways the monomer ring can be opened as shown in Figure 39.
Figure 39.
Nucleophilic ring-opening of asymmetrical heterocycles.
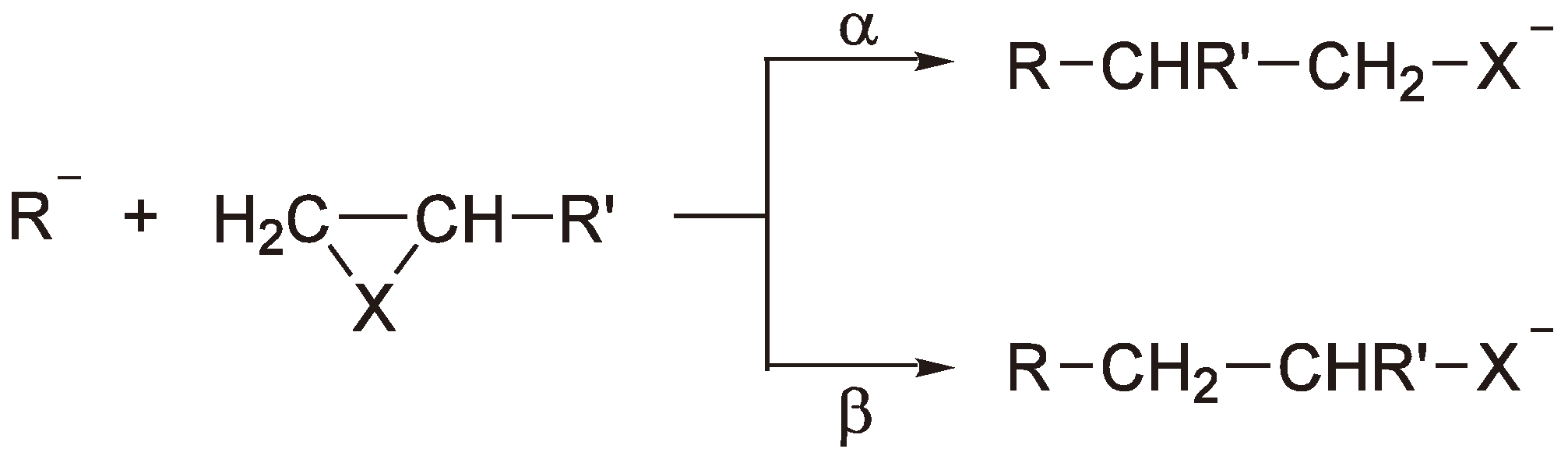
Although not trivial, with careful microstructure and end group analysis the preferred reaction route can be identified [93]. Additionally, simple consideration of the stability of the reaction intermediates can prove helpful. Thus, the carboxylate ion (route β) is preferred, even for lactones (see Figure 40) [113], and the nucleophilic attack takes place at the least substituted carbon atom.
Figure 40.
The AROP of lactones.
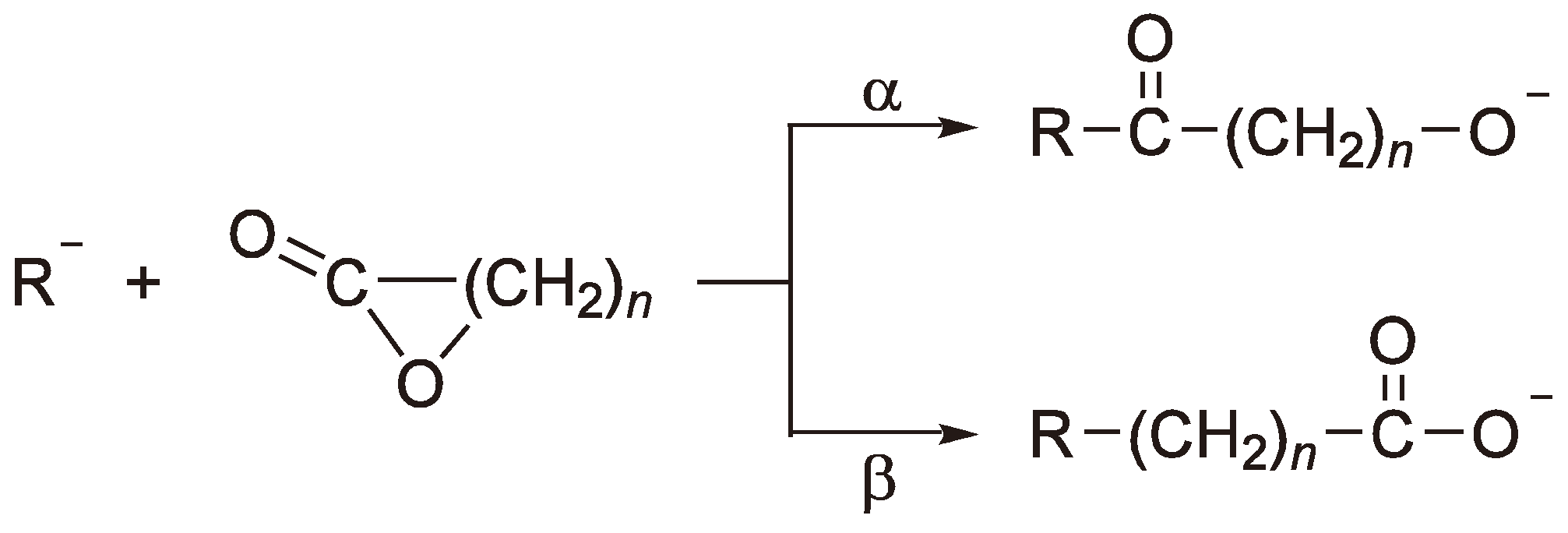
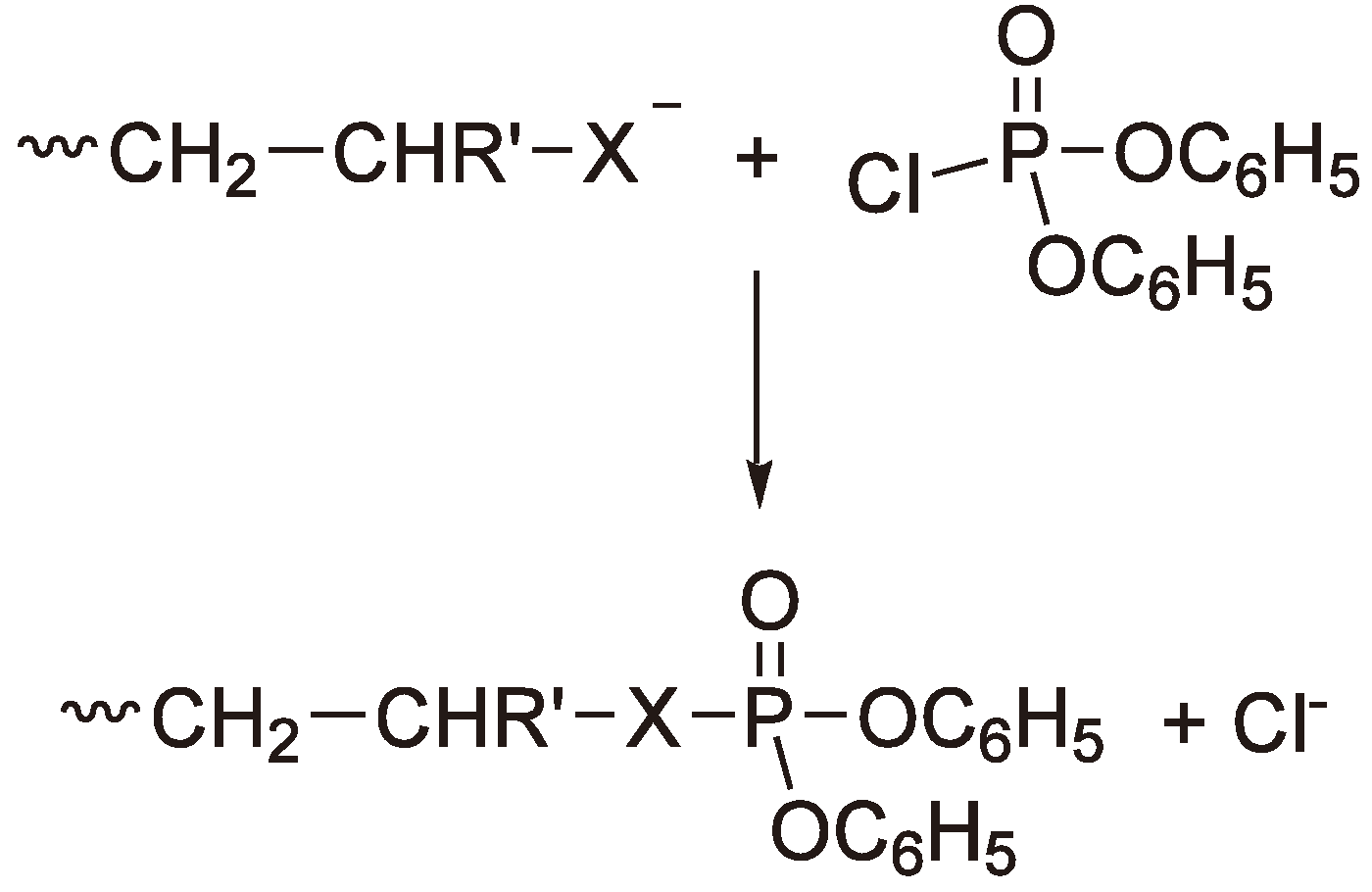
An additional possibility for determining whether route α or β (Figure 40) is preferred is to short stop the polymerization e.g., with diphenyl chlorophosphate (see Figure 41) and then to analyze the solution with 31P NMR [114,115].
Figure 41.
Polymerization short stop with diphenyl chlorophosphate (Alcoholate (X = O) or thiolate (X = S)).
Figure 41.
Polymerization short stop with diphenyl chlorophosphate (Alcoholate (X = O) or thiolate (X = S)).
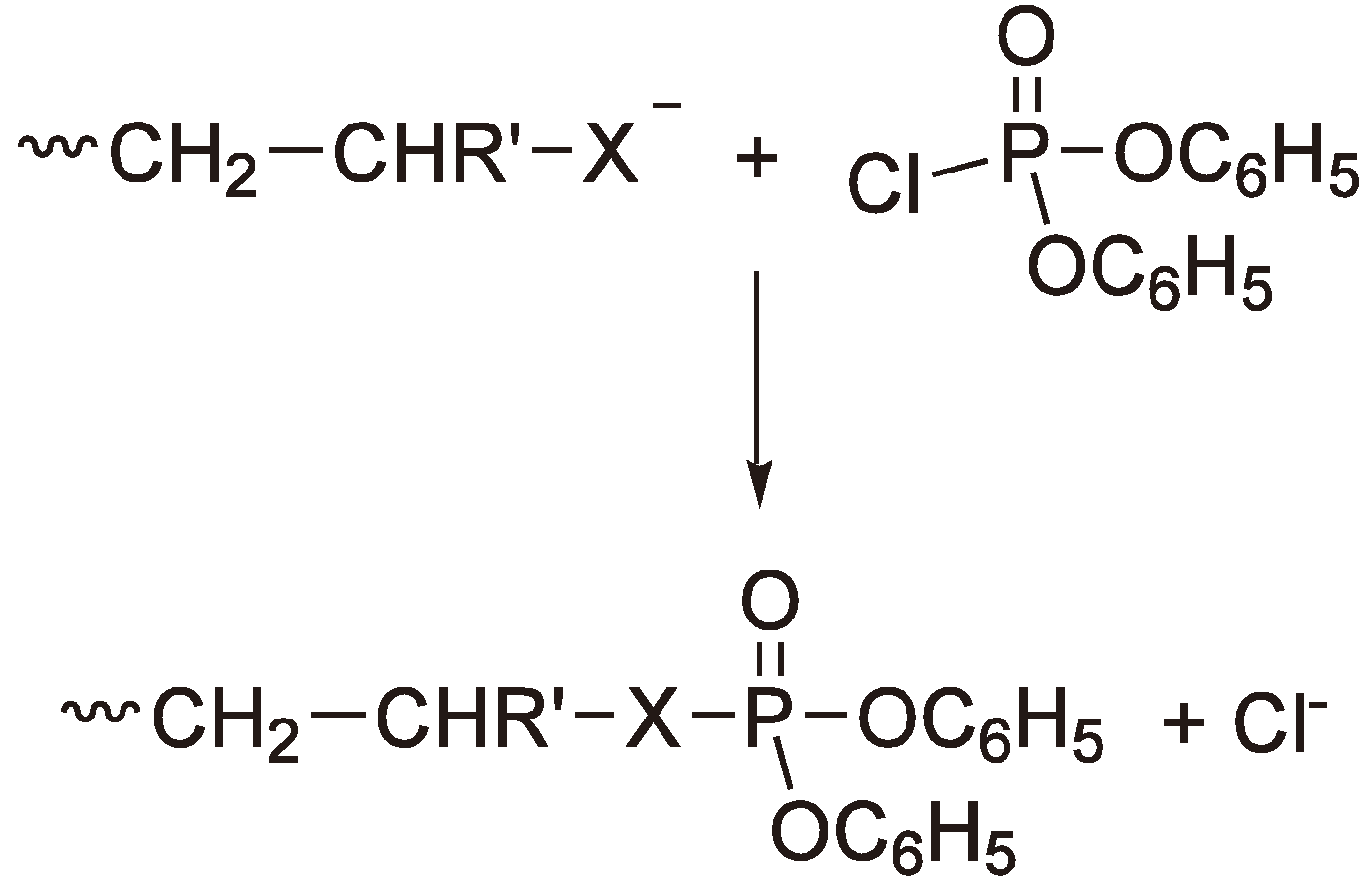
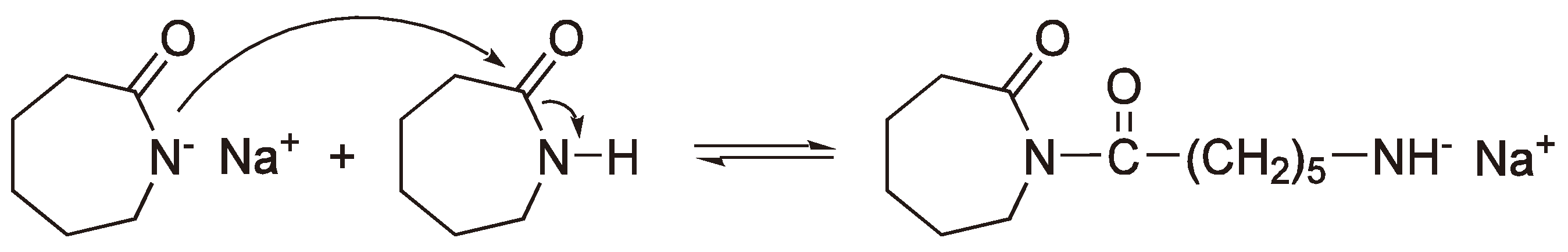
The AROP of lactams can be initiated with strong bases such as the alkali metals, metal hydrides or metal amides which give, as a first step, lactam anions (see Figure 42) [116].
Figure 42.
The formation of a lactam anion from the reaction of sodium with ε-caprolactam.

The lactam ring is then opened in the second step (see Figure 43).
Figure 43.
The opening of a lactam ring by a lactam anion.
The next step is the proton abstraction by the very reactive primary amine anion from another monomer to regenerate a lactam anion (Activated monomer mechanism, see Figure 44).
Figure 44.
Regeneration of the lactam anion.
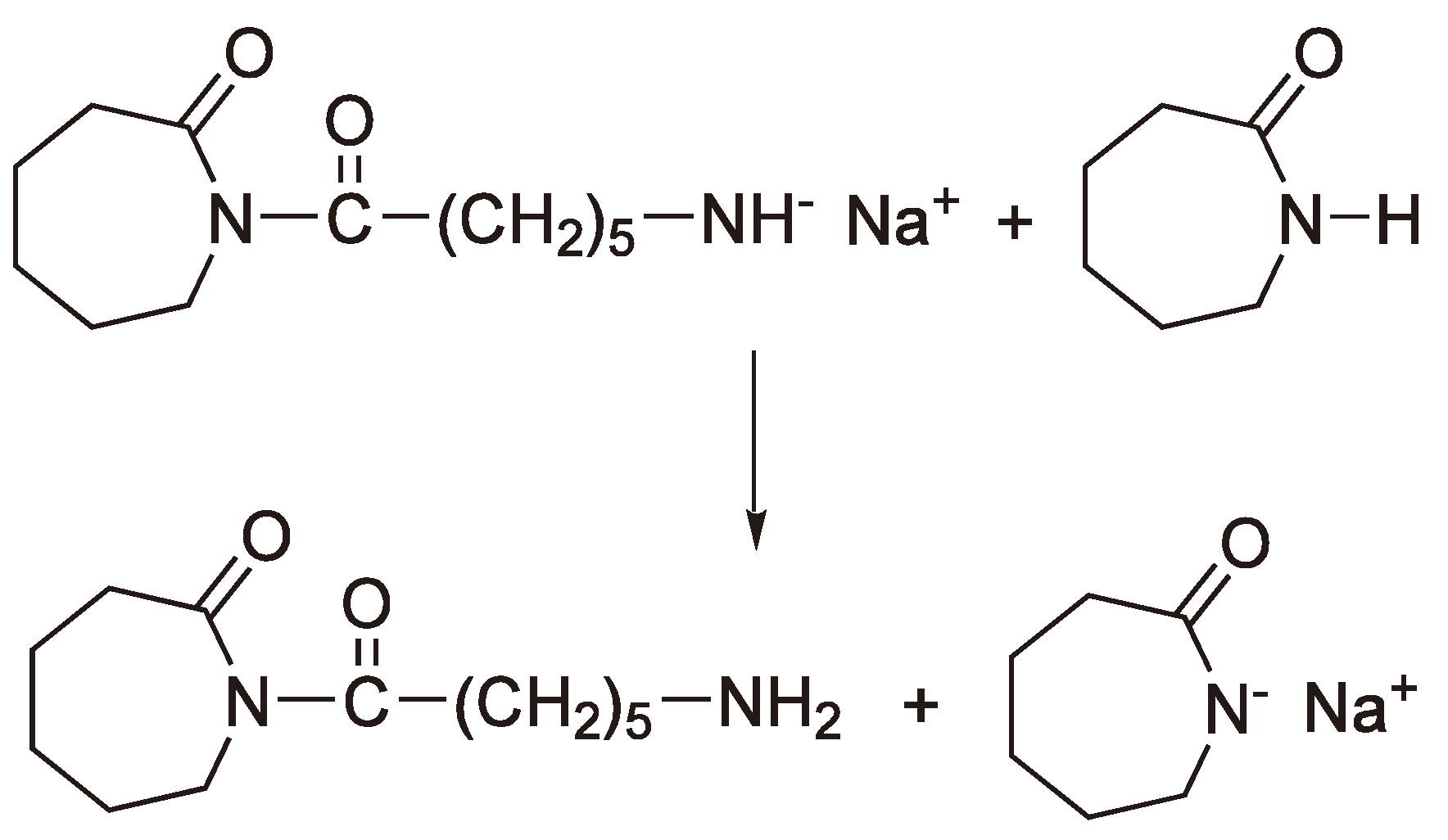
Whereby the terminated dimer can also react with a lactam anion and thus grow.
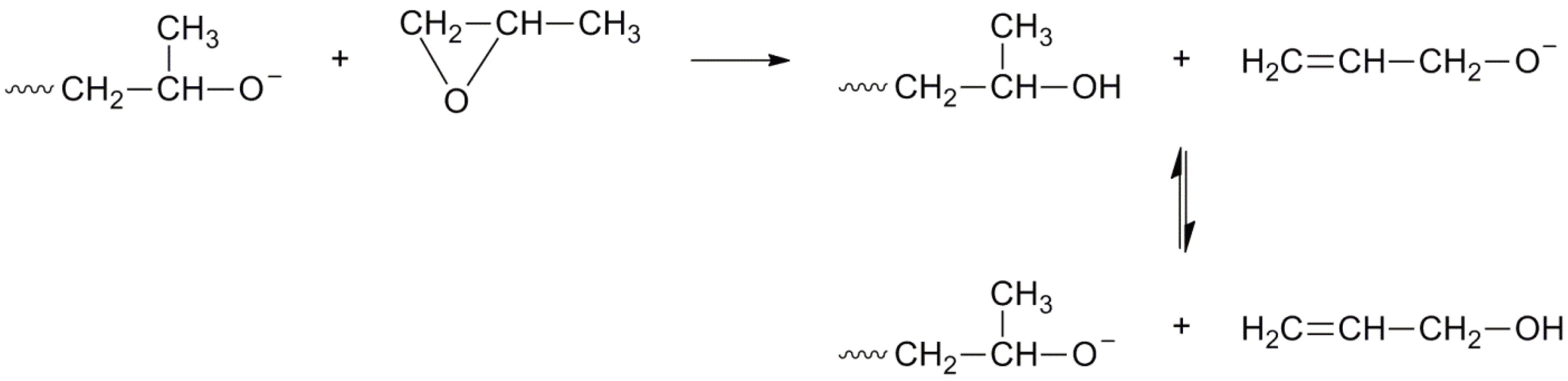
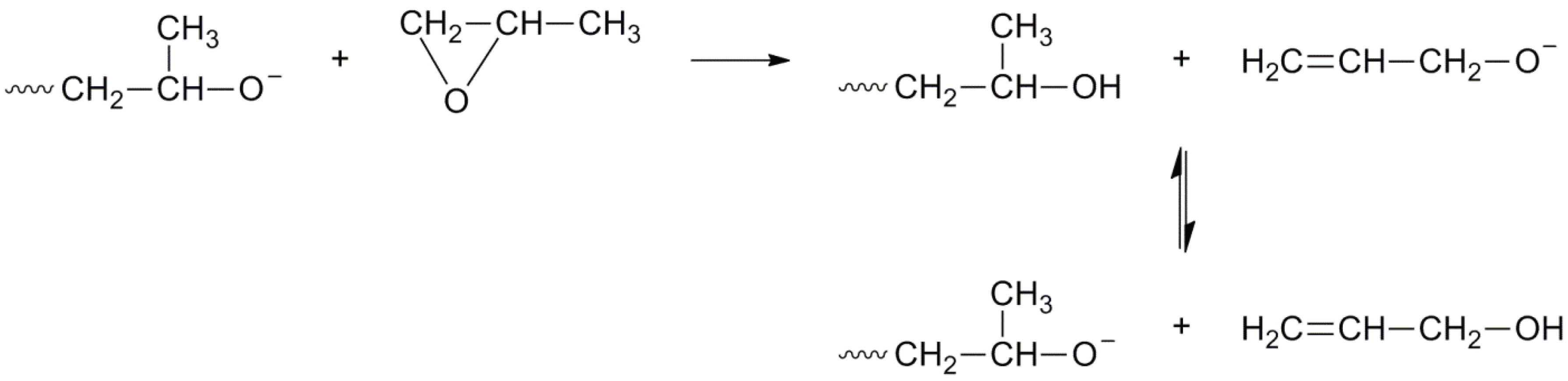
An AROP of methyl oxirane (1,2-propylene oxide) is an industrial process for the preparation of oligomers in combination with oxiranes (ethylene oxide). The extensive chain transfer to monomer, typical of substituted oxiranes prohibits the formation of high polymers [117]; the reaction is shown in Figure 45. It should be noted that the allyl alkoxide ion can also generate propagating chains. This leads to less than ideal alcohol group formation where simple end capping reactions are employed (or diol formation with bifunctional initiators).
Figure 45.
AROP of methyl oxiranes with chain transfer to monomer.
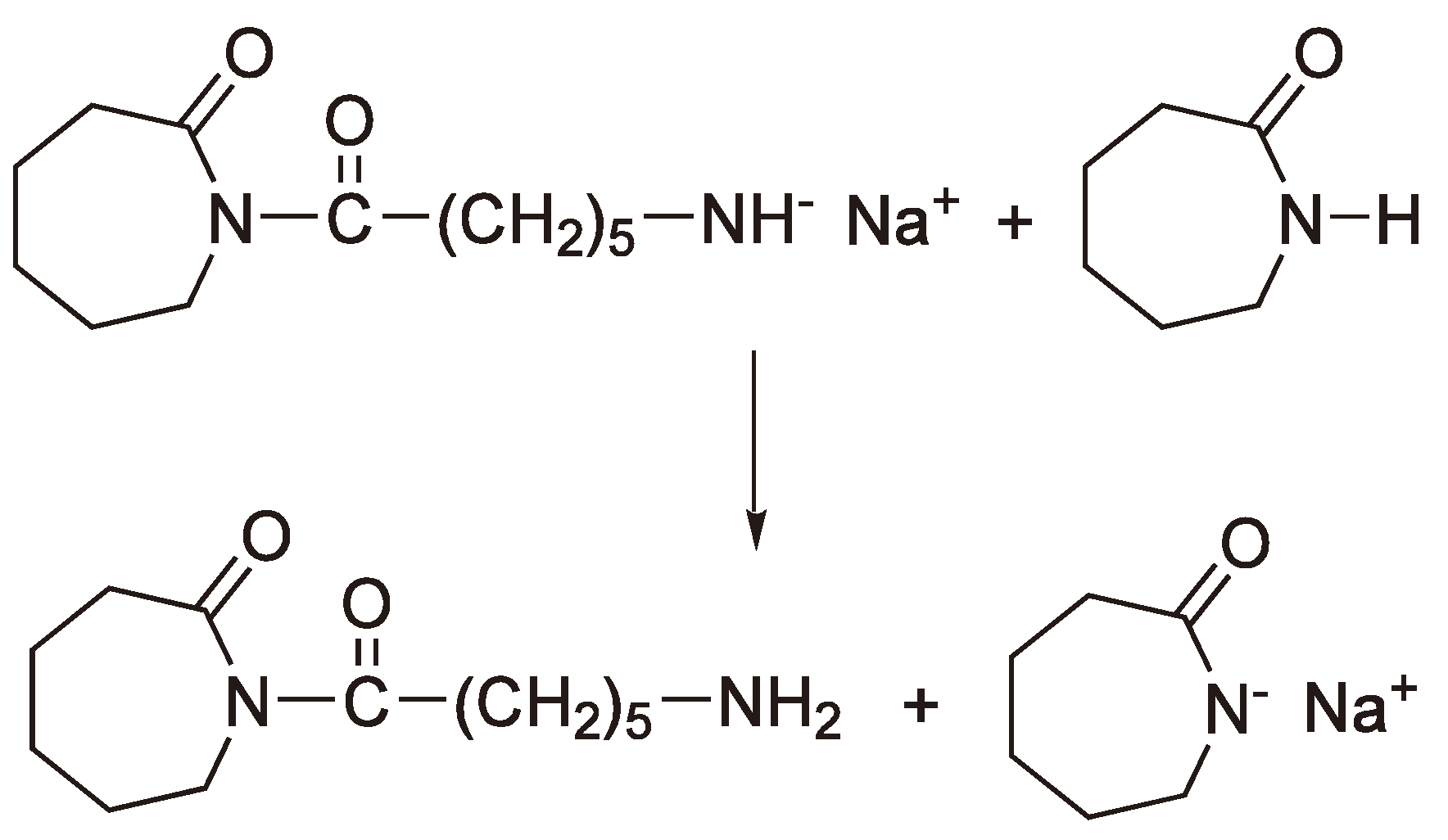
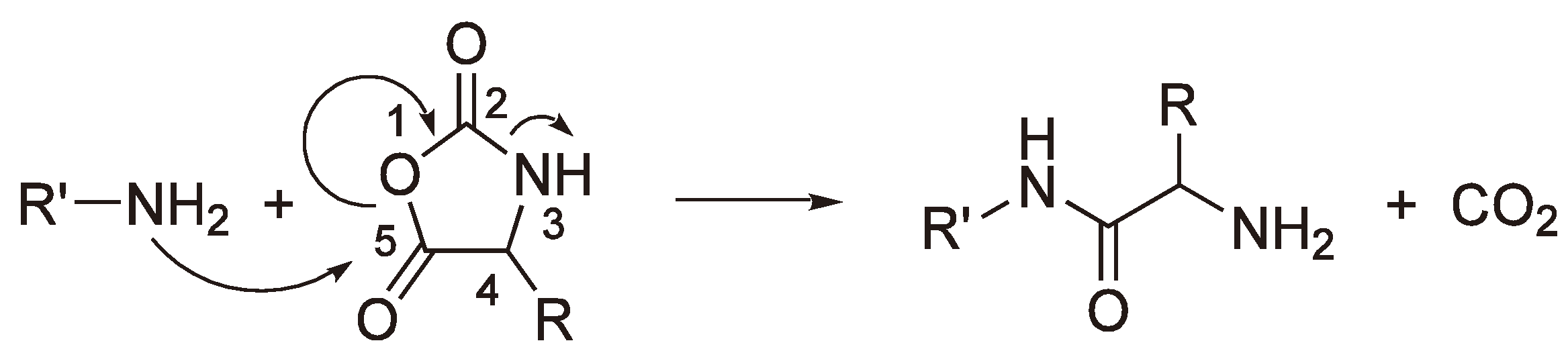
A particularly interesting case of AROP is the condensation ROP of a Leuchs’ anhydride (N-Carboxy-α-amino acid anhydride), which eliminates a molecule of CO2 for each addition step (see Figure 46).
Figure 46.
Formation of “Nylon-2” from a Leuchs’ anhydride.

It could be shown by using 14C labeled monomer that the CO2 contains exclusively the C-2 (Figure 47) carbon atom. Thus, the free electron pair of the primary amine nitrogen attacks the monomer at C-5, the carbon with the lowest electron density and the CO2 elimination results from sequentially breaking the C-5–O-1 and the C-2–N-3 bonds, whereby the amine nitrogen acts as the base.
Figure 47.
Mechanism of CO2 elimination as a result of the attack of a primary amine on a Leuchs anhydride.
Figure 47.
Mechanism of CO2 elimination as a result of the attack of a primary amine on a Leuchs anhydride.
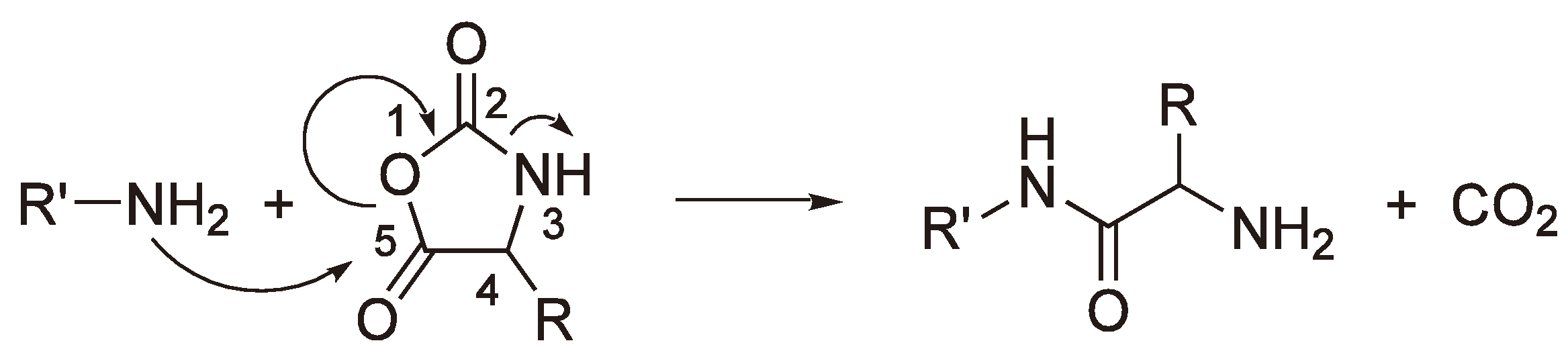
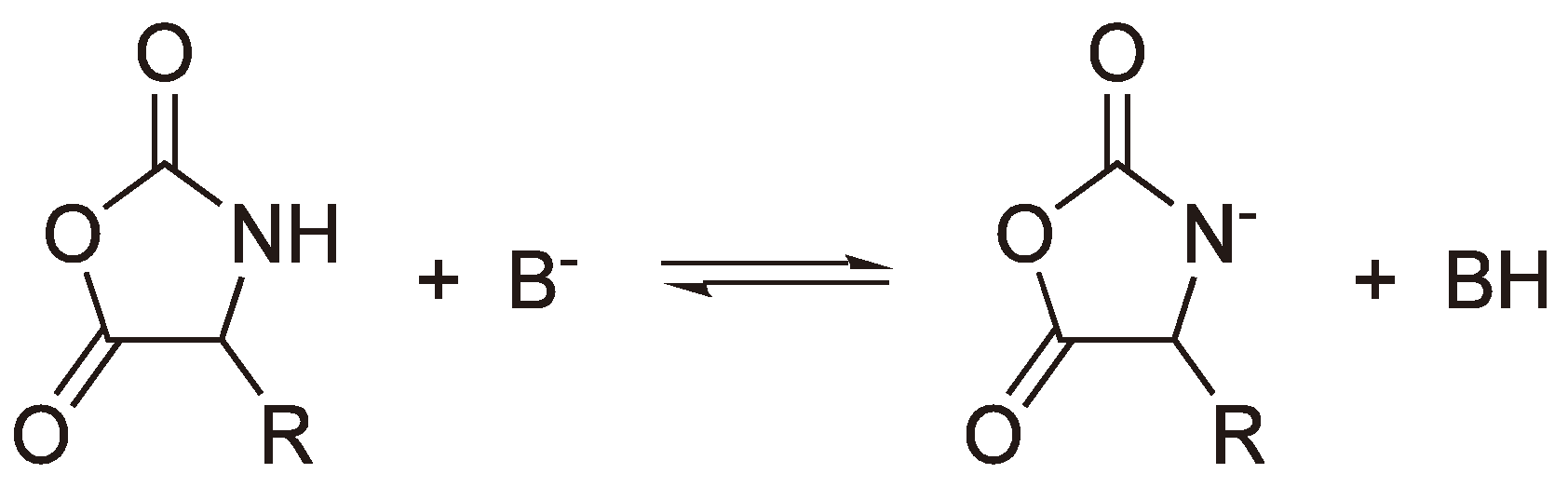
An alternative to a covalent initiator is the use of a base B− (e.g., −OCH3, H−, −OOCCH3 [118]) which generates an anion from oxazolidin-2,5-diones as shown in Figure 48. This anion can then initiate chain growth via a nucleophilic attack on another monomer.
Figure 48.
Formation of an anion from a Leuchs’ anhydride using a base.
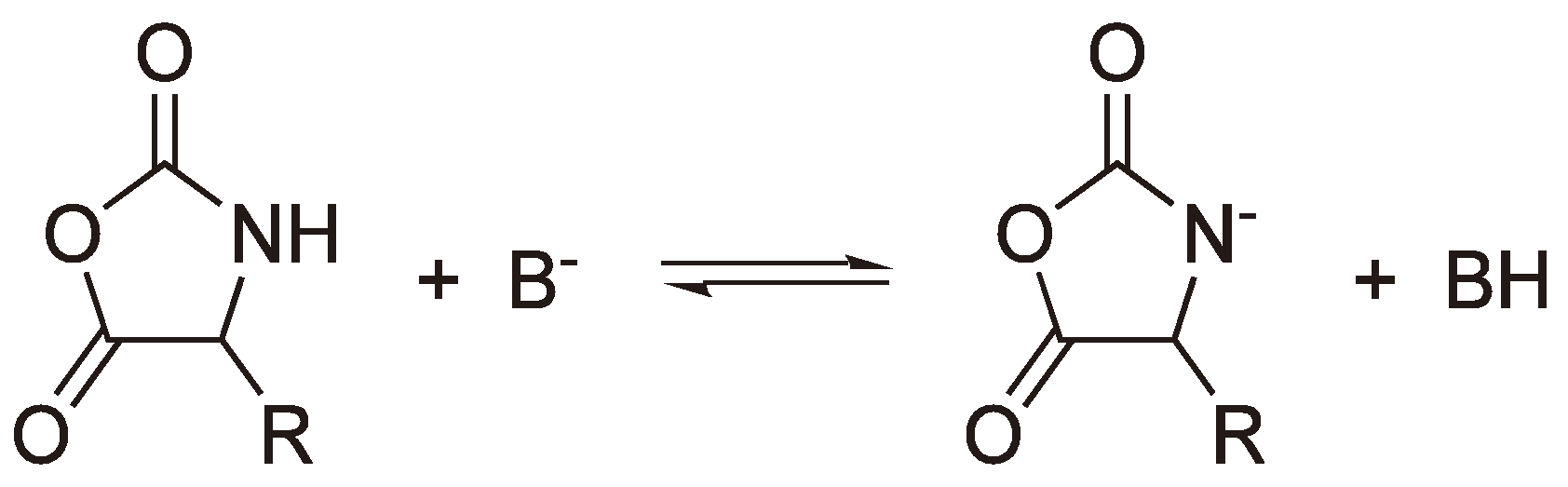
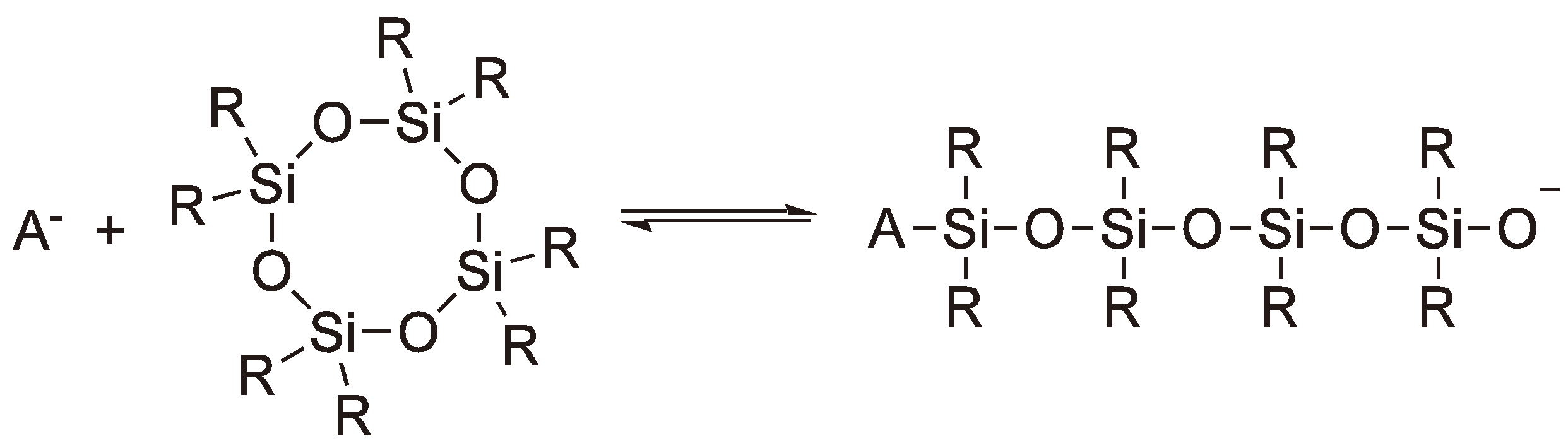
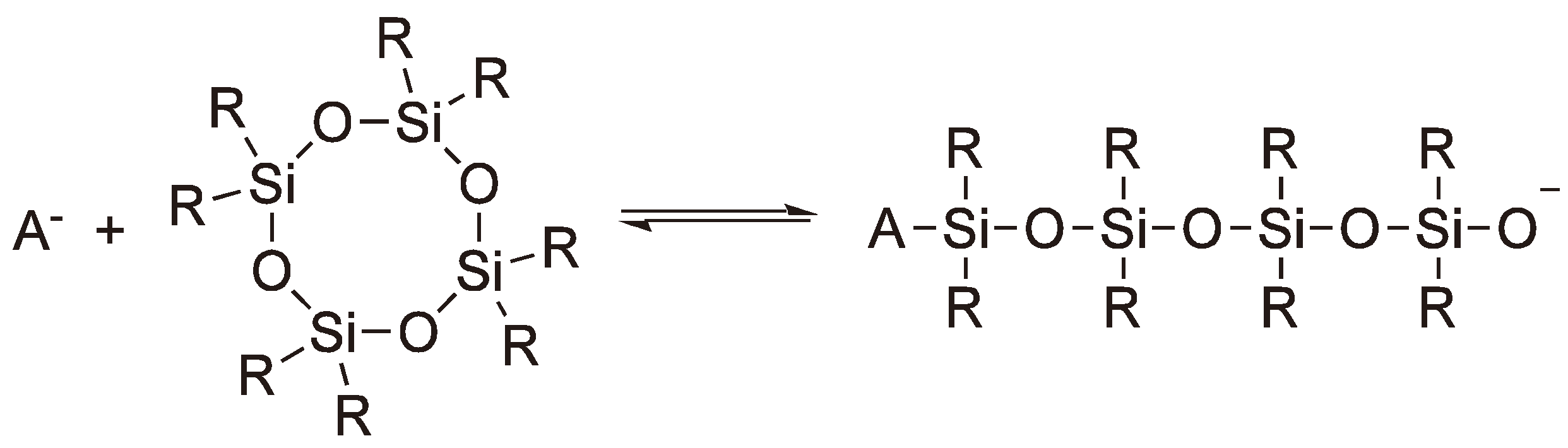
Cyclic siloxanes can also undergo ROP via anionic intermediates employing alkali metal oxides or hydroxides as initiators [119]. On an industrial scale, octamethylenecyclotetrasiloxane (D4) is employed as a starting material. For the propagation step the ΔHp ≈ 0 kJ mol−1, but due to the increased rotational freedom of the side chains the ΔSp = +6.7 J mol−1 polymerization does occur. However, depolymerization competes with polymerization and rings and chains are in equilibrium (see Figure 49). The equilibrium is shifted to the ring form with increasing size of the substituents. If other siloxanes with different (e.g., phenyl or methyl) substituents are mixed with D4 statistical copolymers are obtained [119]. An alternative, employing D3 (Hexamethylenecyclotrisiloxane) as the monomer, has the disadvantage that the synthesis is more elaborate [120,121].
Figure 49.
Equilibrium between rings and chains during the AROP of cyclic siloxanes (A− = ~Si(R)2O−).
Figure 49.
Equilibrium between rings and chains during the AROP of cyclic siloxanes (A− = ~Si(R)2O−).
4.3. Transfer and Termination
As discussed above (e.g., Figure 34) chain termination reactions are more accurately described as transfer reactions as long as additional monomer is available. The active centers of AROP, e.g., alcoholates or carboxylates are not only nucleophilic but act as bases and can abstract protons from the monomer to initiate new chains. Thus, the polymerization of propylene oxide initiated with alkali metal salts does not give polymers with high molar masses (Vide supra). One possibility to increase the molar mass of the products is to add crown ethers as complexing agents for the counter ions to the polymerization system [122]. The free-ion/ion-pair equilibrium is shifted to the free-ion side; the free-ions preferentially add to monomer rather than abstract a proton.
5. Ring-Opening Metathesis Polymerization (ROMP)
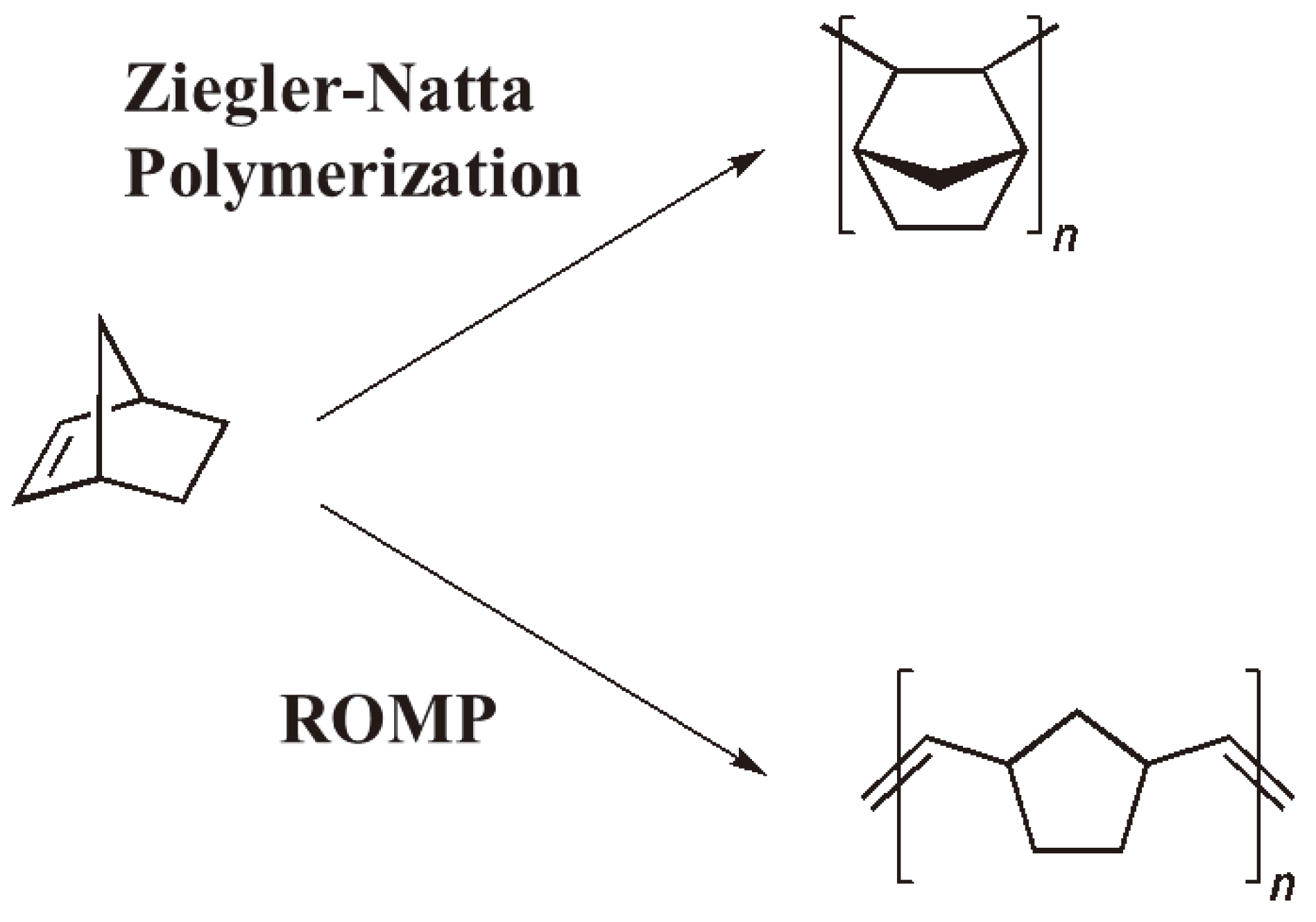
Olefin metathesis is a reaction that can be seen to have influenced both organic and polymer chemistry more than any other during the last few decades. Historically, the ring-opening metathesis reaction has its origin in polymer chemistry: In 1955 Anderson and Merckling (DuPont), working with norbornene and TiCl4/EtMgBr catalysts obtained a polymer containing a highly unsaturated backbone [123] which some years later was identified as the product of a ring-opening polymerization [124] (see Figure 50).
Figure 50.
Ziegler-Natta polymerization and ring-opening metathesis polymerization (ROMP) of norbornene.
Figure 50.
Ziegler-Natta polymerization and ring-opening metathesis polymerization (ROMP) of norbornene.
The development of ROMP is also documented in the academic literature [125,126,127,128]. The driving force for ROMP is the release of the ring strain enthalpy and, since the number of molecules is considerably reduced by polymerization, a positive (−TΔS) must be compensated by the enthalpy term in order for the reaction to proceed according to:
ΔG = ΔH – TΔS
Table 5 lists ΔH-, ΔS- and ΔG-values for a selection of cyclic olefins. The positive ΔG0 confirms that cyclohexene cannot be polymerized by a ROMP at room temperature.
The remaining double bonds in the backbone of the polymer allow backbiting reactions (see Figure 51) to form macrocycles, which often observed as products from the ROMP of cyclic olefins [131].
Figure 51.
The formation of macrocycles via back biting during the ROMP of cyclic olefins.
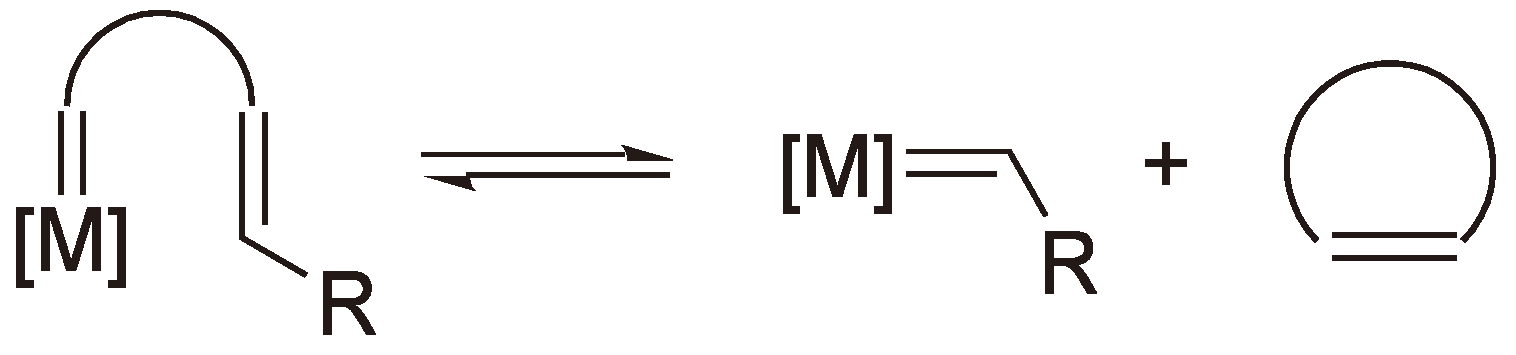
| Monomer | Configuration of the double bond in the polymer | ΔH0/(kJ/mol) | ΔS0/(J/(mol K)) | ΔG0/(kJ/mol) |
|---|---|---|---|---|
| Cyclopentene | cis | −15.4 | −52 | −0.3 |
| trans | −18 | −52 | −2.3 | |
| Cyclohexene | cis | −1–3 | −31 | 6.2 |
| trans | 1–3 | −28 | 7.3 | |
| Cycloheptene | cis | −16 | −20 | −10.0 |
| trans | −20 | −17 | −15.0 | |
| Cyclooctene | cis | −20 | −2 | −19.4 |
| trans | −22 | −2 | −21.5 | |
| 1,5-Cyclooctadiene | cis | −25 | −5 | −23.5 |
| trans | −33 | −5 | −31.5 |
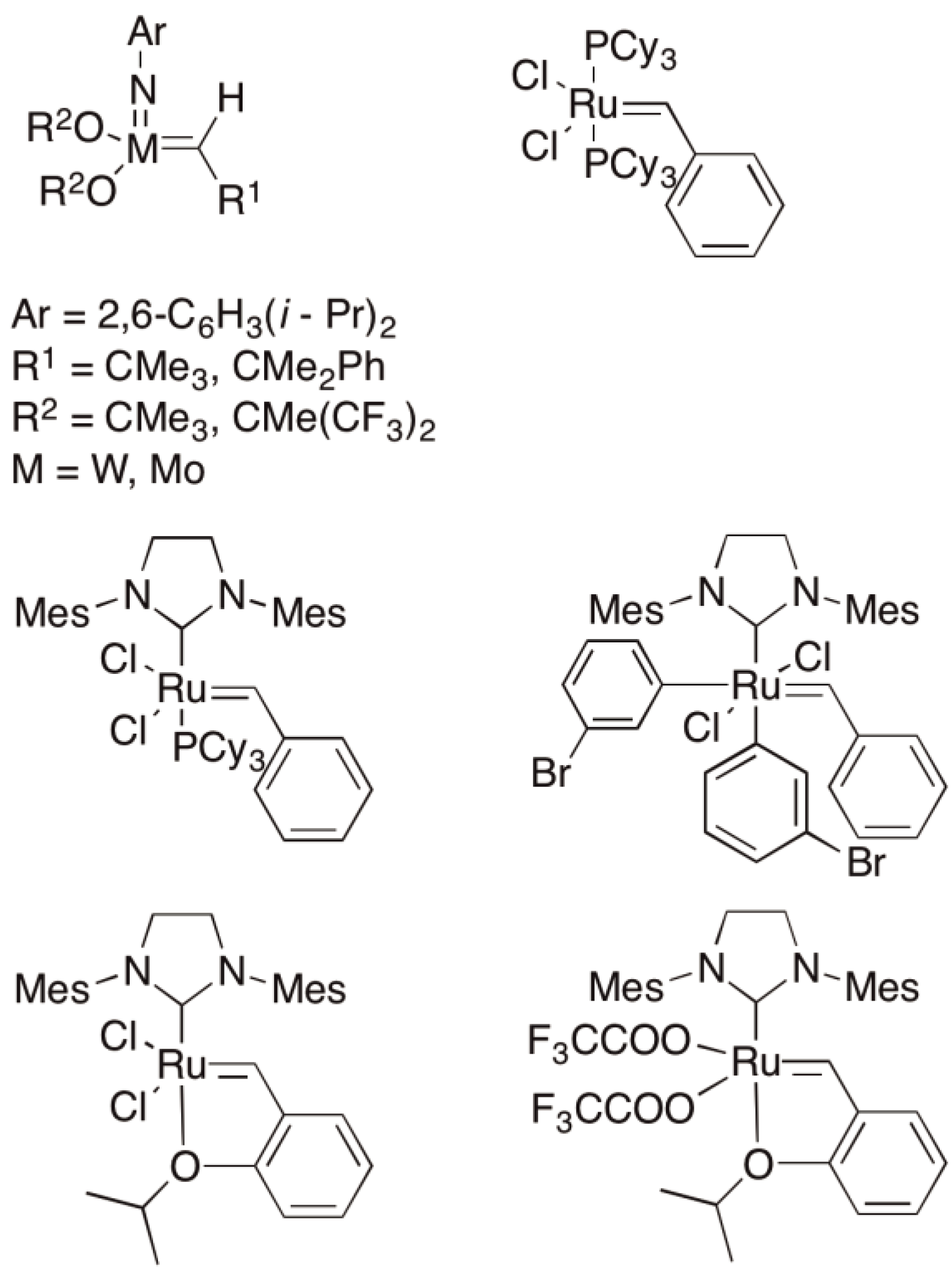
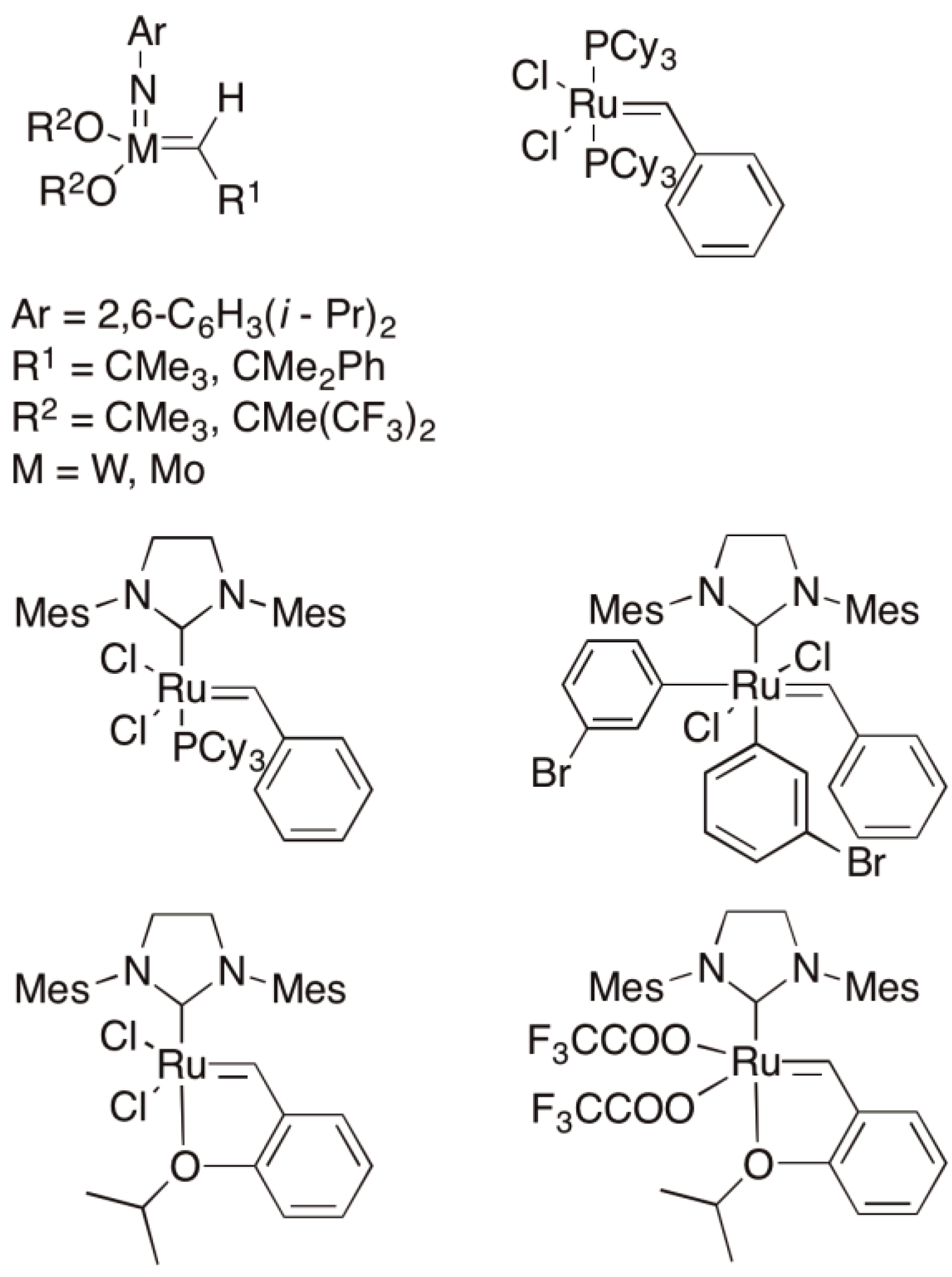
The metathesis of olefins and in particular cyclic olefins can be initiated by homogeneous, heterogeneous and immobilized catalysts. The classical catalysts are the oxides of Group VI transition metals on Lewis acid carriers, which are robust and can be regenerated. Typical examples of heterogeneous catalysts are WO3/Al2O3 and MoO3/Al2O3 [125], as homogeneous catalysts WCl6/SnBu4 und MoCl6/SnPh4 are quoted whereby these and a large number of other systems have been mentioned in previous reviews of the subject [125,126,127,128]. Only with the development of well-defined complexes based on Molybdenum or Ruthenium metals could a tolerance with respect to e.g., acid, ester and amide functionalities be developed and the possibility for a much wider variety of polymer structures opened up. Schrock, Grubbs and Chauvin were honored with a Nobel Prize in 2005 for their synthetic and mechanistic work in this area [132,133,134].
A selection of catalysts is shown in Figure 52.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
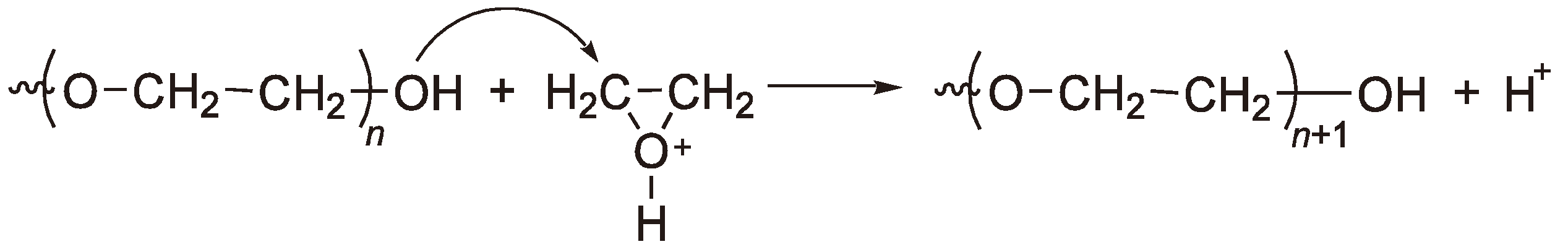{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
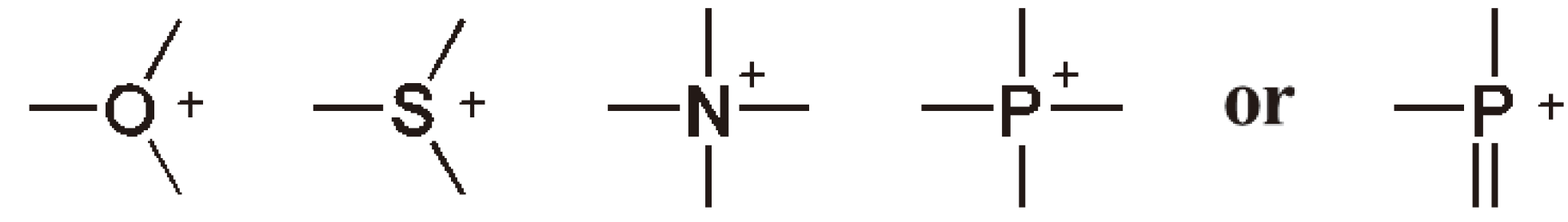{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
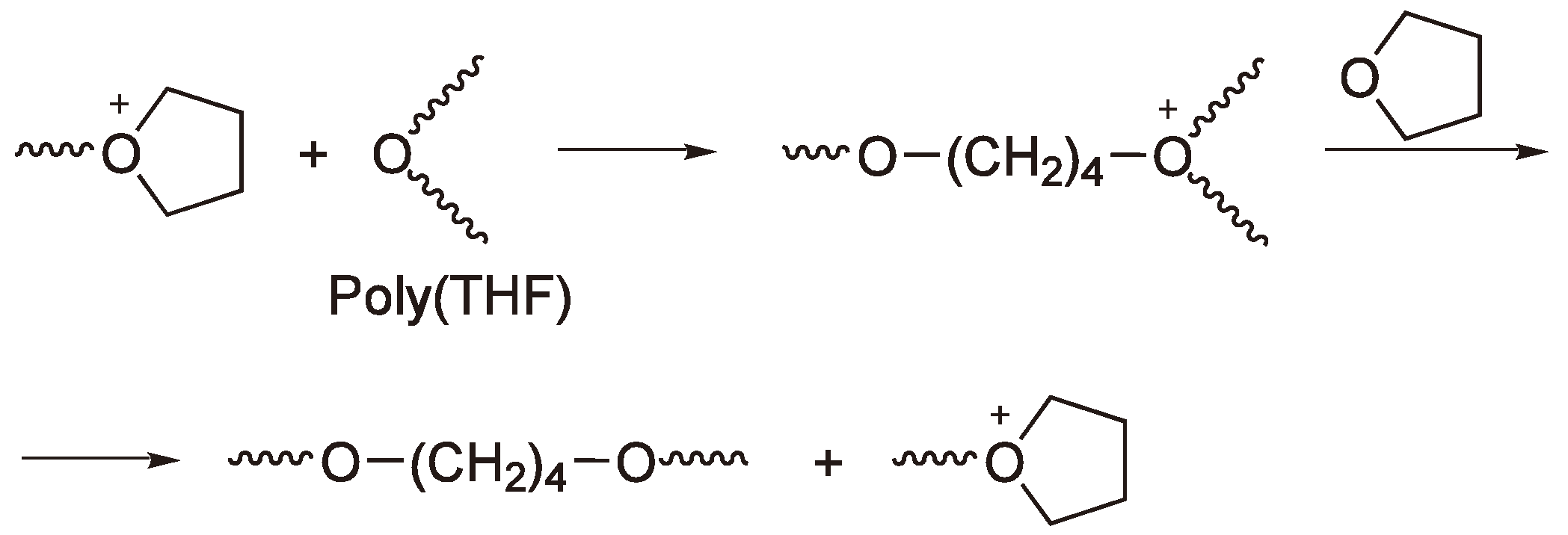{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
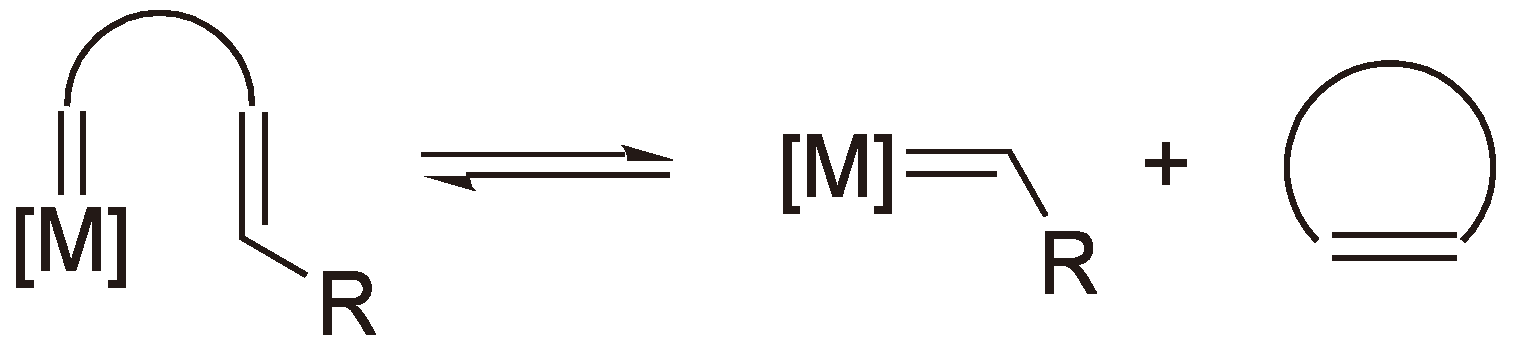{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ROMP is employed to produce a variety of interesting polymer structures of which a selection is shown in Figure 53 together with their reported commercial applications.
Figure 53.
Functional polymers via ROMP: (A) Super absorber, after crosslinking [141]; (B) Ion exchange resin, flocculation aid [141]; (C) High temperature thermoplast [141]; (D) Polymer with liquid crystal side chains (m ≥ 20) [142].
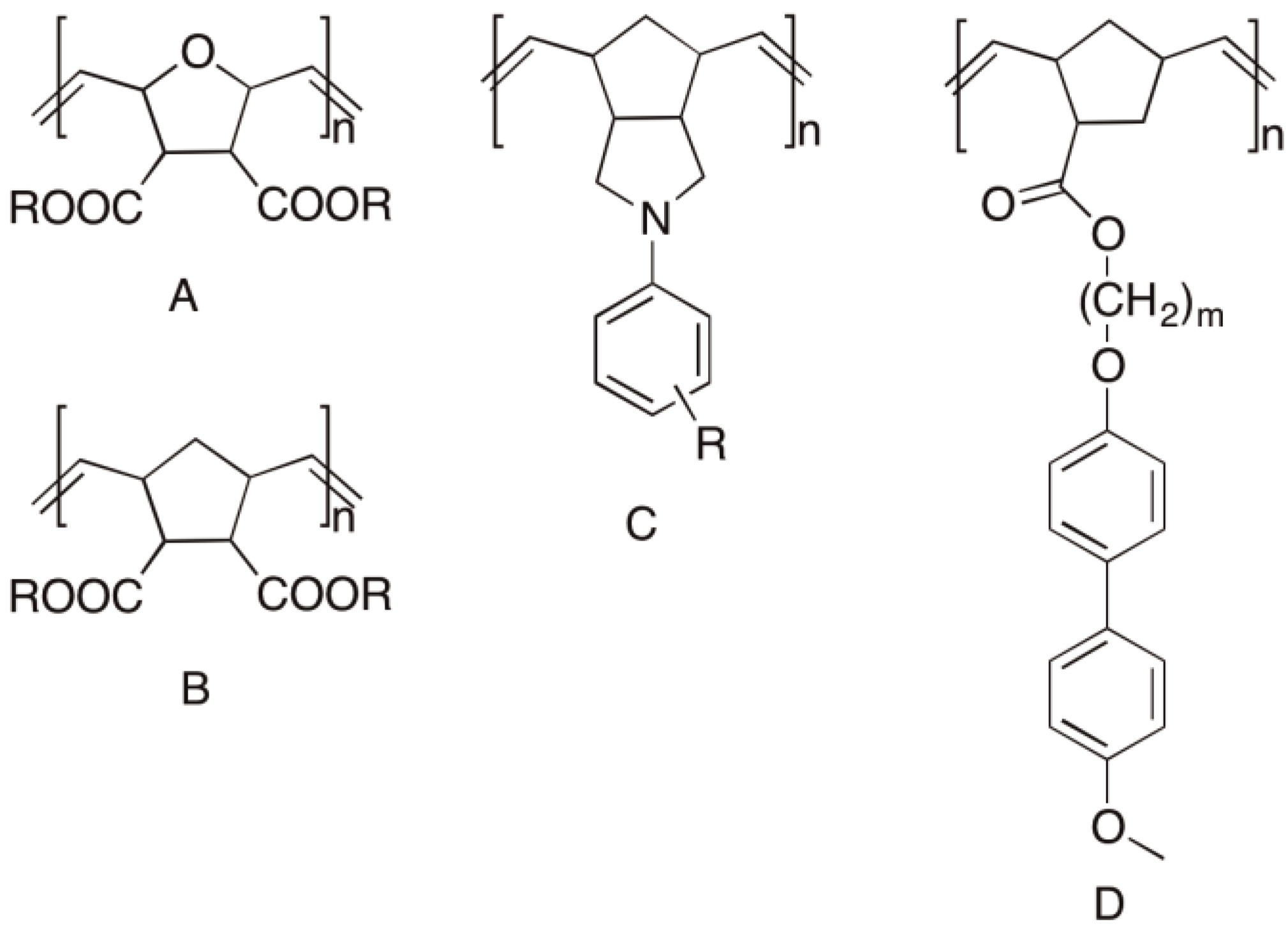
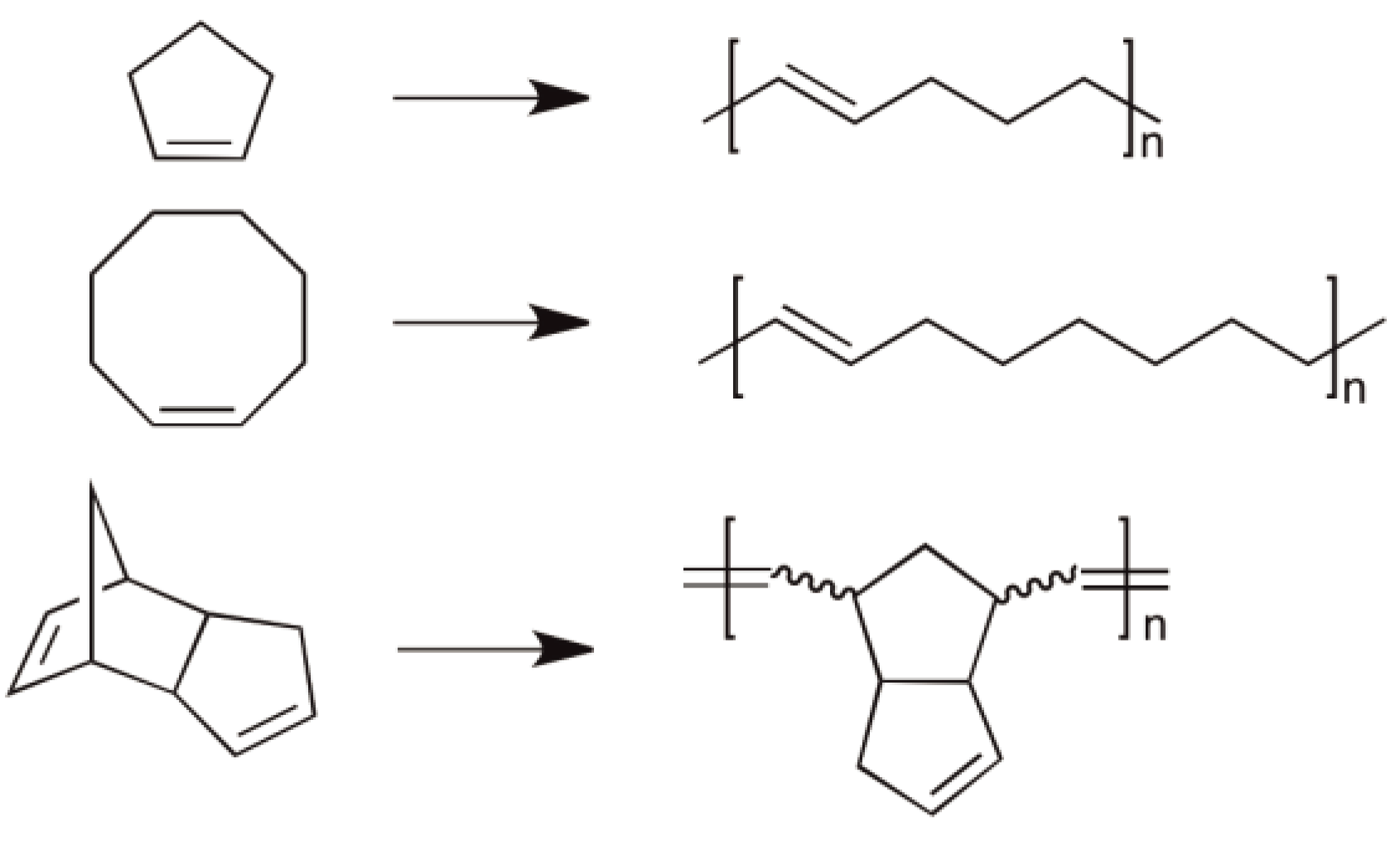
The best known industrial products synthesized via ROMP are: Norsorex® (Polynorbornene, Astrotech Advanced Elastomerproducts GmbH) (see Figure 50), Vestenamer® (Polycyclooctene) (Evonik Degussa GmbH), polycyclopentene and polycyclopentadiene (Metton®) are all well established commercial products (see Figure 54).
Figure 54.
The structures of Norsorex®, Vestenamer® and Polycyclopentadiene.
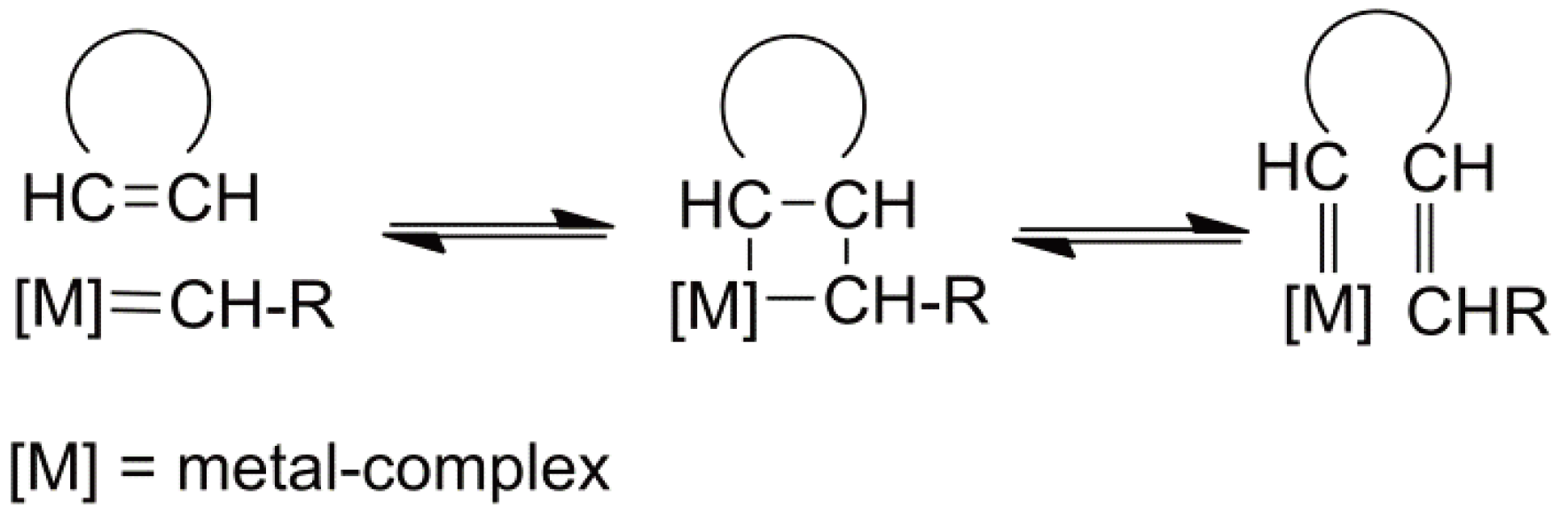
It is now widely accepted that the critical propagation step in ROMP is the formation of a metallocyclobutane intermediate from a coordination of a cycloolefin by the catalytic metal center of the carbine complex followed by a [2+2] cycloaddition. The metallocyclobutane then undergoes a cycloreversion and the metal atom separates from the olefin bond (see Figure 55) [143].
Figure 55.
The mechanism of ROMP including the metallocyclobutane intermediate.
The reactivity of the metal complexes with respect to different functional groups is graphically shown in Figure 56; thus, ruthenium complexes react preferentially with olefins and can, indeed, be used in aqueous media for metathesis reactions whereas the titanium complexes are sensitive to almost all impurities [144].
Figure 56.
The reactivity/tolerance of metathesis catalysts.

One disadvantage of homogeneous metathesis catalysts is that they cannot be reused; one solution is to immobilize them on either inorganic or organic carrier materials [128] as shown in Figure 57.
Figure 57.
Metathesis catalysts immobilized on A silica (Schrock Type) and B polystyrene (Grubbs type).
Figure 57.
Metathesis catalysts immobilized on A silica (Schrock Type) and B polystyrene (Grubbs type).

The opposite of ROMP, i.e., a ring “closure” metathesis polymerization occurs during the metathesis of of α, ω-dialkynes. Thus, controlled backbiting reactions occur leading to interesting new polymers with perfect 5- or 6-membered ring structures which exhibit good electrical conductivity, as shown in Figure 58 [145].
Figure 58.
The ring-closure metathesis polymerization of α, ω-dialkynes.

The good control of polymer structures possible via ROMP has led to many studies concerning the synthesis of polymers with well-defined end groups which have been reviewed in detail recently [146].
6. Other Ring-Opening Polymerizations
In this section the living (also sometimes called “Immortal”) polymerization and ring opening that occur via oxidation/reduction processes are briefly described [147,148].
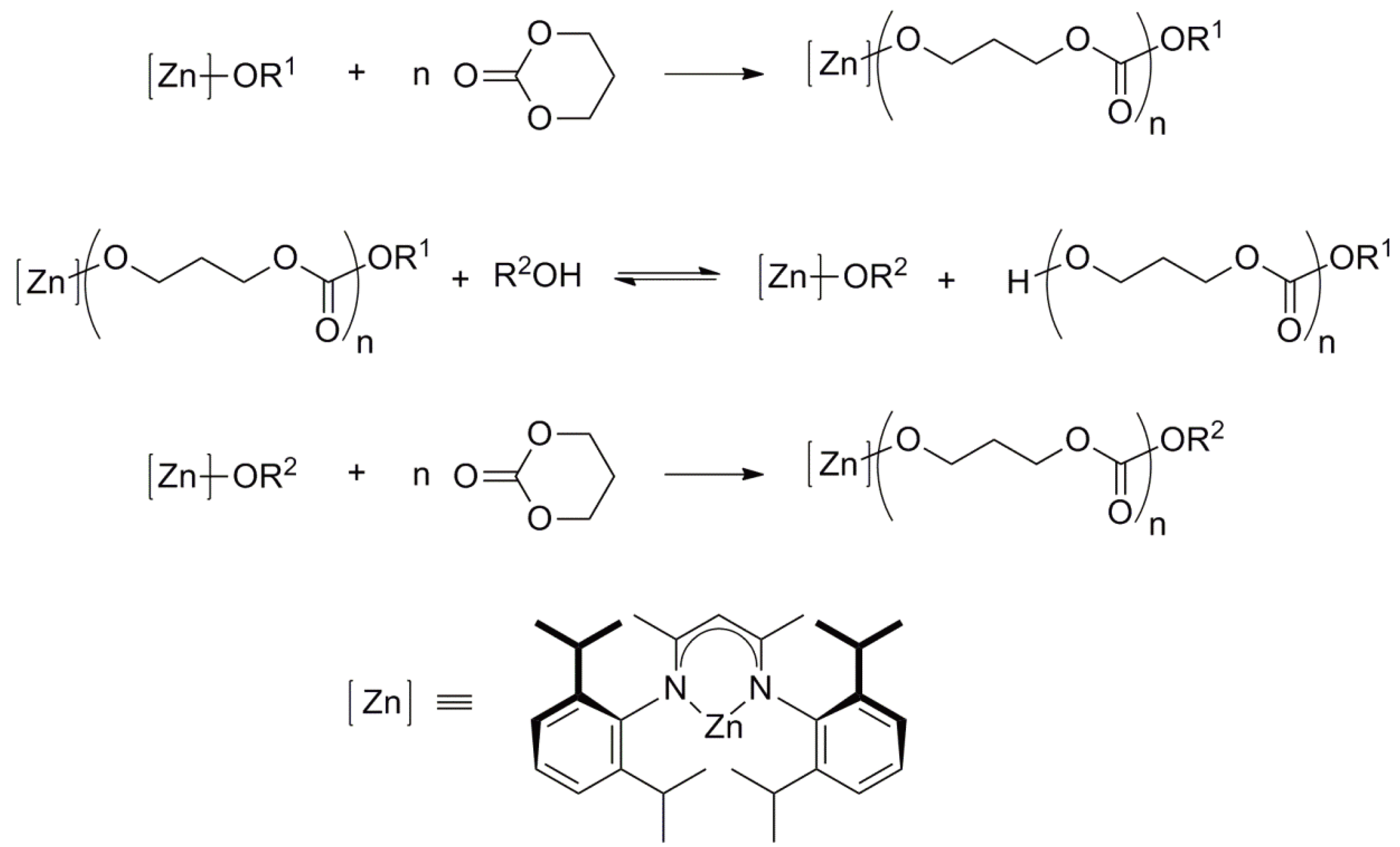
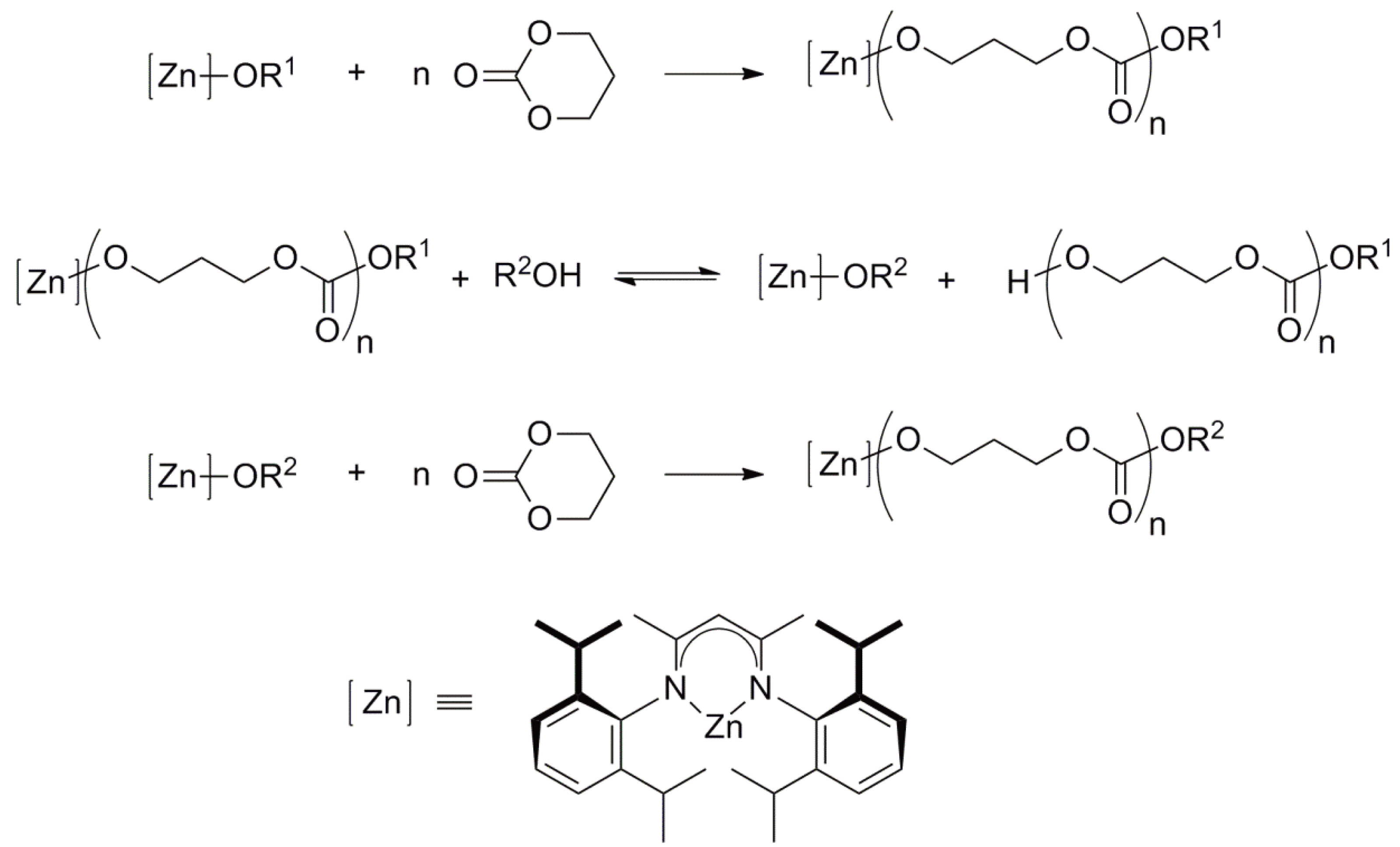
The pioneering work of Inoue [147,149] involving the ROP of oxiranes initiated by aluminium porphyrin/alcohol describes an “immortal” polymerization; a living polymerization during which the growing chains do not “die”. Rapid and reversible chain transfer takes place between the growing chains and alcohol molecules. Thus, only a very small initiator concentration is required. The number of polymer chains is equal to the sum of the number of initiator and transfer molecules. The overall reaction is shown in Figure 59 using the ROP of trimethylene carbonate, initiated by a [Zn]-mediated initiator in the presence of an alcohol (R2OH) as an example [150].
Figure 59.
[Zn] mediated living ring-opening polymerization of trimethylene carbonate.
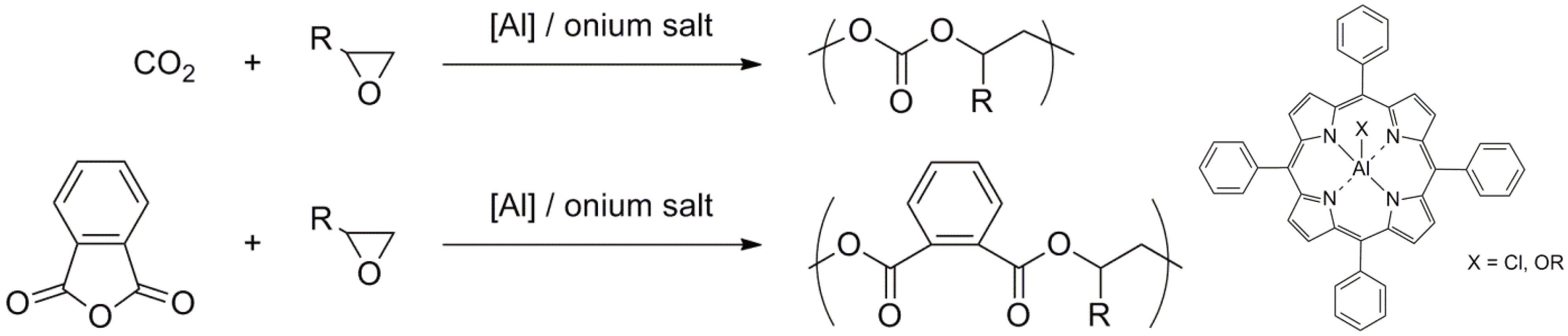
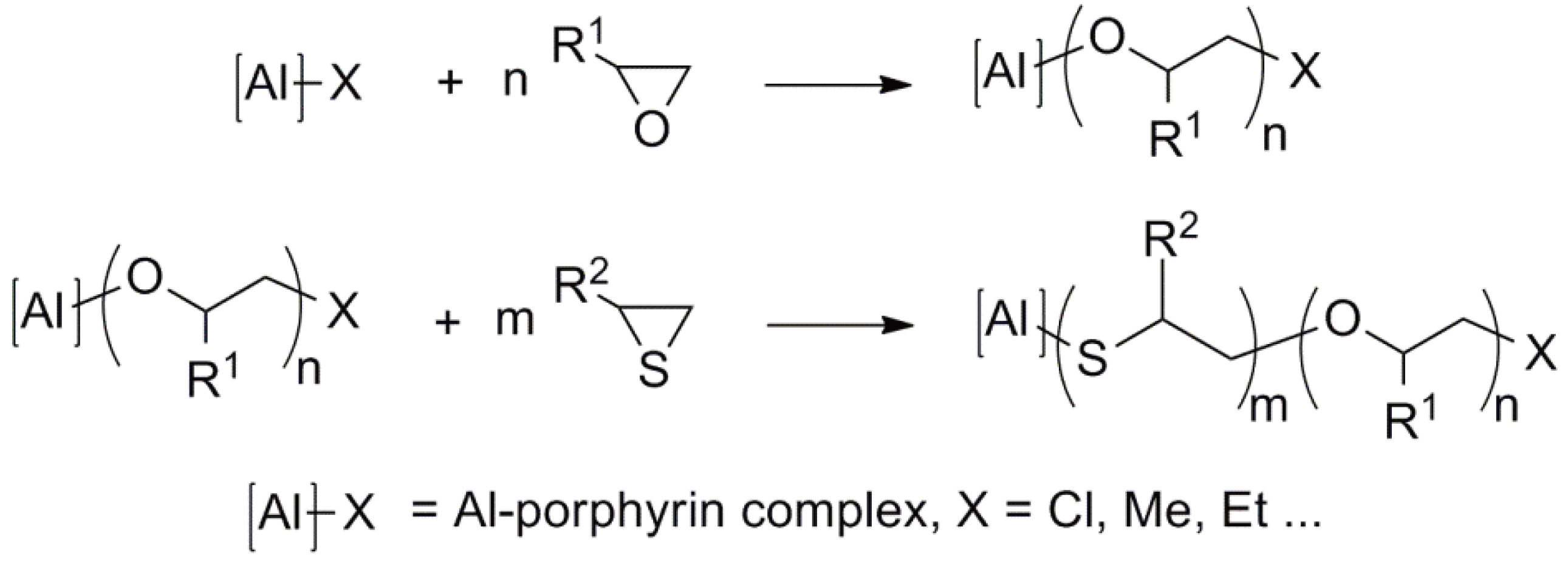
The initiators based on porphyrin complexes or complexes with related structures based on Al, Zn, Y, Lu, In have been used for the polymerization of e.g., oxiranes, thiiranes, lactones, lactides, cyclic carbonates and others [147,149,150,151,152,153,154,155,156]. Early examples of alternating copolymerization are shown in Figure 60 [147].
Figure 60.
Alternating copolymers via a living copolymerization of oxirane with CO2 or phthalic anhydride.
Figure 60.
Alternating copolymers via a living copolymerization of oxirane with CO2 or phthalic anhydride.
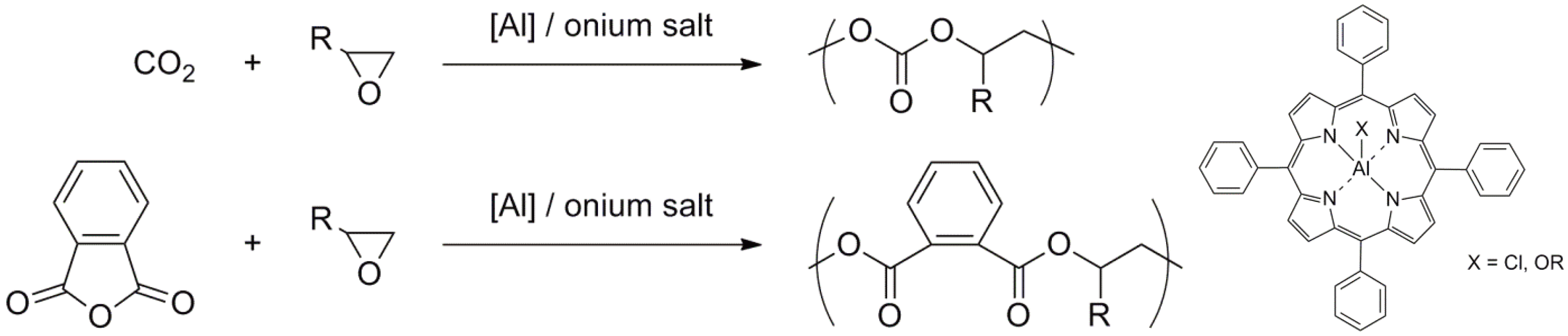
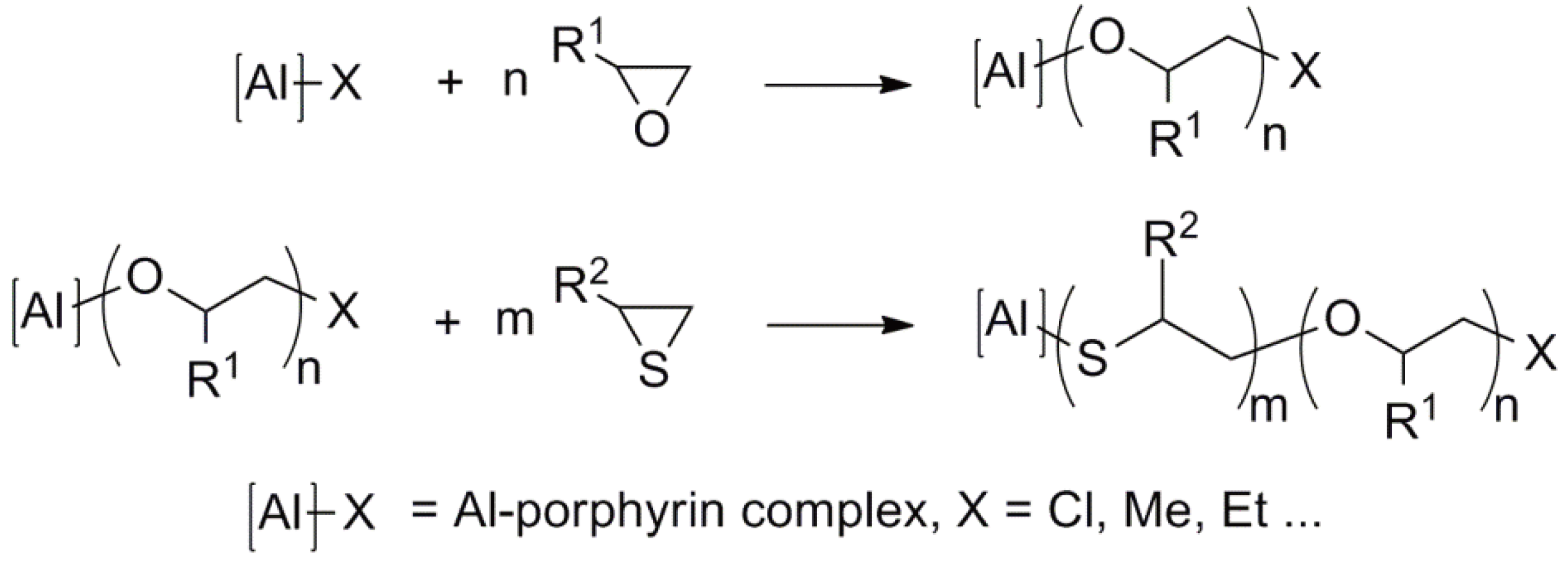
Furthermore, a great variety of block copolymers have been synthesized using this method [147]. One typical example is presented in Figure 61.
Figure 61.
Synthesis of a block copolymer via a living ring-opening polymerization.
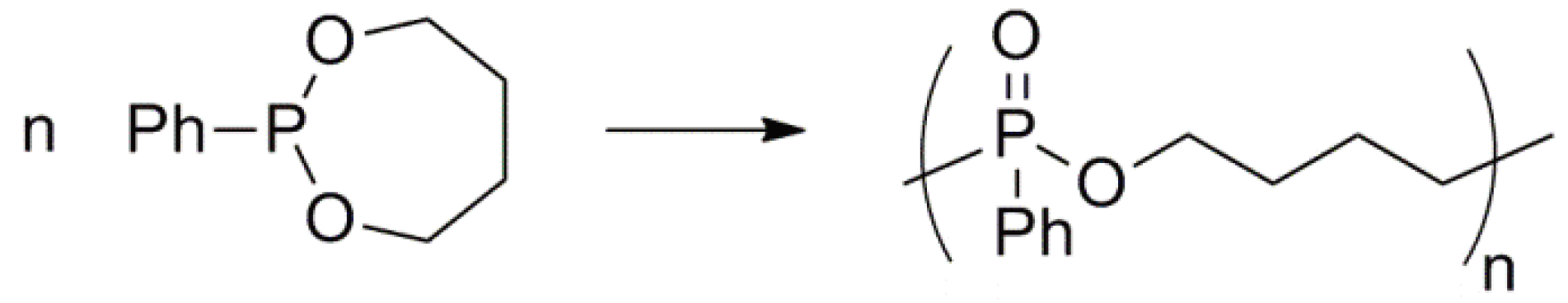
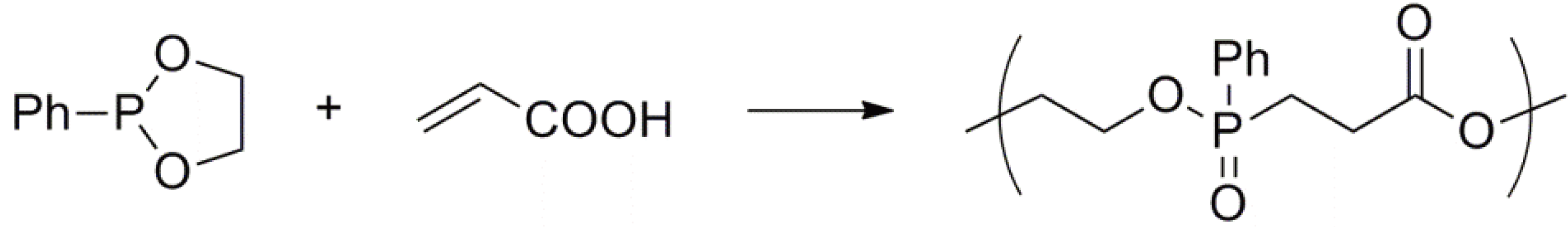
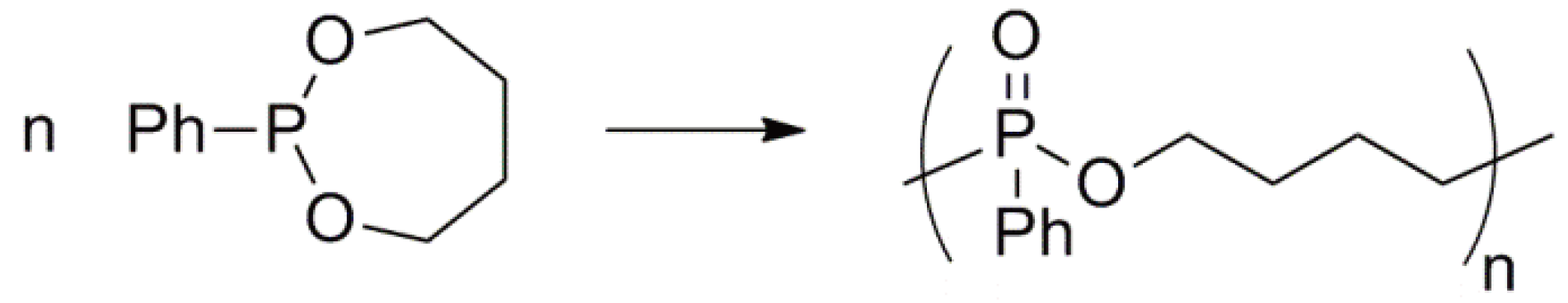
Cyclic phosphonites undergo an electrophilic ring-opening polymerization via an Arbuzov-type reaction to yield a polyphosphonate (Figure 62) [148].
Figure 62.
Electrophilic ring-opening polymerization of a cyclic phosphonite.
During the polymerization the phosphorus atom of the monomer is oxidized (from +3 to +5) and the carbon of the CH2O group is reduced (from −1 to −3) to yield the PCH2 moiety in the polymer; i.e., an intramolecular oxidation-reduction reaction takes place.
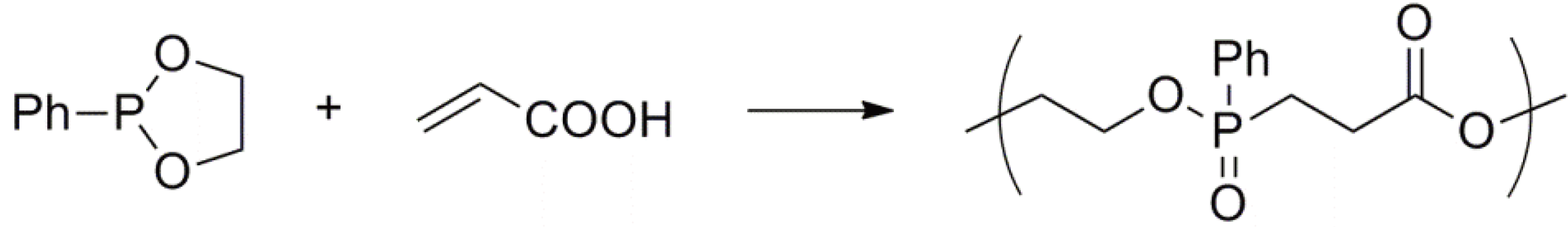
An interesting aspect of this type of ROP is the alternating copolymerization of a cyclic phosphorus (III) compound as reductant with a simple vinyl monomer as oxidant, as depicted in Figure 63 [157].
This copolymerization takes place without any initiator or catalyst and additional details are available in the literature [158].
Figure 63.
Intermolecular reduction-oxidation during the alternating copolymerization of a cyclic phosphorus (III) monomer with acrylic acid.
Figure 63.
Intermolecular reduction-oxidation during the alternating copolymerization of a cyclic phosphorus (III) monomer with acrylic acid.
7. Ring-Opening Polymerization of Phosphazenes
An interesting and promising class of polymers, which is still waiting for a real industrial breakthrough, can be obtained by the ring-opening polymerization of phosphazenes. The literature contains reports on more than 300 polymers on this basis [159,160]. The simple polymerization of (PNCl2)3 (Figure 64) is carried out in sealed vessels under an inert atmosphere by heating to ca. 250 °C for several hours. A conversion of much greater than 50% is avoided to minimize the formation of insoluble material. The polymerized material is then converted, by reaction with a stoichiometric amount of an alcoholate, an amine or an organometallic reagent, to yield a polymer more stable to hydrolytic decomposition [159,161].
Figure 64.
The ROP of hexachloro cyclo-triphosphazene at higher temperatures.
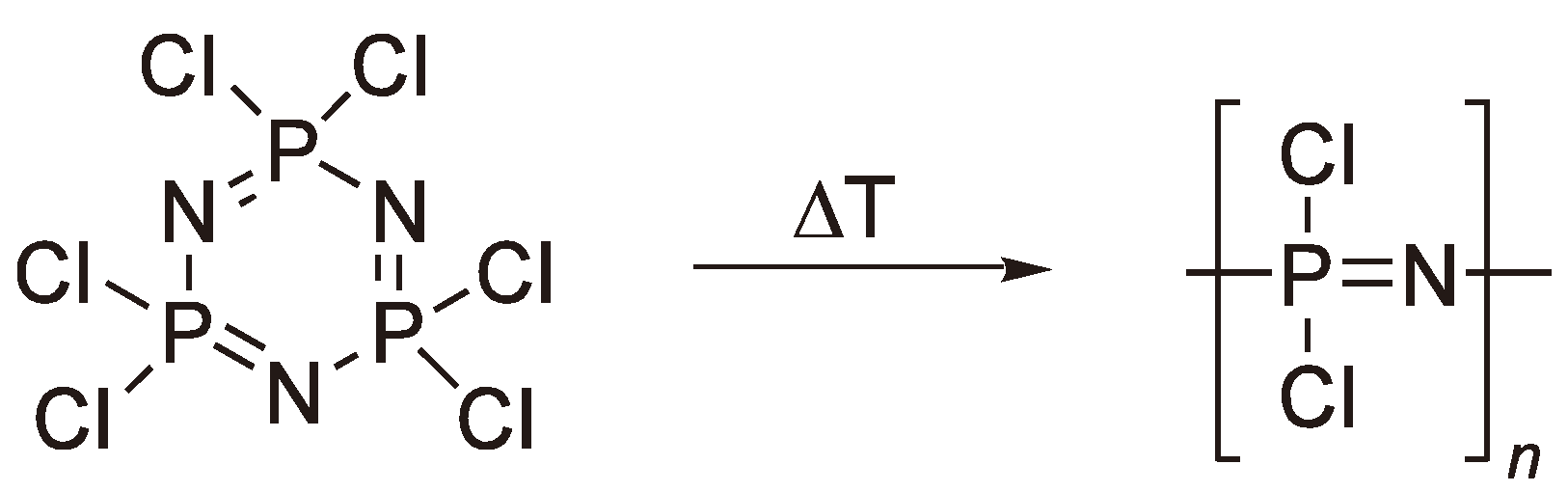
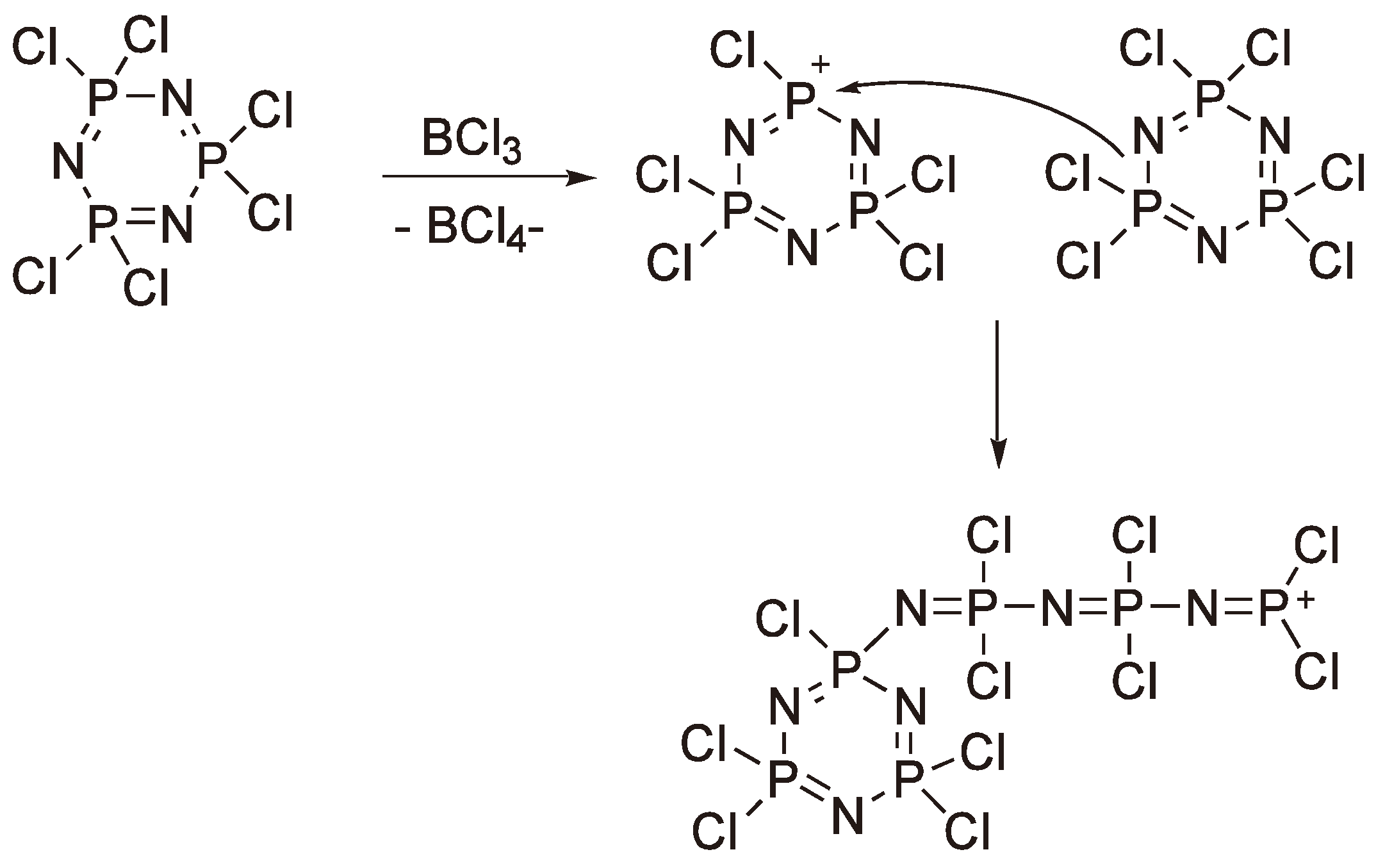
This reaction was first reported at the end of the 19th century by Liebig [162] and Stokes [163] and represents the first example of a synthetic rubber. At higher temperatures the cyclic phosphazene eliminates Cl− to yield a cyclic cation which then reacts with the free electron pair on a nitrogen of another monomer ring (see Figure 65); the P–N single bond in the second ring is broken to yield a linear phosphazene with a –P(Cl)2+ terminus. The reaction can be catalyzed by Lewis acids such as BCl3. As the reaction proceeds charge generation along the chain becomes more prevalent and eventually leads to crosslinked insoluble material at conversions much greater than 50%. The poor yields of soluble polymer have proved a major limitation for the commercialization of polyphosphazenes. Thus, alternative, non-ROP, routes have also been investigated [164,165,166,167], albeit with, as yet, limited commercial success.
Almost one hundred years after polyphosphazenes were first documented, Allcock’s group showed how to convert the base polymer into useful materials (see, e.g., Reference [168]).
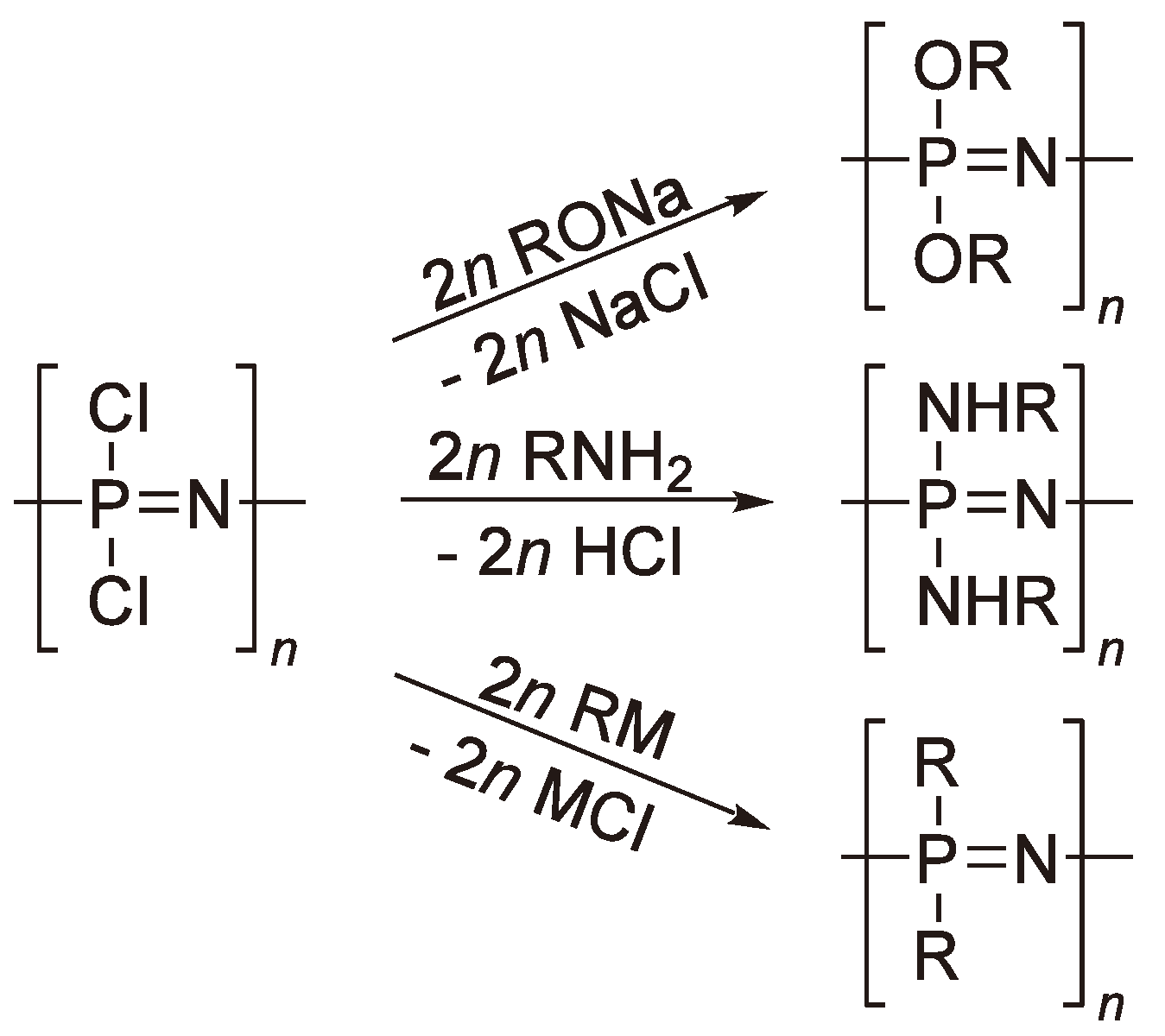
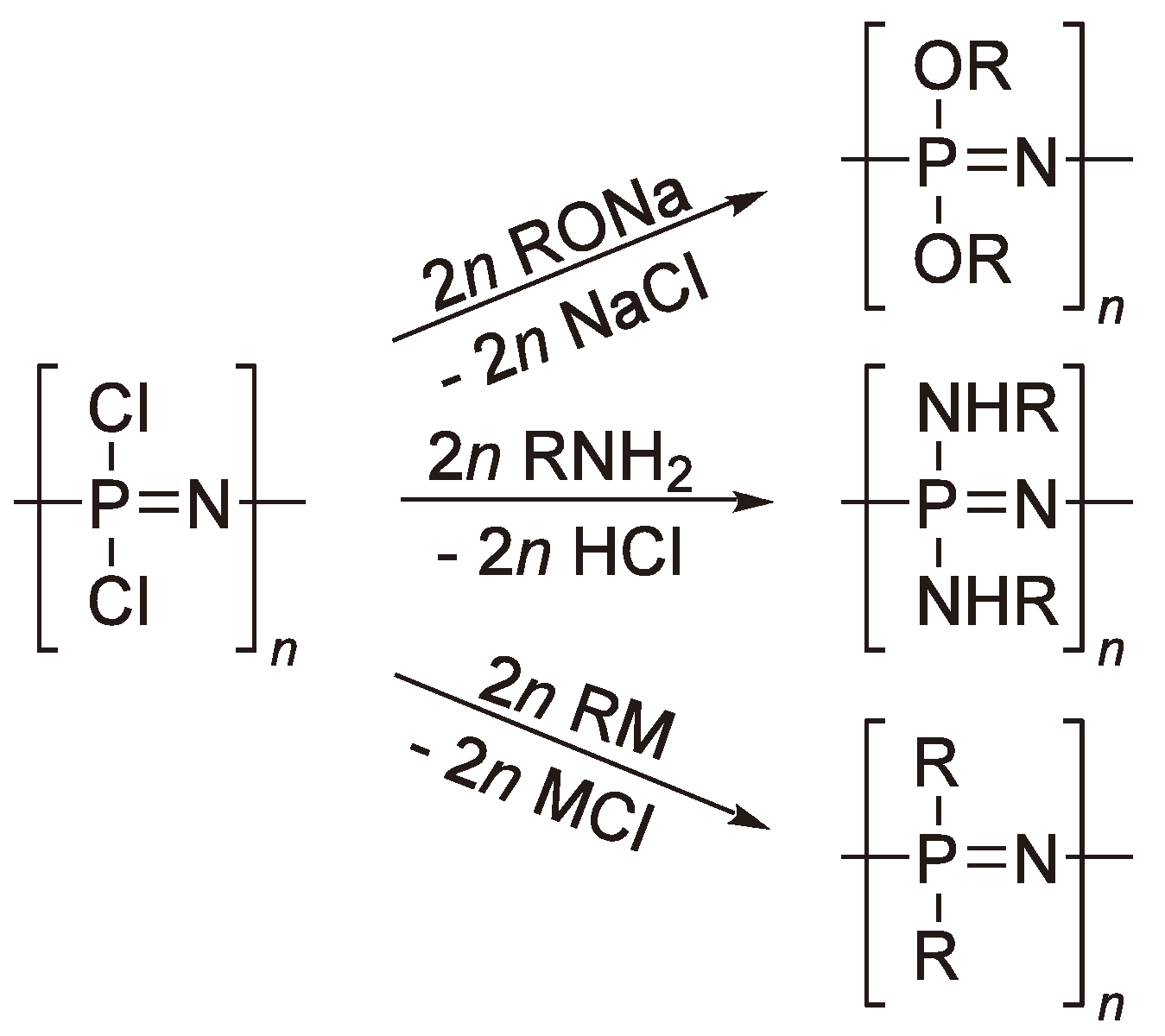
Since the base material ((PNCl2)n) is unstable in moist air, all the Cl-atoms must be substituted e.g., by alkoxy or amino groups to convert the so-called “inorganic rubber” into a material which is stable under practical conditions and thus makes the material suitable for a number of applications (Figure 66) [169]. Polyphosphazenes with perfluorinated alkoxy side chains have been commercialized under the trade name Eypel F® (Ethyl Corp., USA ).
Figure 65.
The cationic polymerization of hexachloro cyclo-triphosphazene.
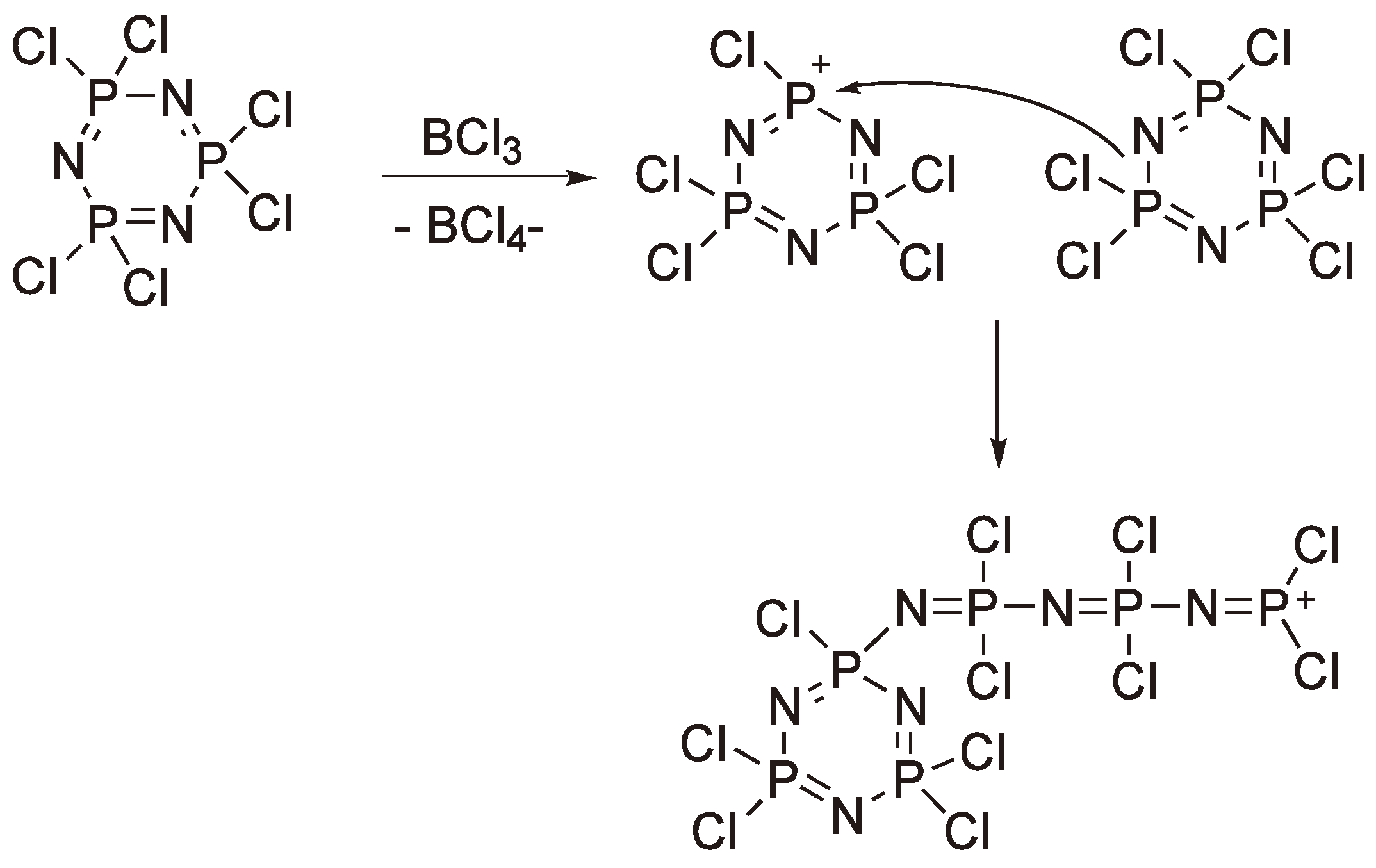
Figure 66.
Some substitution reactions to yield useful polyphosphazenes.
By changing the side group substitution the flexibility, refractive index, the transparency (especially for UV light) and the flammability can be adjusted and the hydrophobic or hydrophilic nature of the polymer determined [168,170,171]. Phosphazene elastomers having −OCH2(CF2)nCF3 side groupsexhibit excellent oil resistance and Tg as low as −60 °C without the addition of plasticizers. They are thus very suitable for special automotive and aerospace applications [172,173,174,175] and have also been suggested for biostable biomedical devices [176]
Polphosphazenes with oligo-ethylene oxy side chains have also been considered for use in lithium-ion polymer batteries [177,178,179] and those with sulfonated aryloxy side groups as membranes for fuel cells [180]. Polyphosphazenes with ethylene oxy side groups can be crosslinked with γ-rays to give hydrogels, which have been investigated as controlled drug release agents [181,182,183,184,185]. Polyphosphazenes with amino acid side chains are biodegradable and can be employed to aid the regeneration of bone tissue in vivo [186,187,188,189]. Polyphosphazenes with aryloxy side groups have also been studied as fire retardants and thermoplastics.
Numerous researchers have tried to substitute the monomer in order to avoid the polymer analogue reactions (Figure 66) but substitution of −Cl in the (PNCl2)3 monomer by −CH3, −OCH2CF3, –OPh or −Ph leads to molecules which simply decompose or undergo ring expansion on heating. If one of the −PCl2-groups in the ring is exchanged for XCl (see Figure 67) the ring strain can be increased and a ring-opening polymerization occurs at lower temperature but necessity of subsequent substitution of the –Cl groups and the crosslinking during polymerization remain as disadvantages [190,191,192].
Figure 67.
Examples of phosphazene analog monomers with increased ring strain (X = C,S).
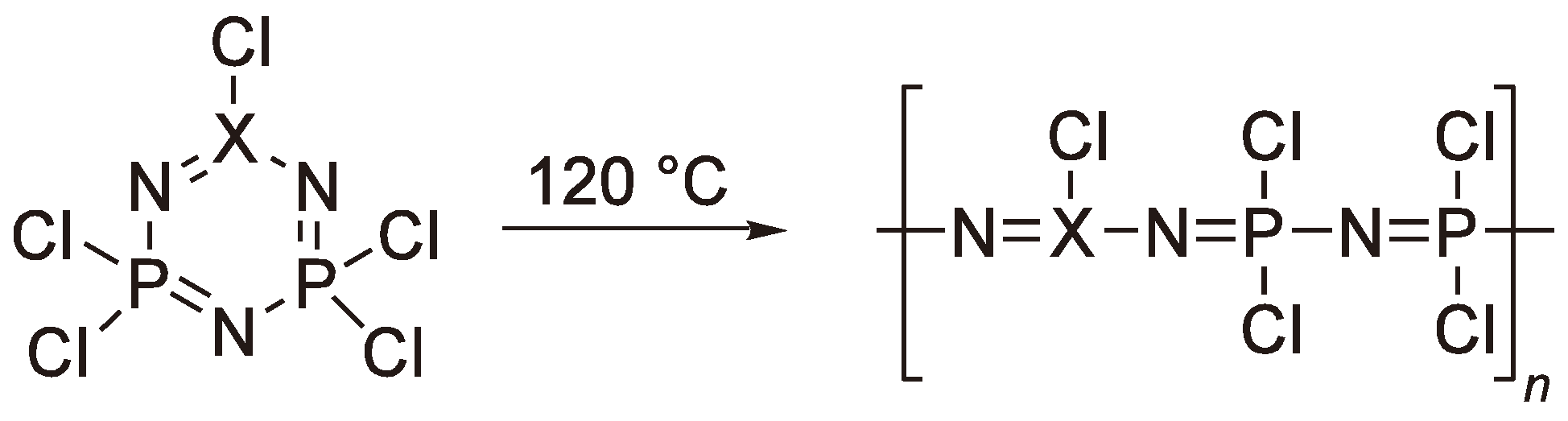
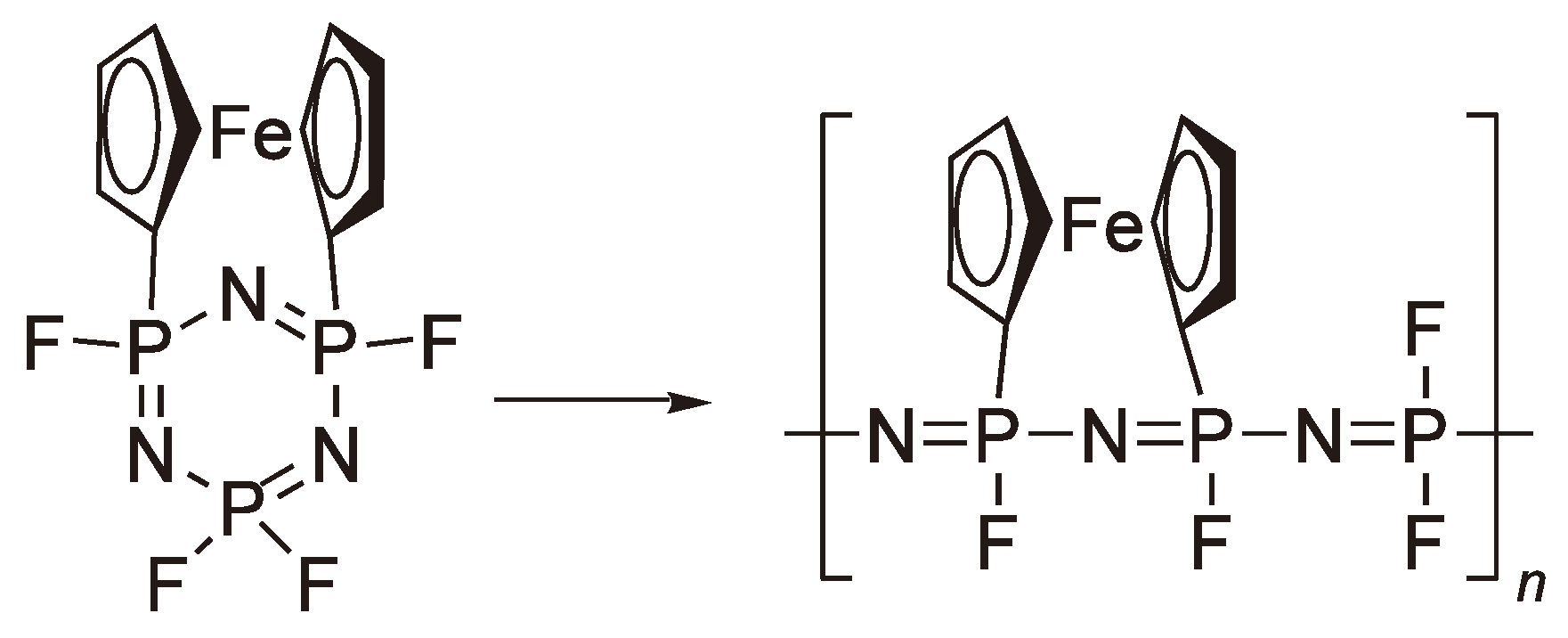
An alternative method to increase the ring strain is shown in Figure 68.
Figure 68.
Synthesis of a polyphosphazene with a ferrocene side chain.
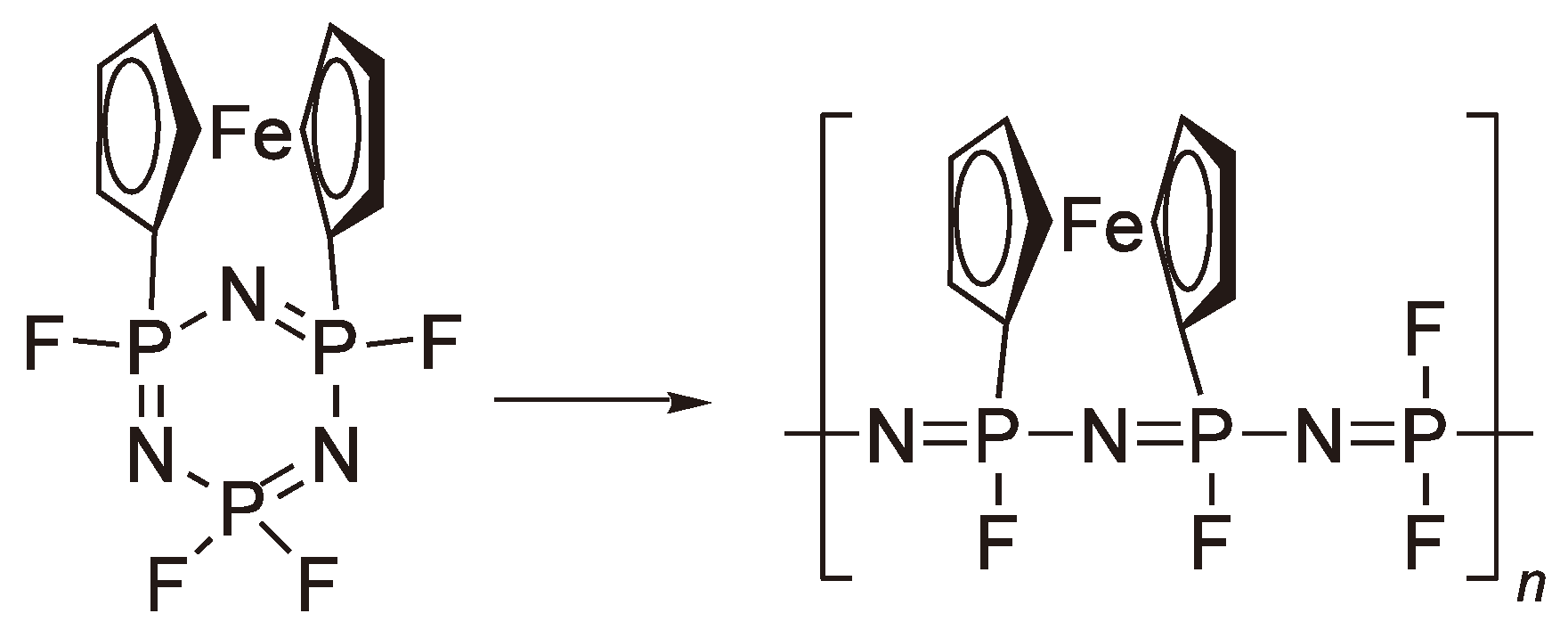
Nevertheless, these polymers still require the exchange of the remaining halogens e.g., with –OCH2CF3 to obtain polymers stable to hydrolysis [193] and this approach has not yet led to a commercial breakthrough for polyphosphazenes.
8. Summary and Outlook
It is hoped that this review has made it very clear that ring-opening polymerization covers a very broad variety of chemistries, monomers and areas of application; ROP enjoys growing interest from both academic and industrial scientists. The focus has been rather on the mechanistic aspects than on the classes of materials; thus, the organization into radical, cationic, anionic and metathesis type ROP.
The potential for novel polymers or novel processes for producing known polymers via ROP is demonstrated by the continuing flood of literature. No excuse is made for being selective but every effort has been made to acknowledge both that literature which has made significant contributions to an understanding of the mechanisms involved and that which describes pioneering synthetic work, which may point to future developments.
In particular, ROMP has developed exponentially in recent years (More than 5000 papers in the last 20 years!). It is believed that this will continue. Catalysts (be they homogeneous, heterogeneous or immobilized), which are more tolerant of functional groups could lead to even more dramatic expansion of the industrial applicability of ROMP and thus the interest in the subject. It is very pleasing to see that contributions in this direction are now coming from all branches of chemistry.
The control of the backbiting reactions as well as the ring/chain equilibria are important aspects for the synthesis of well-defined polymers, e.g., the backbiting reactions, if controlled, can be utilized to generate well-defined hyper branched polymers via ROP [194]. Generally, ROP are rather slow and the development of catalysts or co-catalysts, which enable the rate of polymerization to be increased is another development which would increase the applicability of the reactions to commercially viable systems.
We have chosen to treat polyphosphazenes in a separate section because, on the one hand, the mechanism of ring-opening is not understood in detail. On the other hand, the monomer, hexachloro cyclo-triphosphazene, can be polymerized and modified employing a plethora of reagents to yield a very wide variety of different materials for very different applications from plastics to elastomers and from damping to biomedical elements. The challenge remains to develop an economically viable synthesis.
Of course, the polyphosphazene backbone does not rely on fossil fuels but nor do many of the monomers discussed in this review, especially some oxacyclic monomers can be easily obtained from e.g., (potato) starch or sugar sources, so that with the current emphasis on renewable raw materials it can be expected that the interest in ring-opening polymerization will not wane in the near future.
References
- Duda, A.; Kowalski, A. Thermodynamics and Kinetics of Ring Opening Polymerisation. In Handbook of Ring Opening Polymerisation; Dubois, P., Coulembier, O., Raquez, J-M., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2009; pp. 1–51. [Google Scholar]
- Kubisa, P. Cationic Polymerization of Heterocyclics. In Cationic Polymerizations; Matyjaszewski, K., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 437–553. [Google Scholar]
- Ivin, K.J. Polymer Handbook, 2nd; Brandrup, J., Immergut, E.H., Eds.; John Wiley & Sons: New York, NY, USA, 1975; p. 8. [Google Scholar]
- Brunelle, D.J. Introduction. In Ring-Opening Polymerization; Hanser Publishers: Munich, Germany, 1993; pp. 2–4. [Google Scholar]
- Kubisa, P.; Vairon, J.P. Cationic Ring Opening Polymerization of Cyclic Acetals. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier BV: Amsterdam, the Netherlands, 2012; Volume 4, pp. 183–211. [Google Scholar]
- Endo, T. General Mechanism in Ring-Opening Polymerization. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp. 53–63. [Google Scholar]
- Hall, H.K.; Ykman, P. Addition polymerization of cyclobutene and bicyclobutane monomers. J. Polym. Sci. Macromol. Rev. 1976, 11, 1–45. [Google Scholar] [CrossRef]
- Sarker, P.; Ebdon, J.R.; Rimmer, S. Branched oligovinylcyclopropane by transfer to allylic carbonate oligomers via radical ring-opening polymerization. Macromol. Rapid Commun. 2006, 27, 2007–2013. [Google Scholar]
- Singha, N.K.; Kavitha, A.; Sarker, P.; Rimmer, S. Copper Mediated Controlled Radical Ring-Opening Polymerization (RROP) of Vinylcycloalkane. Chem. Commun. 2008, 3049–3051. [Google Scholar]
- Sanda, F.; Endo, T. Radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 2001, 39, 265–276. [Google Scholar] [CrossRef]
- Moszner, N.; Zeuner, F.; Völkel, T.; Rheinberger, V. synthesis and polymerization of vinylcyclopropanes. Macromol. Chem. Phys. 1999, 200, 2173–2187. [Google Scholar]
- Cho, I.; Kim, J.-B. Exploratory ring-opening polymerization. XIII, Radical ring-opening polymerization of 2-phenyl-3-vinyloxirane: A C–C-Bond scission polymerization of the epoxide ring. J. Polym. Sci. Polym. Lett. 1983, 21, 433–436. [Google Scholar]
- Ochiai, B.; Endo, T. Computational evaluation of radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 2007, 45, 2827–2834. [Google Scholar] [CrossRef]
- Endo, T.; Kanda, N. Syntheses of 2-phenyl-3-vinyloxirane derivatives that undergo radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 1985, 23, 1931–1938. [Google Scholar] [CrossRef]
- Hiraguri, Y.; Endo, T. Synthesis and radical ring-opening polymerization of 1,2-dicarbomethoxy-3-vinylcyclobutane. J. Polym. Sci. Polym. Lett. 1989, 27, 333–337. [Google Scholar]
- Bailey, W.J. Ring-Opening Polymerization. In Comprehensive Polymer Science; Allen, G., Bevington, A.G., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 283–320. [Google Scholar]
- Hiraguri, Y.; Endo, T. Synthesis and radical ring-opening polymerization of 2,2-diphenyl-4-methylene-5-methyl-l,3-dioxolane, alternating copolymer of propylene, and carbon monoxide. J. Polym. Sci. Polym. Chem. 1992, 30, 689–690. [Google Scholar] [CrossRef]
- Evans, R.A.; Rizzardo, E. Free-Radical ring-opening polymerization of cyclic allylic sulfides. Macromolecules 1996, 29, 6983–6989. [Google Scholar] [CrossRef]
- Angermann, J.; Burtscher, P.; Fischer, U.K.; Moszner, N.; Rheinberger, V. Dental Materials Based on Multicyclic Allyl Sulfides. Eur. Patent 1825843 Al, 29 August 2007. [Google Scholar]
- Luck, R.M.; Sadhir, R.K. Monomers that Expand During Polymerization. In Expanding Monomers; Sadhir, R.K., Luck, R.M., Eds.; CRC-Press: Boca Raton, FL, USA, 1992; pp. 21–61. [Google Scholar]
- Endo, T.; Bailey, W.J. Synthesis and radical ring-opening polymerization of spiro orthocarbonates. J. Polym. Sci. Polym. Chem. 1975, 13, 2525–2530. [Google Scholar] [CrossRef]
- Tagoshi, H.; Endo, T. Syntheses of polymers that undergo no shrinkage on crosslinking. J. Polym. Sci. Polym. Lett. 1988, 26, 77–81. [Google Scholar] [CrossRef]
- Bailey, W.J.; Zheng, Z.-F. Synthesis and double ring-opening polymerization of cyclic ketals of γ-methylenelactones. J. Polym. Sci. Polym. Chem. 1991, 29, 437–446. [Google Scholar] [CrossRef]
- Endo, T.; Bailey, W.J. Synthesis and radical ring-opening polymerization of 2-Methylene-l,4,6-trioxaspiro[4,4]nonane. J. Polym. Sci. Polym. Lett. 1980, 18, 25–27. [Google Scholar] [CrossRef]
- Agarwal, S. Chemistry, chances and limitations of the radical ring-opening polymerization of cyclic ketene acetals for the synthesis of degradable polyesters. Polym. Chem. 2010, 1, 953–964. [Google Scholar] [CrossRef]
- Ren, L.; Agarwal, S. Synthesis, characterization, and properties evaluation of poly[(N-isopropylacrylamide)-co-ester]s. Macromol. Chem. Phys. 2007, 208, 245–253. [Google Scholar] [CrossRef]
- Agarwal, S.; Ren, L.; Kissel, T.; Bege, N. Synthetic route and characterization of main chain ester-containing hydrolytically degradable poly(N,N-dimethylaminoethylmethacrylate)-Based polycations. Macromol. Chem. Phys. 2010, 211, 905–915. [Google Scholar] [CrossRef]
- Agarwal, S.; Ren, L. Polycaprolactone-based novel degradable ionomers by radical ring-opening polymerization of 2-methylene-1,3-dioxepane. Macromolecules 2009, 42, 1574–1579. [Google Scholar] [CrossRef]
- Grabe, N.; Zhang, Y.; Agarwal, S. Degradable elastomeric block copolymers based on polycaprolactone by free-radical chemistry. Macromol. Chem. Phys. 2011, 212, 1327–1334. [Google Scholar] [CrossRef]
- Zhang, Y.; Dafeng, C.; Zheng, M.; Kissel, T.; Agarwal, S. Biocompatible and degradable poly(2-hydroxyethyl methacrylate) based polymers for biomedical applications. Polym. Chem. 2012, 3, 2752–2759. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, M.; Kissel, T.; Agarwal, S. Design and biophysical characterization of bioresponsive degradable poly(dimethylaminoethyl methacrylate) based polymers for in vitro dna transfection. Biomacromolecules 2012, 13, 313–322. [Google Scholar] [CrossRef]
- Jin, Q.; Maji, S.; Agarwal, S. Novel amphiphilic, biodegradable, biocompatible, crosslinkable copolymers: Synthesis, characterization and drug delivery applications. Polym. Chem. 2012, 3, 2785–2793. [Google Scholar] [CrossRef]
- Undin, J.; Illanes, T.; Finne-Wistrand, A.; Albertsson, A.-C. Random introduction of degradable linkages into functional vinyl polymers by radical ring-opening polymerization, tailored for soft tissue engineering. Polym. Chem. 2012, 3, 1260–1266. [Google Scholar] [CrossRef]
- Plikk, P.; Tyson, T.; Finne-Winstrand, A.; Albertsson, A.-C. Mapping the characteristics of radical ring-opening polymerization of a cyclic ketene acetal towards the creation of a functional polyester. J. Polym. Sci. A Polym. Chem. 2009, 47, 4587–4601. [Google Scholar] [CrossRef]
- Wei, Y.; Connors, E.J.; Jia, X.; Wang, J. Controlled free radical ring-opening polymerization and chain extension of the living polymer. J. Polym. Sci. A Polym. Chem. 1998, 36, 761–771. [Google Scholar] [CrossRef]
- He, T.; Zou, Y.-F.; Pan, C.-Y. Controlled/“Living” radikal ring-opening polymerization of 5,6-benzo-2-methylene-1,3-dioxepane based on reversible addition fragmentation chain transfer mechanism. Polym. J. 2002, 34, 138–143. [Google Scholar] [CrossRef]
- Yuan, G.-Y.; Pan, C.-Y.; Tang, B.-Z. “Living” free radical ring-opening polymerization of 5,6-benzo-2-methylene-1,3-dioxopane using the atom transfer radical polymerization method. Macromolecules 2001, 34, 211–214. [Google Scholar] [CrossRef]
- Penczek, S.; Kubisa, P. Cationic Ring-Opening Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 13–86. [Google Scholar]
- Vairon, J.-P.; Spassky, N. Industrial Cationic Polymerization: An Overview. In Cationic Polymerizations; Matyjaszewski, K., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 683–750. [Google Scholar]
- Motokucho, S.; Sudo, A.; Endo, T. Living cationic-ring-opening polymerization of five-membered cyclic dithiocarbonate controlled by neighboring group participation of carbamate group. J. Polym. Sci. Polym. Chem. 2007, 45, 4459–4464. [Google Scholar] [CrossRef]
- Suzuki, A.; Sudo, A.; Endo, T. Cationic ring-opening-polymerization of 3-isochromanone through the formation of benzyl cationic intermediate and its friedel-crafts reaction. J. Polym. Sci. Polym. Chem. 2009, 47, 2214–2218. [Google Scholar] [CrossRef]
- Firat, Y.; Jones, F.R.; Plesch, P.H.; Westermann, P.H. The propagating species in the polymerization of 1,3-dioxacycloalkanes by percholoric acid. Makromol. Chem. Suppl. 1975, 1, 203–216. [Google Scholar] [CrossRef]
- Jaacks, V.; Boehlke, K.; Eberius, E. A method of determining the active centers in cationic polymerization of dioxolane, trioxane and formaldehyde. Makromol. Chem. 1968, 118, 354–360. [Google Scholar] [CrossRef]
- Brzezinska, K.; Chwialkowska, W.; Kubisa, P.; Matyjaszewski, K.; Penczek, S. Ion trapping in cationic polymerizations. Makromol. Chem. 1977, 178, 2491–2494. [Google Scholar] [CrossRef]
- Penczek, S. Cationic Ring-Opening Polymerization(CROP) major mechanistic phenomena. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 1919–1933. [Google Scholar]
- Bednarek, M.; Kubisa, P.; Penczek, S. Coexistence of activated monomer and active chain end mechanisms in cationic copolymerization of tetrahydrofuran with ethylene oxide. Macromolecules 1999, 32, 5257–5263. [Google Scholar] [CrossRef]
- Bednarek, M.; Brzezinska, K.; Stasinski, J.; Kubisa, P.; Penczek, S. Heteropolyacids—New efficient initiators of cationic polymerization. Makromol. Chem. 1989, 190, 929–938. [Google Scholar] [CrossRef]
- Ryu, C.Y.; Spencer, M.J.; Crivello, J.V. Involvement of supramolecular complexes in the capture and release of protonic acids during the cationic ring-opening polymerization of epoxides. Macromolecules 2012, 45, 2233–2241. [Google Scholar] [CrossRef]
- Slomkowski, S.; Penczek, S. Kinetics of hydride transfer from 1,3-dioxolane to the free triphenymethyl cation preceeding the true initiation. Chem. Commun. l970, 1347–1348. [Google Scholar]
- Afshar-Taromi, F.; Scheer, M.; Rempp, P.; Franta, E. A new efficient cationic initiator for the polymerization of tetrahydrofuran. Makromol. Chem. 1978, 179, 849–853. [Google Scholar] [CrossRef]
- Szymanski, R.; Penczek, S. Dynamic NMR studies of equilibria involving active species in the polymerization of cyclic acetals. Reaction of methoxycarbenium ion (CH3OCH2)+ with 5, 6 and 7 membered cyclic acetals. Makromol. Chem. 1982, 183, 1587–1602. [Google Scholar]
- Matyjaszewski, K.; Penczek, S. The macroester-macroion equilibrium in the cationic polymerization of THF observed directly by 300 MHz H NMR. J. Polym. Sci. Polym. Chem. 1974, 12, 1905–1912. [Google Scholar] [CrossRef]
- Goethals, E.J.; Drijvers, W. Cationic polymerization of cyclic sulfides. IV. Determination of the structure and concentration of the propagating species during the polymerization of 3,3-dimethoxythietane by 300 MHz NMR spectroscopy. Makromol. Chem. 1973, 165, 329–333. [Google Scholar]
- Goethals, E.J.; Schacht, E.H. Cationic polymerization of cyclic imines. II. 300 MHz 1H NMR of the structure and concentration of propagating species during the polymerization of 1,3,3-trimethylazetidine. J. Polym. Sci. Polym. Lett. Ed. l973, 11, 497–501. [Google Scholar]
- Li, Y.; Yu, B. Glycolization initiated cationic ring-opening polymerization of tetrahydrofuran to prepare neoglycolpolymers. Chem. Commun. 2010, 46, 6060–6062. [Google Scholar]
- Saegusa, T.; Kobayashi, S.; Yamada, A. Kinetics of the isomerization polymerization of 2-methyl-2-oxazoline by benzyl chloride and bromide initiators. Makromol. Chem. 1976, 177, 2271–2283. [Google Scholar]
- Smith, S.; Hubin, A.J. Preparation and chemistry of dicationically active polymers of tetrahydrofuran. J. Macromol. Sci. Chem. 1973, A7, 1399–1413. [Google Scholar]
- Kobayashi, S.; Danda, H.; Saegusa, T. Superacids and their derivatives. IV. Kinetic studies on the ring-opening polymerization of tetrahydrofuran initiated with ethyl trifluoromethanesulfonate by means of 19F and 1H nuclear magnetic resonance spectroscopy. Evidence for the oxonium-ester equilibrium of the propagating species. Macromolecules 1974, 7, 415–420. [Google Scholar]
- Matyjaszewski, K.; Kubisa, P.; Penczek, S. Ion ⇄ Ester equilibria in the living cationic polymerization of tetrahydrofuran. J. Polym. Sci. Polym. Chem. 1974, 12, 1333–1336. [Google Scholar]
- Kennedy, J.P.; Marechal, E. Carbocationic. Polymerization; John Wiley & Sons: New York, NY, USA, 1982; pp. 95–97. [Google Scholar]
- Hoene, R.; Reichert, K.-H. Elucidation of the initation step in the cationic polymerization of tetrahydrofuran with phospherous pentafluoride. Makromol. Chem. 1976, 177, 3545–3570. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Diaryliodonium salts. A new class of photoinitiators for cationic polymerization. Macromolecules 1977, 10, 1307–1315. [Google Scholar]
- Sangermano, M.; Crivello, J.V. Visible and long-wavelength cationic photopolymerization. ACS Symp. Ser. 2003, 847, 242–252. [Google Scholar]
- Crivello, J.V. Cationic photopolymerization of alkyl glycidyl ethers. J. Polym. Sci. Polym. Chem. 2006, 44, 3036–3052. [Google Scholar] [CrossRef]
- Putzien, S.; Louis, E.; Nuyken, O.; Crivello, J.V.; Kühn, F.E. UV curing of epoxy functional hybrid silicones. J. Appl. Polym. Sci. 2012, 126, 1188–1197. [Google Scholar]
- Anger, C.A.; Hinelang, K.; Helbich, T.; Halbach, T.; Stohrer, J.; Rieger, B. Photoinduced polysiloxane architechtures from spirosiloxane precursors via intramolecular hydrosilylation. ACS Macro Lett. 2012, 1, 1204–1207. [Google Scholar]
- Hoefler, T.; Griesser, M.; Gruber, G.; Jakopic, G.; Trimmel, G.; Kern, W. Photo fries rearrangement in polymer media: An investigation on fully aromatic esters containing the naphthyl chromophore. Macromol. Chem. Phys. 2008, 209, 488–498. [Google Scholar]
- Buijsen, P.F.A.; Hacker, N.P. Photochemical reaction of onium salts with N,N-Dimethylaniline; evidence for photo induced electron transfer. Tetrahedron Lett. 1993, 34, 1557–1560. [Google Scholar]
- Jungermann, S.; Riegel, N.; Mueller, D.; Meerholz, K.; Nuyken, O. Novel photo-cross-linkable hole transporting polymers: synthesis, characterization and application in organic light emitting diodes. Macromolecules 2006, 39, 8911–8919. [Google Scholar]
- Schelter, J.; Mielke, G.F.; Koehnen, A.; Wies, J.; Koeber, S.; Nuyken, O.; Meerholz, K. Novel non-conjugated main-chain hole-transporting polymers for organic electronics application. Macromol. Rapid. Commun. 2010, 31, 1560–1567. [Google Scholar]
- Müller, C.D.; Falcon, A.; Reckefuss, N.; Rojahn, M.; Wiederhirn, V.; Rudati, P.; Frohne, H.; Nuyken, O.; Becker, H.; Meerholz, K. Multi-colour organic light emitting displays by solution processing. Nature 2003, 421, 829–833. [Google Scholar]
- Feser, S.; Meerholz, K. Investigation of the photocrosslinking mechanism in oxetane functionalized semicunductors. Chem. Mater. 2011, 23, 5001–5005. [Google Scholar] [CrossRef]
- Makino, A.; Kobayashi, S. Chemistry of 2-oxazolines: A crossing of the cationic ring-opening polymerization and enzymatic ring-opening polyaddition. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1251–1270. [Google Scholar] [CrossRef]
- Novel polyoxazolines polymer drug delivery platform. Available online: http://www.baypat.de/en/industry/technology-offers.html?tech_ang=1684 (accessed on 17 April 2013).
- Kourti, M.-E.; Vougioukalakis, G.C.; Hadjichristidis, N.; Pitsikalis, M. Metallocene-mediated cationic-ring-opening polymerization of 2-methyl and 2-phenyl-oxazoline. J. Polym. Sci. Polym. Chem. 2011, 49, 2520–2527. [Google Scholar]
- Saegusa, T.; Matsumoto, S. Determination of concentration of propagating species in cationic polymerization of tetrahydrofuran. J. Polym. Sci. A 1968, 6, 1559–1565. [Google Scholar]
- Saegusa, T.; Kobayashi, S. Cationic ring-opening polymerization of cyclic ethers. Progr. Polym. Sci. Jpn. 1973, 4, 106–151. [Google Scholar]
- Matyjaszewski, K.; Penczek, S. Ion trapping in cationic polymerization. II, Relative rates of trapping and relative chemical shifts of structural differing phosphines as trapping agents. Makromol. Chem. 1981, 182, 1735–1742. [Google Scholar]
- Brzezinska, R.; Szymanski, P.; Kubisa, P.; Penczek, S. Activated monomer mechanism in cationic polymerization. Ethylenoxide, formulation of mechanism. Makromol. Chem. Rapid Commun. 1986, 7, 1–4. [Google Scholar]
- Penczek, S.; Kubisa, P.; Szymanski, R. Activated monomer propagation in cationic polymerization. Makromol. Chem. Macromol. Symp. 1986, 3, 203–220. [Google Scholar] [CrossRef]
- Qayouh, H.; Lahcini, M.; Six, J.-L.; Kricheldorf, H.R. Polymerizations of hexamethylcyclosiloxane catalyzed by metal sulfonate/acid chloride combinations. J. Appl. Polym. Sci. 2012, 124, 4114. [Google Scholar]
- Sauvet, G.; Lebrun, J.J.; Sigwalt, P. Cationic Polymerization of Cyclosilanes: The Various Processes Involved. In Cationic Polymerization and Related Processes; Goethals, E.J., Ed.; Academic Press: London, UK, 1984; pp. 237–251. [Google Scholar]
- Wilczek, L.; Chojnowski, J. Acidolytic ring-opening of cyclic siloxane and acetal monomers. Role of hydrogen bonding in cationic polymerization initiated with protonic acids. Macromolecules 1981, 14, 9–17. [Google Scholar]
- Richards, D.H.; Kingston, S.B.; Souel, T. Block copolymer synthesis by reaction of living polystyrene with living polytetrahydrofuran. Polymer 1978, 19, 68–72. [Google Scholar]
- Nagai, D.; Sato, M.; Ochai, B.; Endo, T. Synthesis and properties of the polythiourethanes obtained by the cationic ring-opening polymerization of cyclic thiourethanes. J. Polym. Sci. Polym. Chem. 2006, 44, 4795–4803. [Google Scholar]
- Krieg, A.; Weber, C.; Hoogenboom, R.; Becer, C.R.; Schubert, U.S. Block Copolymers of Poly (2-Oxazoline)s and Poly (Methyl) Acrylates; A Crossover between Cationic Ring-Opening Polymerization (CROP) and Reversible Addition-Fragmentation Chain Transfer (RAFT). ACS Macro Lett. 2012, 1, 776–779. [Google Scholar]
- Lefevre, N.; Fustin, C.-A.; Hoogenboom, R.; Schubert, U.S.; Gohy, J.-F. Nanostructured surfaces from block copoly(2-oxazoline)s prepared by microwave-assisted cationic ring-opening polymerization. Polym. Prepr. 2008, 49, 949–950. [Google Scholar]
- Paulus, R.M.; Erdmenger, T.; Becer, C.R.; Hoogenboom, R.; Schubert, U.S. Scale-Up of microwave-assisted polymerizations in continuous flow mode: Cationic ring-opening polymerization of 2-Ethyl-2-oxazoline. Macromol. Rapid Commun. 2007, 28, 484–491. [Google Scholar]
- Hoogenboom, R. A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar]
- Krause, J.O.; Zarka, M.T.; Anders, U.; Weberskirch, R.; Nuyken, O.; Buchmeiser, M.R. Simple synthesis of Poly(acetylene) latex particles in aqueous media. Angew. Chem. Int. Ed. 2003, 42, 5965–5969. [Google Scholar]
- Gall, B.; Bortenschlager, M.; Nuyken, O.; Weberskirch, R. Cascade reaction in polymeric nanoreactors. Mono(Rh) and Bimetallic(Rh, Ir) micellar catalysis in the hydroformylation of octene. Macromol. Chem. Phys. 2008, 209, 1152–1159. [Google Scholar]
- Schönfelder, D.; Fischer, K.; Schmidt, M.; Nuyken, O.; Weberskirch, R. Poly(2-oxazoline) functionalized with palladium carbene complexes: soluble amphiphilic polymer supports for C–C coupling reactions in water. Macromolecules 2005, 8, 254–262. [Google Scholar]
- Slomkowski, S.; Duda, A. Anionic Ring-Opening Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 88–128. [Google Scholar]
- Ostrovskii, V.E.; Khodzhemirov, V.A.; Barkova, A.P. Heat of ethylene oxide polymerization and kinetics of polymerization on solid potassium hydroxide. Dokl. Akad. Nauk. SSSR 1970, 191, 1095–1098. [Google Scholar]
- Nicco, A.; Boucheron, R.G. Anionic polymerization of thiirane. Eur. Polym. J. 1970, 6, 1477–1490. [Google Scholar]
- Richards, D.H.; Eastmond, G.C.; Stewart, M.J. Anionically Prepared Telechelic Polymers. In Telechelic Polymers: Synthesis and Applications; Goethals, E.J., Ed.; CRC Press: Boca Rotan, FL, USA, 1989; p. 43. [Google Scholar]
- Duda, A.; Penczek, S. Thermodynamics of L-Lactide polymerization. Equilibium monomer concentration. Macromolecules 1990, 23, 1636–1639. [Google Scholar]
- Lebedev, B.V.; Mukhina, N.N.; Kulagina, T.G. Thermodynamics of Poly(dimethyldisiloxane) in the Range of 0–350 K. Vysokomol. Soed. Ser. A 1978, 20, 1297–1303. [Google Scholar]
- Morton, M.; Kammereck, R.F. Nucleophilic substitution at bivalent sulfur, reaction of alkyllithium with cyclic sulfides. J. Am. Chem. Soc. 1970, 92, 3217–3218. [Google Scholar]
- Richards, D.N.; Szwarc, M. Block polymers of ethylene oxide and its analogs with styrene. Trans. Faraday Soc. 1959, 55, 1644–1650. [Google Scholar]
- Boileau, S.; Champetier, G.; Sigwalt, P. Initiation mechanism for the polymerization of episulfides by sodium naphthalene. J. Polym. Sci. Polym. Symp. 1967, 16, 3021–3031. [Google Scholar]
- Boileau, S. Use of cryptates in anionic polymerization of heterocyclic compounds. ACS Symp. Ser. 1981, 166, 283–305. [Google Scholar]
- Deffieux, A.; Boileau, S. Anionic polymerization of heterocyclic compounds. Polymer 1977, 18, 1047–1050. [Google Scholar]
- Hofman, A.; Slomkowski, S.; Penczek, S. Structure and active centers and mechanism of the anionic polymerization of lactones. Makromol. Chem. 1984, 185, 91–101. [Google Scholar]
- Koinuma, H.; Naito, K.; Hirai, H. Anionic polymerization of oxiranes and cyclic siloxanes initiated with potassium salt-crown ether systems. Makromol. Chem. 1982, 183, 1383–1392. [Google Scholar]
- Sosnowski, S.; Slomkowski, S.; Penczek, S. Kinetics of ε-caprolactone polymerization and formation of cyclic monomers. Makromol. Chem. 1983, 184, 2159–2171. [Google Scholar]
- Morton, M.; Kammereck, R.F.; Fetters, L.J. Polymerization and block copolymerization of cyclic sulfides. Br. Polym. J. 1971, 3, 120–128. [Google Scholar]
- Stehlicek, J.; Labsky, J.; Sebenda, J. Alkaline polymerization of 6-caprolactam XXV. The effect of structure of the acyl on polymerization activated by acylcaprolactams or diacrylamines. Collect. Czech. Chem. Commun. 1987, 32, 545–557. [Google Scholar]
- Hamitou, A.; Ouhadi, T.; Jerome, R.; Teyssie, P. Soluble bimetallic μ-oxoalkoxides: VII. Characteristics and mechanism of ring-opening polymerization of lactones. J. Polym. Sci. Polym. Chem. 1977, 15, 865–873. [Google Scholar]
- Vion, J.M.; Jerome, R.; Teyssie, P.; Aubin, H.; Prud’homme, R. Synthesis and characterization and miscibility of caprolactone copolymers. Macromolecules 1986, 19, 1828–1838. [Google Scholar]
- Hofman, A.; Slomkowski, S.; Penczek, S. Polymerization of ε-caprolactone with kinetic suppression of macrocycles. Makromol. Chem. Rapid Commun. 1987, 8, 387–391. [Google Scholar]
- Duda, A.; Florjanczyk, Z.; Hofman, A.; Slomkowski, S.; Penczek, S. Living pseudoanionic polymerization of ε-caprolactone. Poly(caprolactone) free of cyclics and with controlled end groups. Macromolecules 1990, 23, 1640–1646. [Google Scholar]
- Inoue, S.; Tsukuma, I.; Kawaguchi, M.; Tsurata, T. Synthesis of optically active polymers by asymmetric catalysts VI. Behavior of organozinc catalyst systems in the stereoselective polymerization of propylene oxide. Makromol. Chem. 1967, 103, 151–163. [Google Scholar]
- Sosnowski, S.; Duda, A.; Slomkowski, S.; Penczek, S. Determination of the structure of active centers in the anionic polymerization by phosphorus-31 NMR, introducing a phosphorus containing end group. Makromol. Chem. Rapid. Commun. 1984, 5, 551–557. [Google Scholar] [CrossRef]
- Michalski, J.; Skowronska, A.; Bodalski, R. Mechanisms of Reactions of Phosphorus Compounds. In Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; Wiley VCH: New York, NY, USA, 1987; p. 255. [Google Scholar]
- Sebenda, J. Anionic Ring-Opening Polymerization: Lactams. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 511–530. [Google Scholar]
- Yu, G.-E.; Heatley, F.; Booth, C.; Blease, T.G. Anionic polymerization of propylene oxide; isomerization of allylic ether to propenyl ether end groups. J. Polym. Sci. A Polym. Chem. 1994, 32, 1131–1135. [Google Scholar]
- Kricheldorf, H.R. Anionic Ring-Opening Polymerization: N-Carboxyanhydrides. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 531–551. [Google Scholar]
- Duda, A.; Kowalski, A. Thermodynamics and Kinetics of Ring-Opening Polymerization. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulombier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; p. 8. [Google Scholar]
- Ackermann, J.; Damroth, V. Chemie und Technologie der Silikone II. Herstellung und Verwendung von Siliconpolymeren. Chemie in unserer Zeit 1989, 23, 86–99. [Google Scholar] [CrossRef]
- Choijnowski, J. Kinetically controlled ring-opening polymerization. J. Inorg. Organomet. Polym. 1991, 1, 299–323. [Google Scholar] [CrossRef]
- Boileau, S. Anionic Ring-Opening Polymerization: Epoxides and Episulfides. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 467–487. [Google Scholar]
- Anderson, A.N.; Merckling, N.G. Polymeric Bicyclo[2.2.1]hept-2-ene. U.S. Patent 2,721,189, 18 October 1955. [Google Scholar]
- Eleuterio, H.S. Polymerization of Cyclic Olefins. U.S. Patent 3,074,918, 22 January 1967. [Google Scholar]
- Ivin, K.J.; Mol, J.C. Metathesis and Metathesis Polymerization; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Buchmeiser, M.R. Homogeneous metathesis polymerization by well-defined group VI and Group VII transition metal alkylidenes: Fundamentals and applications in the preparation of advanced materials. Chem. Rev. 2000, 100, 1565–1604. [Google Scholar]
- Frenzel, U.; Müller, B.K.M.; Nuyken, O. Metathesis Polymerization of Cycloolefins. In Handbook of Polymer Synthesis, 2nd; Kricheldorf, H.R., Nuyken, O., Swift, G., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 381–426. [Google Scholar]
- Nuyken, O.; Schneider, M.; Frenzel, U. Metathesis Polymerization. In Encyclopedia of Polymer Science and Technology; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Lebedev, B.; Smirnova, N. Thermodynamics of cycloalkenes, of their bulk polymerization in the presence of metathesis catalysts and of polyalkenes. Macromol. Chem. Phys. 1994, 195, 35–63. [Google Scholar] [CrossRef]
- Cheridnichenko, V.M. The equilibrium polymerization of cyclic olefins. Polym. Sci. USSR 1978, 20, 1225–1233. [Google Scholar] [CrossRef]
- Andrews, J.M.; Jones, F.R.; Semlyen, J.A. Equilibrium ring concentration and the statistical conformations of polymeric chains. 12. Cycles in molten and solid Nylon-6. Polymer 1974, 15, 420–424. [Google Scholar] [CrossRef]
- Schrock, R.R. Multiple metal-carbon bonds for catalytic metathesis reactions (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef]
- Grubbs, R.H. Olefin metathesis catalysts for the preparation of molecules and materials (Nobel lecture). Angew. Chem. Int. Ed. 2006, 45, 3760–3765. [Google Scholar] [CrossRef]
- Chauvin, Y. Olefin metathesis: The early days (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3741–3747. [Google Scholar] [CrossRef]
- Schrock, R.H. On the trail of metathesis catalysts. Organomet. Chem. 1986, 300, 249–262. [Google Scholar]
- Schwab, P.; France, M.B.; Ziller, J.W.; Grubbs, R.H. A series of well-defined metathesis catalysts of [RuCl2(=CHR′)(PR3)2] and their reactions. Angew. Chem. Int. Ed. 1995, 34, 2039–2041. [Google Scholar]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Love, J.A.; Morgan, J.P.; Tmka, T.M.; Grubbs, R.H. A practical and highly active ruthenium-based catalysts that effects the cross metathesis of acrylonitrile. Angew. Chem. Int. Ed. 2002, 41, 4035–4037. [Google Scholar] [CrossRef]
- Kingsbury, J.; Harrity, J.; Bonnitatebus, P.; Hoveyda, A.H. A recycable Ru-based metathesis catalysts. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Halbach, T.S.; Mix, S.; Fischer, D.; Maechling, S.; Krause, J.O.; Sievers, C.; Blechert, S.; Nuyken, O.; Buchmeiser, M.R. Novel ruthenium-based metathesis catalysts containing electron withdrawing ligands, synthesis, immobilization and reactivity. J. Org. Chem. 2005, 70, 4687–4694. [Google Scholar] [CrossRef]
- Schrock, R.R. Ring-Opening Metathesis Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; p. 141. [Google Scholar]
- Trimmel, G.; Riegler, S.; Fuchs, G.; Slugovc, C.; Stelzer, F. Liquid crystalline polymers by metathesis polymerization. Adv. Polym. Sci. 2003, 176, 43–87. [Google Scholar]
- Herisson, J.L.; Chauvin, Y. Catalysis of olefin transformation by tungsten complexes, telomerization of cyclic olefins in the presence of acyclic olefins. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Grubbs, R.H. The development of functional group tolerant ROMP catalysts. J. Macromol. Sci. Chem. 1994, A31, 1829–1833. [Google Scholar]
- Krause, J.O.; Nuyken, O.; Wurst, K.; Buchmeiser, M.R. Synthesis and reactivity of homogeneous and heterogoneous ruthenium-based metathesis catalysts containing electron withdrawing ligands. Chem. Eur. J. 2004, 10, 777–784. [Google Scholar] [CrossRef]
- Hilf, S.; Kilbinger, A.F.M. Functional endgroups for polymers prepared using ring-opening metathesis polymerization. Nat. Chem. 2009, 1, 537–546. [Google Scholar] [CrossRef]
- Inoue, S.; Aida, T. Catalysts for Living and Immortal Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 197–215. [Google Scholar]
- Kobayashi, S. Ring-Opening Polymerization Involving Oxidation-Reduction-Processes. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 337–350. [Google Scholar]
- Inoue, S. Immortal polymerization: The outset, development, and application. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2861–2871. [Google Scholar] [CrossRef]
- Helou, M.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Ultraproductive, Zinc-Mediated, Immortal Ring-Opening Polymerization of Trimethylene Carbonate. Chem. Eur. J. 2008, 14, 8772–8775. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Liu, X.; Cui, D. Facile synthesis of pendant- and α,ω-chain-end- functionalized polycarbonates via immortal polymerization by using salan lutetium alkyl precursor. Chem. Commun. 2012, 48, 4588–4590. [Google Scholar] [CrossRef]
- Ajellal, N.; Carpentier, J.-F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trionov, A. Metal-catalyzed immortal ring-opening of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef]
- Guillaume, S.M.; Carpentier, J.-F. Recent advances in metallo/organo-catalyzed immortal ring-opening polymerization of cyclic carbonates. Catal. Sci. Technol. 2012, 2, 898–906. [Google Scholar] [CrossRef]
- Amgoune, A.; Thomas, C.M.; Carpentier, J.-F. Yttrium complexes as catalysts for living and immortal polymerization of lactide to highly heterotactic PLA. Macromol. Rapid Commun. 2007, 28, 693–697. [Google Scholar] [CrossRef]
- Endo, M.; Aida, T.; Inoue, S. Immortal Polymerization of epsilon-caprolactone, initiated by aluminum porphyrin in the presence of alcohol. Macromolecules 2002, 20, 2982–2988. [Google Scholar] [CrossRef]
- Xu, C.; Yu, I.; Mehrkhodavandi, P. Highly controlled immortal polymerization of β-butyrolactone by a dinuclear indium catalyst. Chem. Commun. 2012, 48, 6806–6808. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tokunoh, M.; Saegusa, T. Cationic ring-opening polymerization of 2-phenyl-1,3,2-dioxaphosphepane, a seven-membered cyclic phosphonite. Macromolecules 1986, 19, 466–469. [Google Scholar] [CrossRef]
- Kobayashi, S.; Saegusa, T. Alternating Copolymerization Involving Zwitterions. In Alternating Copolymers; Cowie, J.M.G., Ed.; Plenum: New York, NY, USA, 1985; pp. 189–239. [Google Scholar]
- Allcock, H.R. Ring-Opening Polymerization in Phosphazene Chemistry. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 217–237. [Google Scholar]
- Allcock, H.R. Current status of polyphosphazene chemistry. ACS Symp. Ser. 1988, 360, 250–267. [Google Scholar] [CrossRef]
- Allcock, H.R. Phosphorus-Nitrogen-Compounds; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- v.Liebig, J. Über Phosphorstickstoff. Ann. Chem. 1834, 11, 139. [Google Scholar]
- Stokes, H.N. Chloronitrides of phosphorus. Am. Chem. J. 1897, 19, 782–796. [Google Scholar] [CrossRef]
- D’Halluin, G.; de Jaeger, R.; Potin, P. Polydichlorophosphazenes: Synthèse à Partir De Cl3Pnp(O)Cl2. Bull. Soc. Chim. Belg. 1989, 98, 653–666. [Google Scholar]
- Matyjaszewski, K.; Cypryk, M.; Dauth, J.; Montague, R.; White, M. New synthetic routes towards polyphosphazenes. Makromol. Chem. Macromol. Sym. 1992, 54–55, 13–30. [Google Scholar]
- Matyjaszewski, K.; Franz, U.; Montague, R.A.; White, M.L. Synthesis of polyphosphazenes from phosphoranimines and phosphine azides. Polymer 1994, 35, 5005–5011. [Google Scholar] [CrossRef]
- Franz, U.; Nuyken, O.; Matyjaszewski, K. Synthesis and characterization of poly(phenyl-p-tolylphosphazene), prepared via in situ polymerization of phenyl-p-tolylphosphine azide. Macromol. Rapid Comm. 1994, 15, 169–174. [Google Scholar] [CrossRef]
- Allcock, H.R. Chemistry and Applications of Polyphosphazenes; Wiley-Interscience: New York, NY, USA, 2003. [Google Scholar]
- Neilson, R.H.; Wisian-Neilson, P. Poly(alkyl/arylphosphazenes) and their precursors. Chem. Rev. 1988, 88, 541–562. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kugel, R.L.; Valan, K.J. High molecular weight Poly(alkoxy and aryloxy-phosphazenes). Inorg. Chem. 1966, 5, 1709–1715. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kugel, R.L. High molecular weight Poly(diaminophosphazenes). Inorg. Chem. 1966, 5, 1716–1718. [Google Scholar] [CrossRef]
- Rose, S.H. Synthesis of phosphonitrilic fluoroelastomers. J. Polym. Sci. 1968, 6, 837–839. [Google Scholar] [CrossRef]
- Singler, R.E.; Schneider, N.S.; Hagnauer, G.L. Polyphosphazenes: Synthesis—Properties—Applications. Polym. Eng. Sci. 1975, 15, 321–338. [Google Scholar] [CrossRef]
- Kolich, C.H.; Klobucar, W.D.; Books, J.T. Process for surface treating phosphonitrilic fluoroelastomers. U.S. Patent 4,945,139 A, 31 July 1990. [Google Scholar]
- Tate, D.P. Polyphosphazene elastomers. J. Polym. Sci. 1974, 48, 33–45. [Google Scholar]
- Gettleman, L.; Farris, C.L.; Rawls, H.R.; LeBouef, R.J. Soft and firm denture liner for a composite denture and method of fabricating. U.S. Patent 4,432,730, 21 February 1984. [Google Scholar]
- Blonsky, P.M.; Shriver, D.F.; Austin, P.E.; Allcock, H.R. Polyphosphazene solid electrolytes. J. Am. Chem. Soc. 1984, 106, 6854–6855. [Google Scholar] [CrossRef]
- Allcock, H.R.; O’Connor, S.J.M.; Olmeijer, D.L.; Napierala, M.E.; Cameron, C.G. Cation complexation and conductivity in crown ether bearing polyphosphazenes. Macromolecules 1996, 23, 7544–7552. [Google Scholar]
- Fei, S.-T.; Allcock, H.R. Methoxyethoxyethoxyphosphazenes as Ionic Conductive Fire Retardant/Additives for Lithium Battery Systems. J. Power Source 2010, 19, 2082–2088. [Google Scholar]
- Tang, H.; Pintauro, P.N. Polyphosphazene membranes. IV. Polymer morphology and proton conductivity in sulfonated poly[bis(3-methylphenoxy)phosphazene] films. J. Appl. Polym. Sci. 2001, 79, 49–59. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kwon, S.; Riding, G.H.; Fitzpatrick, R.J.; Bennett, J.L. Hydrophilic Polyphosphazenes as Hydrogels: Radiation Crosslinking and Hydrogel Characteristics of Poly[bis(methoxyethoxyethoxy)phosphazene]. Biomaterials 1988, 9, 509–513. [Google Scholar] [CrossRef]
- Kim, J.; Chun, C.; Kim, B.; Hong, J.M.; Cho, J.-K.; Lee, S.H.; Song, S.-C. Thermosensitive/magnetic poly(organophosphazene) hydrogel as a long-term magnetic resonance contrast platform. Biomaterials 2012, 33, 218–224. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Turner, M.L.; Fitzpatrick, R.J. Poly(organophosphazenes) with Poly(alkyl ether) Side Groups: A Study of Their Water Solubility and the Swelling Characteristics of Their Hydrogels. Macromolecules 1992, 25, 5573–5577. [Google Scholar] [CrossRef]
- Allcock, H.R.; Fitzpatrick, R.J.; Visscher, K.B. Thin layer grafts of Poly[bis(methoxyethoxy-ethoxy)-phosphazene] on organic polymer surfaces. Chem. Mater. 1992, 4, 775–780. [Google Scholar] [CrossRef]
- Allcock, H.R.; Ambrosio, A.M. A. synthesis and characterization of pH-senstitive poly(organophosphazene) hydrogels. Biomaterials 1996, 17, 2295–2302. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[amino acid ester)phosphazenes] as substrates for the controlled release of small molecules. Macromolecules 1994, 6, 516–524. [Google Scholar]
- Deng, M.; Kumbar, S.G.; Wan, Y.; Toti, U.S.; Allcock, H.R.; Laurencin, C.T. Polyphosphazene polymers for tissue engineering: An analysis of material synthesis, characterization, and applications. Soft Matter 2010, 6, 3119–3132. [Google Scholar] [CrossRef]
- Deng, M.; Kumbar, S.G.; Nair, L.S.; Arlin, L.; Weikel, A.L.; Allcock, H.R.; Laurencin, C.T. Biomimetic structures: Biological implications of dipeptide-substituted polyphosphazene–polyester blend nanofiber matrices for load-bearing bone regeneration. Adv. Funct. Mater. 2011, 21, 2641–2651. [Google Scholar] [CrossRef]
- Allcock, H.R.; Morozowich, N. Bioerodible polyphosphazenes and their medical potential. Polym. Chem. 2012, 3, 578–590. [Google Scholar] [CrossRef]
- Manners, I.; Renner, G.; Nuyken, O.; Allcock, H.R. Poly(carbophosphazenes): A new class of inorganic-organic macromolecules. J. Am. Chem. Soc. 1989, 111, 5478–5480. [Google Scholar] [CrossRef]
- Allcock, H.R.; Coley, S.M.; Manners, I.; Renner, G.; Nuyken, O. Poly[(aryloxy)carbophosphazenes]: Synthesis, properties, and thermal transition behavior. Macromolecules 1991, 24, 2024–2028. [Google Scholar] [CrossRef]
- Dodge, J.A.; Manners, I.; Allcock, H.R.; Renner, G.; Nuyken, O. Poly(thiophospazenes): New inorganic macromolecules with backbone composed of phosphorus, nitrogen and sulfur atoms. J. Am. Chem. Soc. 1990, 112, 1268–1269. [Google Scholar] [CrossRef]
- Manners, I.; Riding, G.H.; Dodge, J.A.; Allcock, H.R. Role of ring strain and steric hindrance in a new method of the synthesis of macrocycles and high polymer phosphazenes. J. Am. Chem. Soc. 1989, 111, 3067–3069. [Google Scholar] [CrossRef]
- Voit, B. New developments in hyperbranched polymers. J. Polym. Sci. Polym. Chem. 2000, 38, 2505–2525. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Nuyken, O.; Pask, S.D. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 361-403. https://doi.org/10.3390/polym5020361
AMA Style
Nuyken O, Pask SD. Ring-Opening Polymerization—An Introductory Review. Polymers. 2013; 5(2):361-403. https://doi.org/10.3390/polym5020361
Chicago/Turabian StyleNuyken, Oskar, and Stephen D. Pask. 2013. "Ring-Opening Polymerization—An Introductory Review" Polymers 5, no. 2: 361-403. https://doi.org/10.3390/polym5020361