Bio-Decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics


Abstract
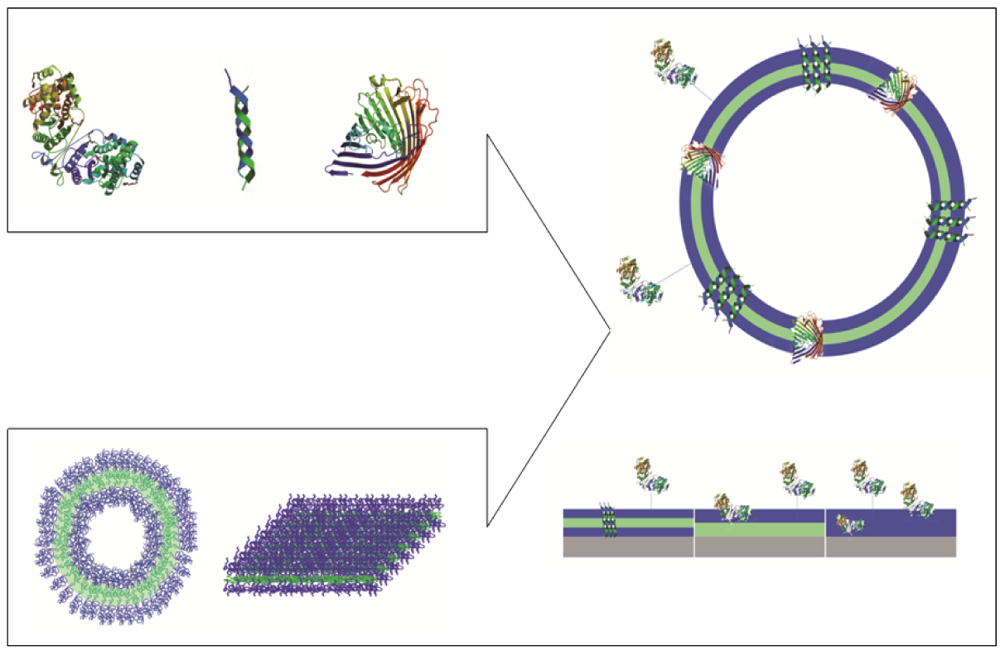
: Today, demand exists for new systems that can meet the challenges of identifying biological entities rapidly and specifically in diagnostics, developing stable and multifunctional membranes, and engineering devices at the nanometer scale. In this respect, bio-decorated membranes combine the specificity and efficacy of biological entities, such as peptides, proteins, and DNA, with stability and the opportunity to chemically tailor the properties of polymeric membranes. A smart strategy that serves to fulfill biological criteria is required, whereby polymer membranes come to mimic biological membranes and do not disturb but rather enhance the functioning and activity of a biological entity. Different approaches have been developed, exemplified by either planar or vesicular membranes, allowing insertion inside the polymer membrane or anchoring via functionalization of the membrane surface. Inspired by nature, but incorporating the strength provided by chemical design, bio-decorated polymer membranes represent a novel concept with great potential in diagnostics and therapeutics.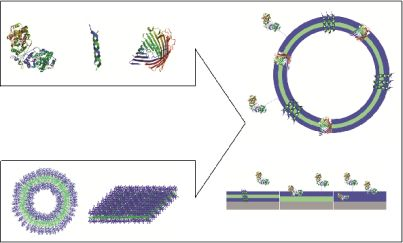
1. Introduction
In nature, membranes play important roles such as separating cell compartments, adsorbing molecules, transporting nutrients and relaying information, catalyzing substrates at surfaces, stabilizing cells, and recognition of molecules/other cells, just to name a few. The cell membrane has a thickness of several nanometers and contains a wide variety of biological compounds including proteins, sterols, sugars, and lipids. The selective lipid bilayer permits molecules and ions to traverse the cell membrane, while larger molecules can trigger structural and conformational changes in the membrane itself. Information can be relayed via membrane proteins inserted in the cell membrane. In order to understand and to simulate natural conditions and interactions to an acceptable degree, it was first necessary to use lipidic membranes as models of cell membrane complexity, both in terms of structure and function.
Lipid membranes, used to serve as simpler models of biological membranes, have been extensively reviewed, and therefore we will not insist on further review here, but we will draw upon the necessary studies to indicate the differences between lipidic and synthetic membranes when designing synthetic membranes [1,2]. For example, lipids such as diacyl-glycero phosphatidylcholine (DCPC) have been used extensively to mimic biomembranes because their bilayer structure is similar to that of cell membranes. Various reviews present the incorporation or/and the attachment of biological molecules to lipidic membranes [3,4], even though the exact mechanism of incorporation is not yet fully understood [5]. However, relative membrane instability, problems with reproducibility, and difficulties in carrying out chemical modification to lipid structures led to the search for another class of materials: the polymers.
Recently, copolymers have been introduced as platforms to mimic biological membranes for the purpose of coping with the complex challenges of reconstituting pores in membrane proteins, immobilizing enzymes for sensing technology, and to serve as templates for other biomolecules such as DNA. In this review we will concentrate on polymeric membranes and we will explain how they can be decorated with biomolecules and their applications. Such polymer membranes can be obtained either as planar membranes formed on support surfaces via polymerization, grafting or spreading, or as vesicular structures formed by self-assembly if the copolymers contain the appropriate hydrophobic-hydrophilic domains. A biological entity can be added to the system before, during or after the formation of the polymer membrane. As formulated by Ahuja et al. [6], optimum immobilization of biomolecules on a conductive polymeric surface must include: i. stable, efficient immobilization; ii. a procedure that does not disrupt the biomolecule; iii. selection of biomolecules that do not affect the properties of the polymer; and iv. allowance of direct interaction of substrates with the biomolecule in cases where it is catalytically active. As proposed for biosensors, theses requirements can be generally applied to bio-polymer hybrid systems, taking into account the specificity of the biomolecule as well as the synthetic polymer system. However, they represent serious limitations in choosing the chemical nature of polymeric systems, the dissolving solvents, and the environmental conditions for the biological molecule to preserve its native structure and function. Matching a totally synthetic system (polymer membrane) to a biological entity would imply a more complex scenario than is the case with lipidic membranes. However, the advantages of using polymeric membranes to achieve, for example, higher stability or possible chemical functionalization represent a firm base on which to develop these new hybrid materials, as we will explain in our review. Here, we will not present the means to obtain protein- and peptide-polymer conjugates, as these have been reviewed recently [7].
2. Polymeric Membranes
Different aspects of block copolymer membranes have recently been summarized in various reviews that provide a good overview of the current state of the art [8-12]. Therefore, in the following, we will primarily focus on selected aspects and newer developments related to bio-decorated membranes.
As a result of higher molar mass, copolymers frequently form larger aggregates than lipids, with lower dynamics. For example, membrane structures derived from amphiphilic block copolymers may be up to 10-times thicker than those of natural bilayer-forming phospholipids. The polymer membranes are significantly more stable to lysis by classical surfactants than are liposomes, due to the lower entropy that exists after mixing the polymers. In addition, it is possible to choose copolymers that are biocompatible in order to maintain low immunogenicity [13,14].
The mechanical properties of block copolymer membranes are primarily controlled by their hydrophobic domains and their ‘phase behavior’ (e.g., fluid or glassy). In addition, the vast number of available blocks make it possible to tune membrane properties, such as membrane thickness, polarity, toxicity or sensor-responsivity [15,16]. The membrane thickness plays an important role [17] in membrane-crossing biomolecules. Compared to lipids, which do not allow a significant change in bilayer thickness [2], polymers are flexible to the extent that, when a biomolecule is inserted, a thinner membrane can form in that specific area, allowing for proper biomolecule functioning.
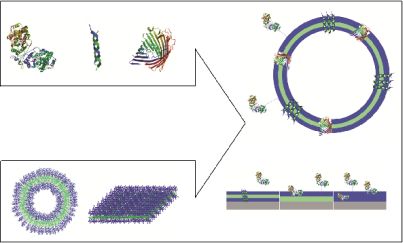
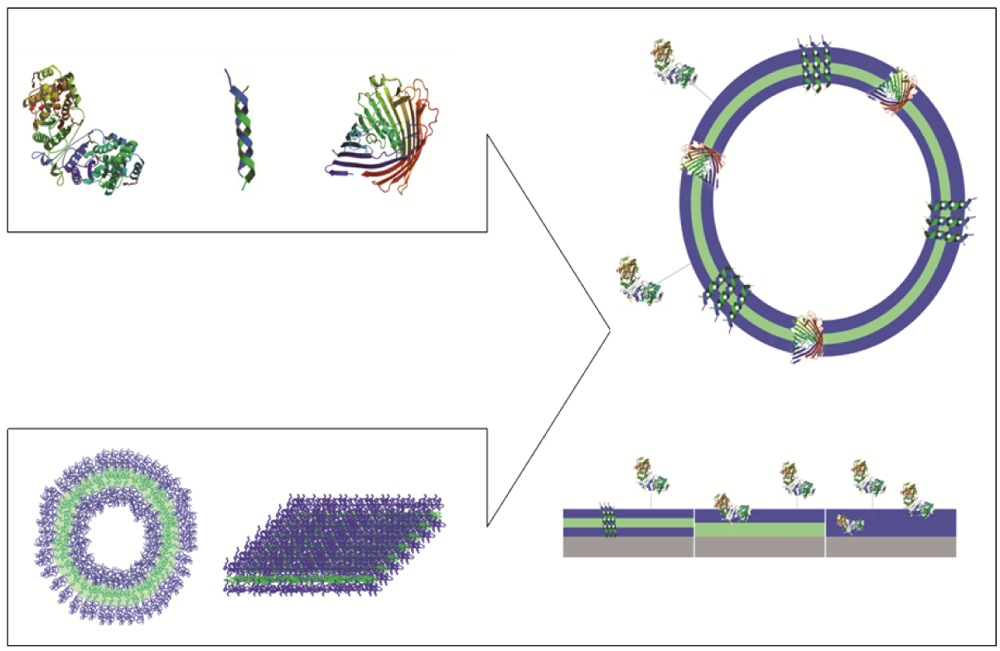
Polymeric membranes that serve as domains to accommodate biological molecules facilitate two possible approaches: i. immobilization of biomolecules at the surface or inside the membrane without perforation; and ii. trans-membrane insertion of membrane proteins or biological pores (Figure 1).
A variety of copolymers have been used to design bio-polymer hybrid membranes (Table 1). Polymer membranes that are used to immobilize or incorporate biomolecules should advantageously feature properties that facilitate the interaction/structure and function of a biological moiety, as well as the intended application. To reconstitute biomolecules in a copolymer membrane, it should mimic the bilayer structure of a normal cell membrane. Therefore, only block copolymers can be used to develop a polymeric membrane, as they have hydrophobic and hydrophilic blocks that will allow the generation of a bilayer similar to lipids [18]. In an aqueous solution, as is relevant for biological molecules, the hydrophobic region is on the inside, whereas the hydrophilic region faces outward. Depending of the specificity of the biomolecule (charge, hydrophilicity, solubility), it should be possible to anchor it to the surface of the hydrophilic domain, or to insert it in the hydrophobic domain. When a polymer membrane is formed on any surface it is not essential to have two different domains; a hydrophilic region alone is sufficient to serve for the immobilization of biomolecules.
The immobilization of the biomolecule on, or in the polymeric membrane, can be performed either before or after the polymer matrix is created. However, the preferred way is to add biomolecules on/into the polymer membrane afterwards, due to the fragility of the biomolecules under conditions that are different from those in nature [19].
It has been shown that amphiphilic triblock copolymers self-assemble into vesicular structures whose membranes are similar to lipidic membranes [13]. Meier and coworkers developed various methods to prepare nanovesicles of triblock copolymers poly(2-methyl-2-oxazoline)-block-poly(dimethyl siloxane)-block-poly(2-methyl-2-oxazoline) (PMOXA-PDMS-PMOXA), and were able to reconstitute membrane proteins [32] such as OmpF (outer membrane protein F) [34] and FhuA [35]. Another approach is to consider enzymes as hydrophilic blocks and, together with a hydrophobic polymer domain, these generate a bilayer [39].
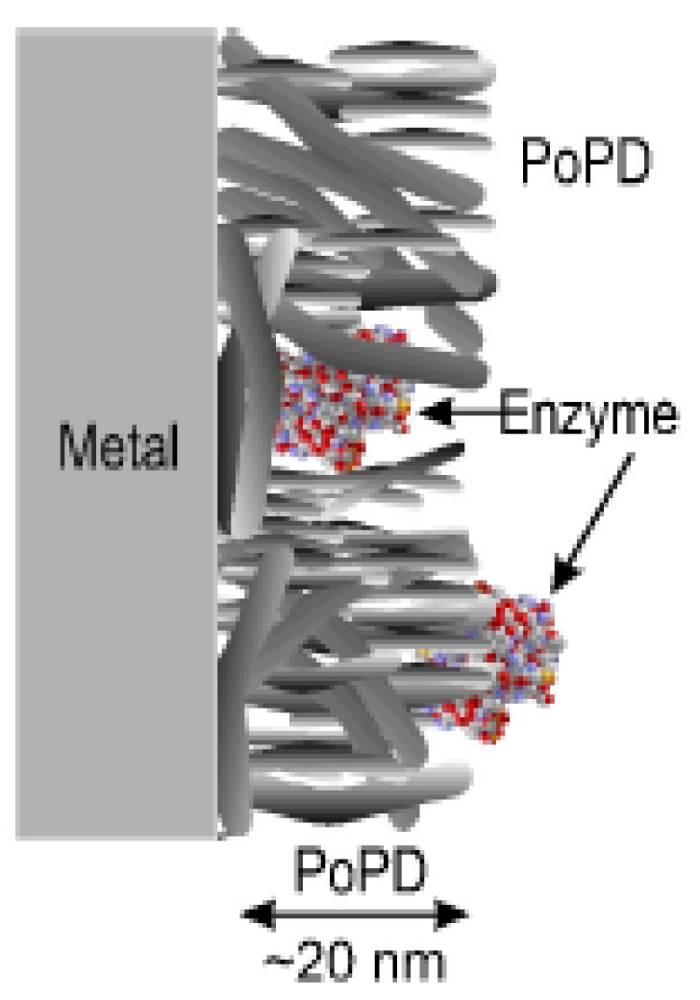
Diblock copolymers have also been used to immobilize enzymes, as in the case of a poly(hydroxyethylmethacrylate) grafted-poly(glycidylmethacrylate) p(HEMA-g-GMA) membrane with laccase inserted [40], or a poly(ethylene oxide)- poly(ether esters) membrane interacting with alamethicin [31]. There are several examples of polymeric membranes on solid surfaces in which polymer films have been used instead of block copolymers: poly(ortho-phenylenediamine) (PoPD) [19], poly(acrylic acid) [20], poly(N-methyl pyrrole) [21], poly 3,4-ethylenedioxythiophene [22]. The ways in which the immobilization was performed was based on entrapping the biomolecule in the polymer membrane, adsorption by hydrophobic or ionic interactions, or covalent binding to the polymer membrane. The latter is generally thought to be the strongest means for immobilization, but has the drawback of potentially inducing a loss of biological activity in the biomolecule [41].
3. Immobilization of Biomolecules on Polymer Membranes
Immobilization of biomolecules can be achieved by attachment at the surface of polymer membranes, by insertion within the membrane, or by a combination of attachment and insertion, depending on the intended application. Two directions have been developed to immobilize biomolecules on polymeric membranes: i. generation of polymeric vesicles whose membranes serve as insertion domains; and ii. formation of polymeric membranes by grafting, spreading or growing on a solid surface.
Delaittre et al. created vesicles in which an enzyme represents the hydrophilic domain, while the polymer represents the hydrophobic domain of the bilayer structure [39]. The polymer (polystyrene or poly(methyl methacrylate)) was attached by click chemistry to the azido-derived cofactor of the horseradish peroxidise (HRP) enzyme. Enzyme-polymer hybrids have generated giant amphiphilic vesicles by self-assembly, and these were able to function as nanoreactors when a second enzyme was encapsulated in their aqueous cavities; the first enzyme, HRP, was able to support a cascade reaction in which the second enzyme was involved, due to its role as the hydrophilic domain of the polymer membrane. These kinds of giant amphiphiles, in which an enzyme serves as a hydrophilic domain, have also been obtained by making use of the biotin-streptavidin coupling, or by covalent coupling of the polymer to a specific location on the surface of the protein [42].
A step beyond this, occurred when enzyme-containing nanoreactors were attached to a modified surface via an anchoring effect due to biomolecules. The strong interaction of biotin-functionalized vesicles with streptavidin served to immobilize the entire assembly on a solid surface. PMOXA-PDMS-PMOXA block copolymers were partially end-functionalized with biotin molecules that were exposed at the surface after vesicle formation. Interaction with streptavidin served to immobilize the vesicles on the surface, an advance that could lead to the development of a biosensor [23]. Another approach was to functionalize the surface of polymeric vesicles with complexing agents, for example nitrilotriacetic acid, and to use these agents to coordinate metal ions that serve to bind His-tag proteins. Polybutadiene-block-poly(ethylene oxide) functionalized with nitrilotriacetic acid or tris(nitrotiacid acid) were able to expose copper(II) or nickel(II) at the surface after vesicles formation. Various His-tag proteins, such as red fluorescent protein, His-tagged green fluorescent protein, and maltose binding proteins have been linked via metal coordination points [24]. Both biotin-streptavidin and metal-His-tag protein couplings are examples of molecular recognition, which represents the most specialized interactions in biological systems. In addition, Ni-nitrilotriacetic acid (Ni-NTA)– functionalized polybutadiene-block-poly(ethylene oxide) formed monolayers that were able to bind His-tags proteins on a solid surface [43]. Small-angle X-ray scattering indicated an increase in membrane thickness upon protein binding, while atomic force microscopy provided information on proteins attached to these selectively immobilized membrane surfaces. These types of metal-functionalized polymer surfaces are of great relevance in 2D protein crystallization and in biosensor technology.
When a polymeric membrane deposited on a surface is enhanced with a biomolecule, so-called “activated surfaces” are generally created. In these cases the biomolecule is chemically bonded, electrostatically attached, or inserted in the polymer membrane. An interesting situation is created when enzymes are used to generate a catalytically active surface. The rapid, specific catalytic activity of enzymes on such a surface permits a highly specialized informational reply from the surface. When the enzyme is embedded in the polymer membrane, it is important to preserve its native structure/function and to allow the diffusion of specific substrates into the polymeric membrane. These enzyme-polymer hybrid materials are of great interest to technology and medical applications [6] because they serve as very rapid, extremely selective detectors. Such detectors can be used for diagnostic purposes, for example.
In the newest generation of biosensors, the product, which is created by the enzymatic conversion, is directly recognized by the polymer, which transfers the signal to an electronic device. An efficient biosensor was developed by immobilizing an enzyme, glucose oxidase (GOx), on a permselective membrane of poly(ortho-phenylenediamine) [19]. An optimal biosensor for glucose was achieved by co-immobilizing 1 mg mL−1 GOx with 300 mM oPD dissolved in distilled water, a process not previously reported for biosensor fabrication. This co-immobilization represents a significant improvement in the functioning of sensors as compared to the old generation of biosensors, which required the diffusion of the reaction products to the electronic device in order to create a signal. Since the electronic device requires a certain threshold for detection, diffusion is not an optimal solution to transfer a signal from the enzyme to the electrode, due to the delay in registering the products and their low probability of arriving at the electrode. In this respect, conductive polymers can help improve the collection of the reaction products and therefore create a more detectable signal in the biosensor [6]. It may also be possible to use conformational changes, as done by enzymes, to improve the sensitivity of enzyme-polymer biosensors. Changes in conformation were observed by Förster resonance energy transfer (FRET) microscopy [44]. Depending on the resolution of the FRET microscopy, even single enzymatic conversion can be detected, which allows direct follow-up of the reaction without the requirement of monitoring the diffusion of the reaction products.
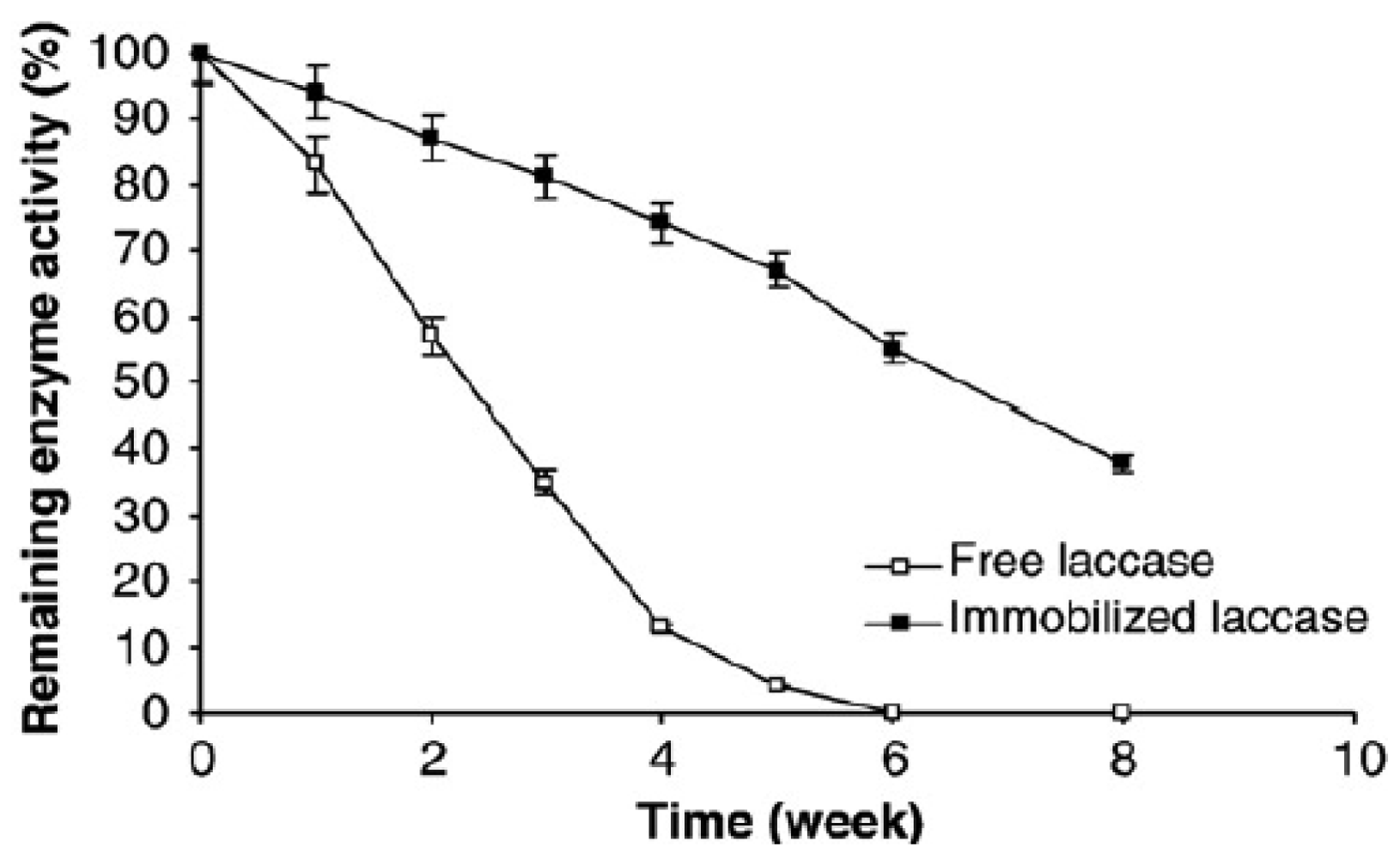
Besides sensitivity, the life-time of a hybrid material is of great importance in technological or medical applications. In the case of enzyme-based hybrid materials, the life-time can be ascertained by enzymatic activity. In the case of laccase immobilized on p(HEMA-g-GMA) copolymer films, the activity of the enzyme is preserved for a longer period of time as compared to free enzyme (Figure 2) [40]. The increased enzyme stability in a hybrid material (high activity recovery of about 71% compared to free enzyme) is explained by multipoint interactions of the polycationic hydrophilic fibrous polymers that serve to keep the enzyme in a stable conformation and protect it from attack by proteases when adsorbed on polymeric membranes. Up to 81% removal of p-chlorophenol by the active hybrid surface has been achieved, and therefore hybrid material consisting of immobilized laccase on polymeric films was proposed for application in the treatment of waste water that contains phenols.
Reversible adsorption of GOx on a polyacrylonitrile/polyaniline-4, PAN/PANI-4 copolymer membrane via interactions between the enzyme and the polymer membrane has been reported [25]. This reversible adsorption was modulated by changing the pH of the solution, which allowed reloading of the polymer membrane with new enzymes. The immobilization of GOx on this synthetic membrane resulted in an increase in thermal and storage stability compared to its free counterpart. The enzyme adsorption approach is simpler and less expensive than the covalent enzyme binding approach, while retaining high catalytic activity and providing the possibility of reusing the support after the inactivation of the immobilized enzyme. However, enzyme adsorption on the polymer membrane has the drawback of the possible unintended release of enzyme, which would decrease the activity of the surface.
Poly 1,2-diaminobenzene has been reported as an efficient polymer membrane to immobilize various species of oxidases, such as glucose oxidase, lactate oxidase or L-amino acid oxidase, and to develop highly sensitive, stabile and interference-free biosensors for hydrogen peroxide [26]. A possible extension of the system would be to design a surface that supports a cascade reaction mediated by immobilized enzymes (assuming that the product of the first enzyme reaction can be converted by the second enzyme). The concept of a cascade reaction at the polymer surface could allow highly sensitive, rapid detection of the substrates of the second enzyme, which might otherwise be difficult to observe directly. However, to the best of our knowledge there is no reported system involving cascade reactions mediated by enzymes immobilized on surfaces.
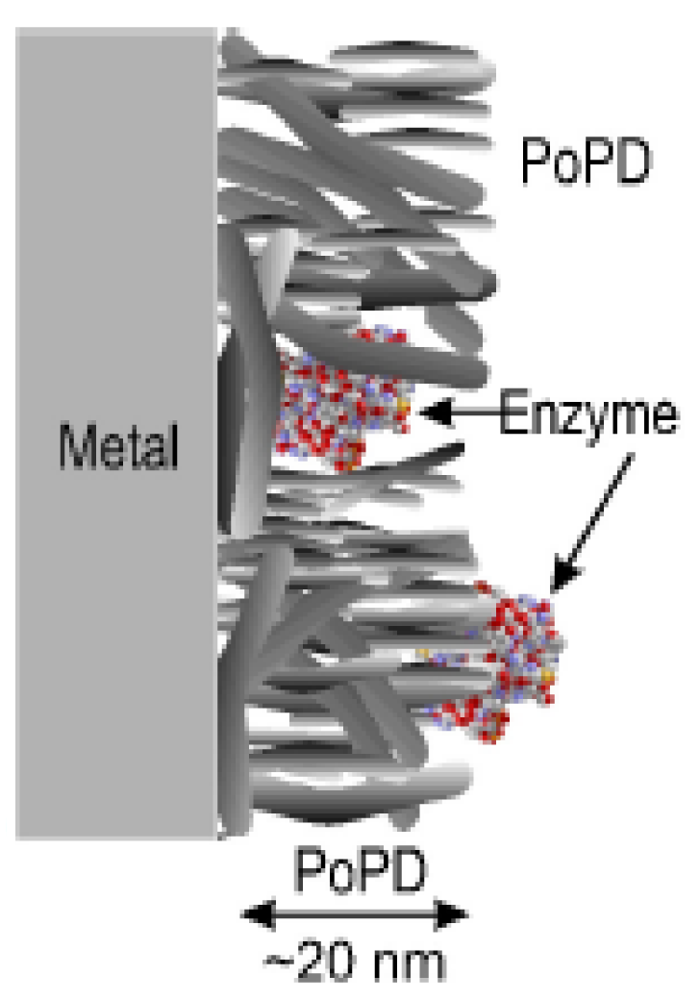
During the immobilization process, not all of the initially added enzymes are attached to the surface and this affects the efficiency of the system. Different immobilization strategies for GOx on PoPD membranes were tested by Rothwell et al. GOx was immobilized on the membrane before the polymerization, during the polymerization, and after the polymerization by dip-evaporation (Figure 3). The best enzyme immobilization was obtained during the polymerization: The enzyme was physically trapped inside a polymer matrix that allowed the substrate to diffuse to the location of the enzyme.
An example of a “purification surface” using ribonuclease A (RNase A) immobilization on a silicon-grafted-poly(acrylic acid) polymer membrane has been reported. This system was developed for the removal of RNA in DNA experiments [20]. RNase A, as a one-unit enzyme, cleaves single-stranded RNA. If the RNase is immobilized, a purification step is eliminated because the enzyme is present only on the polymer surface.
Other types of biomolecules, such as antibodies [27] or DNA [28], were also successfully immobilized on a polymer membrane with the aim of developing highly active surfaces. A rabbit anti-goat antibody was successfully covalently immobilized on a nylon-grafted-glycidyl methacrylate polymer. Nylon itself is a biocompatible, economical, and easily modifiable surface, but grafting with GMA makes the immobilization of biomolecules more stable and efficient. In this respect the use of antigen-antibody interactions represents a very sensitive approach allowing for the development of responsive, activated surfaces with a very specific reply that mimics natural responsiveness.
Molecular recognition based on a DNA hybridization process represents another way to apply nature-specific interactions to the immobilization of biological molecules on surfaces. An aminopropyltritoxysilane-glutaraldehye surface functionalized with single-stranded DNA was able to recognize the complementary strand, which allowed for the development of a highly specific active surface with possible applications in medical domains [28]. Single-stranded nucleotides retain their coil conformation, whereas in the double-stranded form, the helical conformation is preferred. If other molecules are linked to the DNA strand, they can be detected with the help of the complementary strand.
The release of immobilized biomolecules, such as keratinocyte growth factor protein (KGF) that is involved in biological processes including growth and repair of epithelia, from polymer membranes can facilitate wound healing. The release of KGF in a biologically relevant concentration was obtained by immobilizing it in a mixture of (lactic-co-glycolic acid) and poly(l-lactic acid) polymers [45]. The protein is released as a consequence of polymer degradation due to non-enzymatic hydrolysis [46].
4. Biopores in Polymeric Membrane
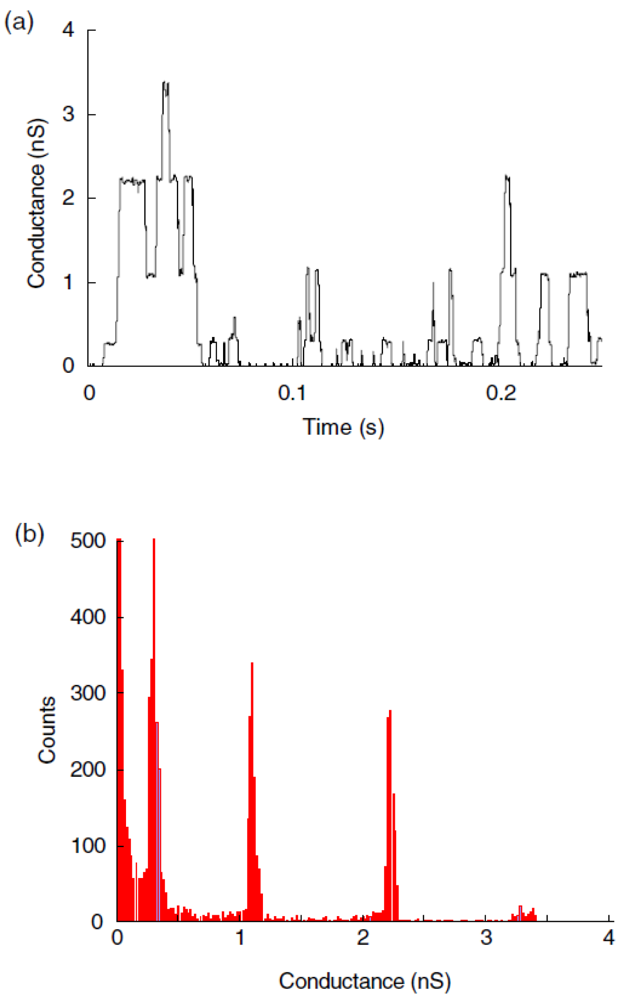
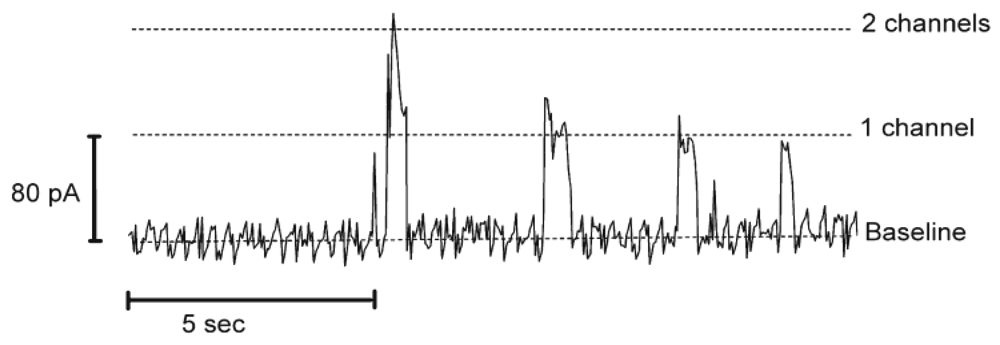
Control of stability and permeability in biological membranes is essential for proper cell functioning. The selective and controlled permeability of a cell membrane, which connects the inside of a cell to the outside, represents a key factor in the proper operation of sensing and signaling pathways. Thus, the investigation of interactions between biologically active molecules and membranes is a highly interesting field of research. In order to simplify the components of a biomimetic membrane, amphiphilic block-copolymers in combination with pore-forming peptides were proposed. As this is an emerging field, the use of a synthetic membrane with peptide-based biopores for transport of ions has been reported, but at the model stage only. The advantages of biopores based on peptides is that they are easier to manipulate and to understand compared to complex membrane proteins, while retaining the ability to transport small molecules, and thus to develop applications based on signaling or sensing processes. In addition, they offer a simple model for studying biopore formation with respect to the interactions/stabilization of the pore in a completely synthetic environment, such as a polymer membrane. If a polymer membrane has a defined thickness and represents the shell of a polymer compartment, such as a vesicular structure, it is also possible to incorporate biopores that serve to transfer signals across the membrane. It has been shown that transmembrane pores regulate ion permeability in cell membranes, and these ion pores then incorporated themselves in polymer membranes [29-31]. The ion channels can be mechanically gated, voltage-gated or pH-gated, depending on the desired application. One of the best known ion-channels is gramicidin, which is used as a model to study membrane organization, dynamics and functioning of membrane-spanning channels [47-49]. Gramicidin is a linear pentadecapeptide antibiotic produced by Bacillus brevis and consisting of alternating L- and D-amino acids with a molecular weight of ∼1,900 Da [50]. In membranes, gramicidin assembles into dimeric channels with a hydrophobic length of 22 Å [51]. The channel pore diameter is about 4 Å and it allows the passage of monovalent cations [49] Gramicidin was incorporated in functional form in a PMOXA7-PDMS60-PMOXA7 membrane, as proved by conductivity experiments (Figure 4), although the membrane thickness was larger than the hydrophobic length of the gramicidin dimmer [29]. As a concept, this system has great potential in protein screening diagnostics.
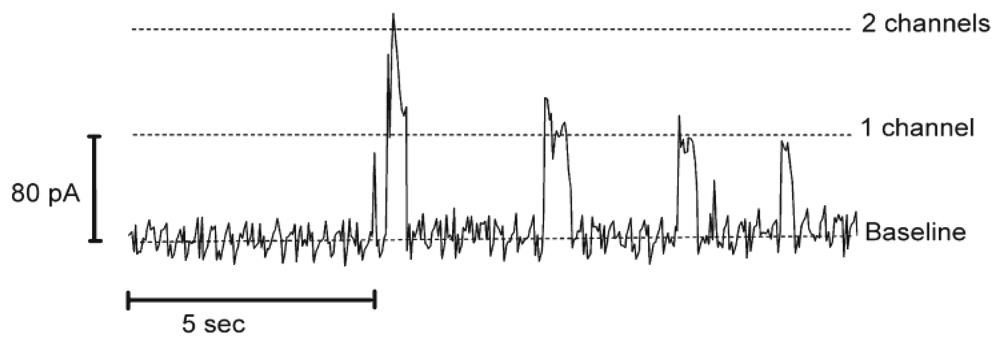
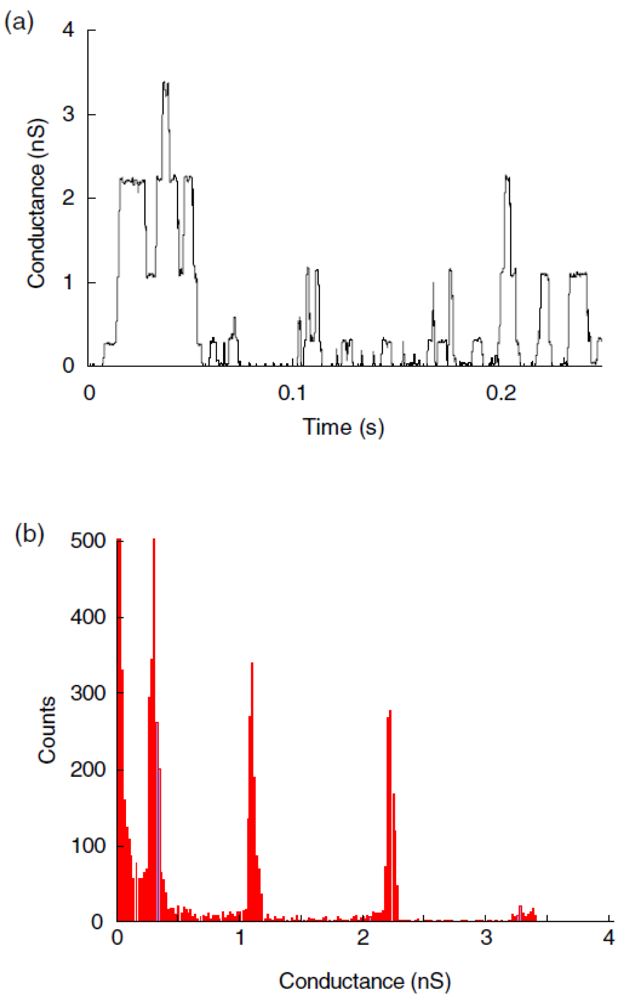
Another example of a biopore is alamethicin, which was inserted into polymeric membranes by aggregation of monomers. Alamethicin, consisting of 20 amino acids, was isolated from the fungus Trichoderma viride and, based on its water solubility, it was possible to insert it into lipid membranes spontaneously [52]. By varying the oligomer number and size it was shown that alamethicin forms voltage-dependent ion conducting pores [53]. The same behavior was observed when alamethicin was incorporated into a polymer membrane, which indicates that the polymer sufficiently mimics a lipid bilayer membrane, providing the essential environment for biopore insertion [30,54]. Alamethicin was also incorporated in a PMOXA12-PDMS54-PMOXA12 tribolock copolymer membrane, as proved by the existence of multiple levels of conductance [55] (Figure 5).
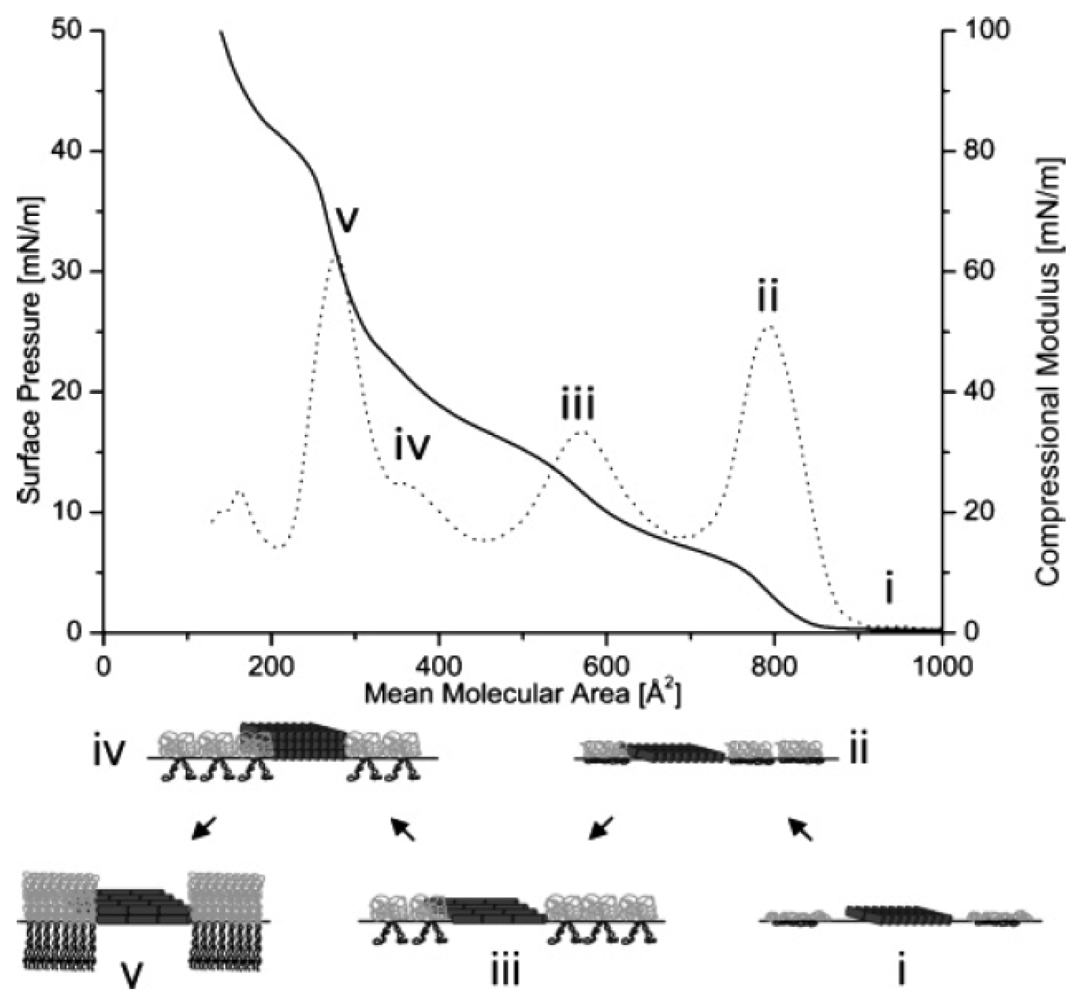
Although the peptide was much smaller than the thickness of the polymer bilayer, alamethicin was able to rupture it [31]. A detailed phase behavior study was carried out by Haefele et al. [54], showing the influence of alamethicin on the ABA triblock-copolymer membrane at an air-water interface (Figure 6). Making use of the advantages of a copolymer membrane, in combination with the voltage-gated properties of alamethicin, holds great promise in terms of applying voltage-gated channels to targeted drug therapeutics.
The nicotinic acetylcholine receptor (nAChR), a ligand-gated ion channel, was incorporated into a lipid/polymer matrix by Salzer et al. and represents a different approach compared to that of using only polymeric membranes [56]. The nicotinic acetylcholine receptor consists of pentameric subunits with two binding sites for acetylcholine. Upon binding of acetylcholine, the nAChR changes its internal configuration, which opens the internal pore (having an apparent entrance diameter of 1 nm) and it becomes selective for Na+, K+ and other small molecules. Functionality was maintained after insertion of nAChR into the lipid-polymer matrix. The concept of using biopores as channels for polymer membranes results in significant mechanical and thermal stability, making the hybrids ideally suited for advanced drug diagnostics.
Polymyxin, a peptide, known to make cell membranes of bacteria permeable, was used to interact with a planar block copolymer bilayer, and succeeded in modifying its transport-related properties, as shown by a decrease in the electrochemical resistance of the membrane [57]. This decrease shows that both peptides interacted with the membrane and were able to increase the permeability in a manner similar to a cell membrane.
The development of other gated channels, such as pH-gated ion channels, in polymeric membranes has still not been investigated, although it has been shown, for example, that a pH-dependent gating process can be implemented in a lipid bilayer [58]. This leads to the assumption that, by using a polymer membrane, the applicability of such a system can be expanded to therapeutics. Biopores incorporated in polymeric membranes represents an emerging domain with great potential to develop future therapeutics and in vivo diagnostic tools. There are plenty of combinations possible using different block-copolymers and peptides, and these are likely to solve specific problems in drug delivery and therapeutics. Because peptides combined with polymers form a convenient system with which to work, various systems can be designed to target or deliver high antimicrobial peptide concentrations, or to act as specific antimicrobial cell surfaces.
5. Reconstituting Membrane Proteins in Polymeric Membranes
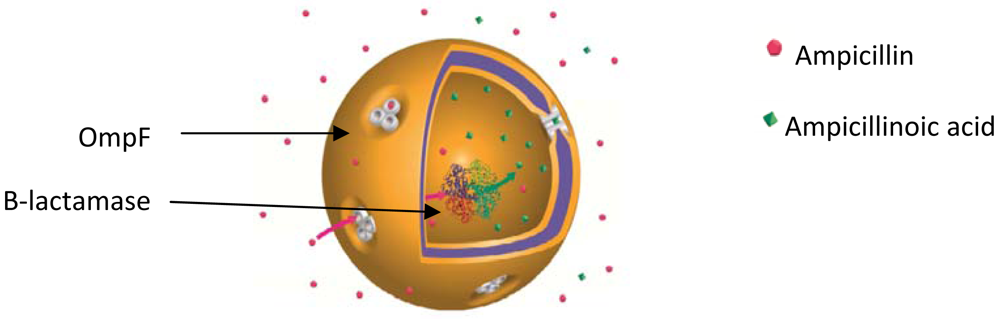
Larger pores, specifically their protein membrane channels as taken from biological membranes, were reconstituted by Meier and coworkers for the first time in the polymeric PMOXA-PDMS-PMOXA membrane [32]. The PMOXA-PDMS-PMOXA membrane is impermeable to small molecules, including water [36], and its permeability was increased by reconstituting channel proteins, allowing the transport of various molecules through the polymeric membrane (Figure 7).
Similarly, other membrane proteins have been successfully reconstituted in PMOXA-PDMS-PMOXA membranes: LamB, FhuA, Tsx, AqpZ, Complex I [34,36,37,59], as shown by the movement of different molecules (water, NADH, nucleotides, DNA, enzyme substrates) through the polymeric membrane. F0F1-ATP synthase and bacteriorhodopsin reconstituted in poly(2-ethyl-2-oxazoline)-b-poly(dimethylsiloxane)-b-poly(2-ethyl-2-oxazoline) (PEtOz-PDMS-PEtOz) polymeric vesicles are further examples of transmembrane proteins that retained their activity after reconstitution in polymer membranes [38]. For an overview, please see Table 1.
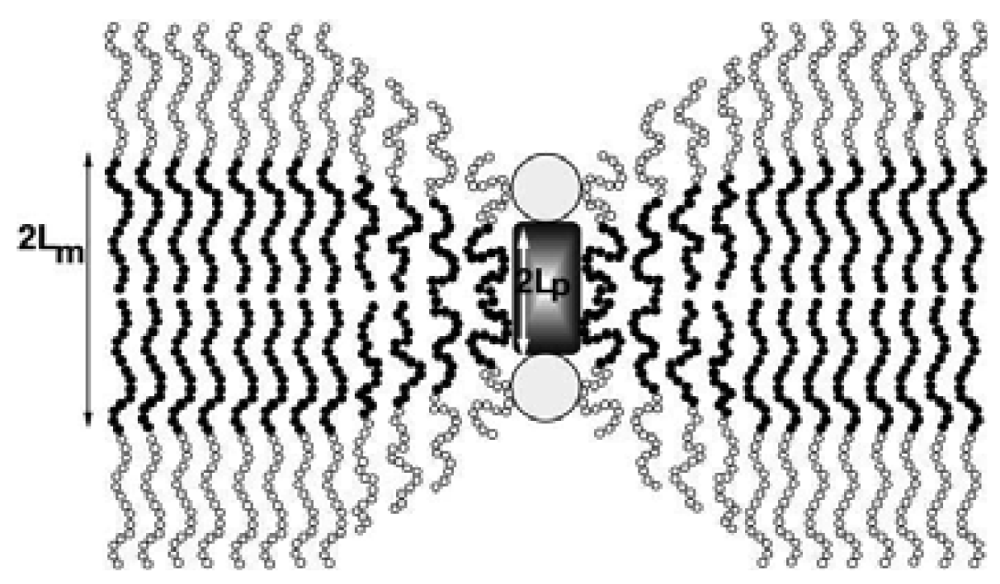
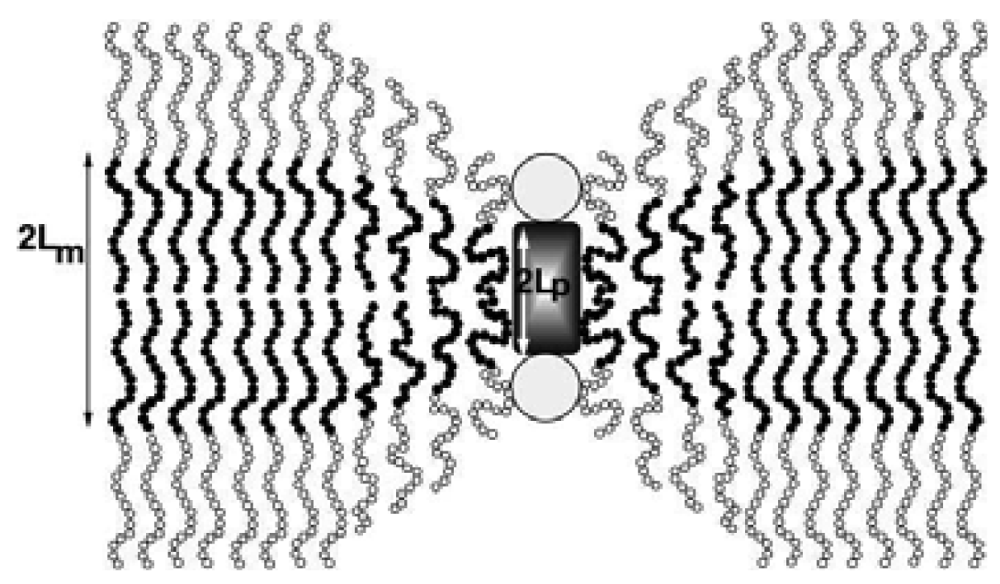
Incorporation of membrane proteins in liposomes has been extensively studied [60] and the interactions between the proteins and the membrane have been correlated with the properties of the membranes [61]. On the other hand, reconstitution of membrane proteins in polymer membranes is far from being understood. Membrane proteins insert into polymeric membranes and they maintain their functions in spite of their thicknesses (∼10 nm, [32]). Theoretical models explain this in the following way: The insertion of proteins is possible due to the compressibility of polymeric membranes, which is not the case for lipid membranes, as shown in Figure 8 [62]. Of course, beyond a certain thickness a mismatch of membrane perturbation energy will not allow any further protein insertion, but this limit has not yet been determined. Another possible explanation for the ability of proteins to reconstitute in polymeric membranes is the polydispersity of polymers; a local segregation of polymers with shorter chains in the vicinity of the proteins may be at play, allowing their insertion into polymer membranes [62,63].
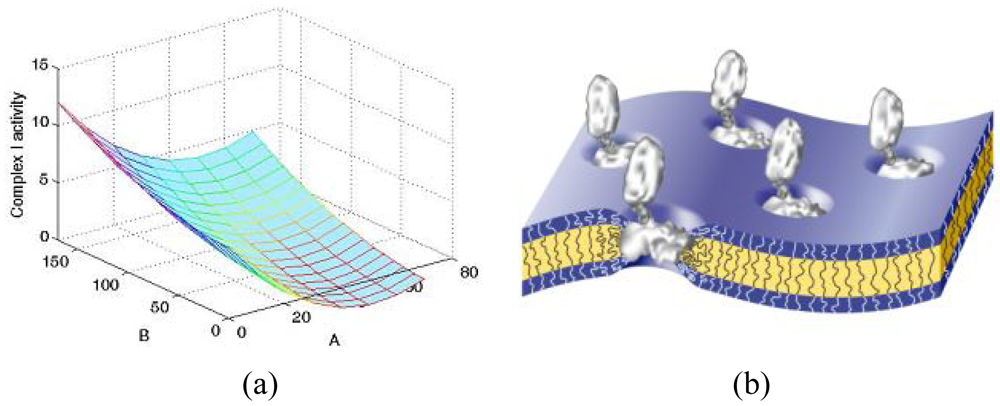
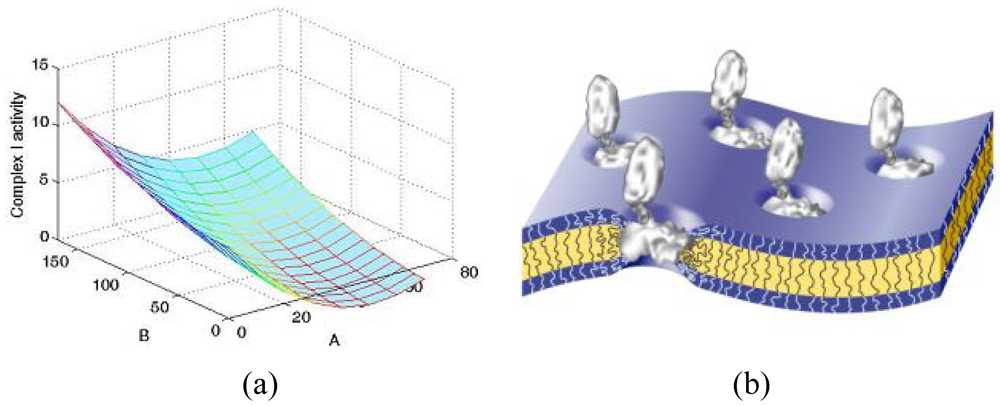
To better understand the influence of the block copolymer on the activity of inserted membrane proteins, studies on the complex I enzyme have been carried out on a library of block copolymers (PMOXA-PDMS-PMOXA) with different hydrophobic and hydrophilic block lengths. It is interesting to note that delipidated protein recovered its activity in the presence of polymeric bilayers, which proves that the synthetic membrane represents a useful mimic of biological membranes. Complex I activity exhibited obvious dependence on the molecular composition of the block copolymer, with significant influence being observed in the hydrophobic domain (Figure 9(a)) [37]. This indicates that the polymer membrane was able to adapt to and stabilize the complex I arm, which is naturally inserted in the membrane (Figure 9(b)).
Another parameter that has been considered when reconstituting membrane proteins in polymeric membranes is the number of proteins being reconstituted. Increasing the amount of protein added to block copolymers increased the permeability of the membrane as measured by enzymatic conversion inside polymeric vesicles, for example due to reconstituted OmpF channel protein. A ratio of 1:10, OmpF to PMOXA-PDMS-PMOXA block copolymer, resulted in a higher activity of the nanoreactors as compared to a 1:100 ratio [34]. Different behavior was seen in the case of AqpZ insertion in polymeric membranes. A ratio of 1:50, protein to block copolymer, has been found to yield the highest permeability to the polymeric vesicles [36]. The results using the PMOXA-PDMS-PMOXA block copolymer, but with different membrane proteins (OmpF, Aqpz respectively) reinforce the general conclusion that quantitative insertion of membrane proteins in membranes depends on the nature of the protein.
Polymer membranes made of PMOXA-PDMS-PMOXA have been proven to be ideal candidates as synthetic scaffolds for membrane proteins. To our knowledge, no other block copolymers that allow reconstitution of membrane proteins in a manner similar to lipids have been reported in literature. It is worth mentioning that studies on folding and insertion of channel proteins in liposomes composed of different types of lipids have shown that the nature of the lipid influences the degree of protein insertion [60]. Therefore, one would expect the chemistry of the block copolymer to be an important factor that supports or fails to support an intended insertion of proteins. So far, however, this premise has not been ascertained by experimental results.
The rising use of polymeric vesicles in biomedical and biotechnological applications requires an ever more flexible approach that allows for easy tuning of membranes according to the types of molecules that need to be produced or delivered. In this respect polymer vesicles that reconstitute membrane proteins offer a resourceful option to be used in diagnostics, therapeutics or bio-sensing. The concept can be tuned with respect either to the membrane protein being reconstituted or to the enzyme being encapsulated in the polymeric vesicle. Up to date, the main approach to use polymersomes in drug delivery is based on the encapsulation of the active compound, ranging from small molecules to proteins/enzymes, and their release in biological compartments upon the degradation of the polymer [10,64-67]. While offering the advantage of a more stable carrier compared to liposomes, the use of polymersomes to release the loaded compounds represents the conventional approach of drug delivery with well known drawbacks, such as an uncontrolled release, or possible degradation in other compartments than the desired ones. A step further was marked by the introduction of polymeric nanoreactors based on polymersomes serving as cages for enzymes/proteins that act in situ without being released [68]. To allow the enzyme to act inside the inner cavity, these nanoreactors possess a membrane permeable in specific molecules, such as to ions, which can penetrate from the environment and arrive at the enzyme location. However, the polymer membrane does not allow the passage of bigger molecules, such as substrates or products, and in this respect the insertion of biopores/channels proteins is essential for the design of efficient therapeutic systems that allow the encapsulated drug to act in situ. By using membrane proteins to facilitate transport across membranes, it is also possible to control the molecular efflux using engineered membrane proteins [12]. Polymer vesicles with engineered membrane proteins for controlled transport advance the optimal use of polymeric vesicles, especially for applications related to sensitive biological entities as, for example, sRNA, or enzymes. Despite numerous examples of polymersomes in use for drug delivery [64-66], only a few of them are bio-decorated [34,67,69]. All of them, presented here, were designed in the context of developing nanoreactors, or synthetic organelles, which serve to provide protected enzymes/proteins in the desired biological compartment. The bio-decorated polymeric shield increases significantly the stability of the encapsulated enzyme while allowing the molecules efflux via the reconstituted bio-gates.
When reconstituting membrane proteins in polymeric membranes, one must consider that mild preparation conditions must be used in order to preserve their activities [23,32]. Another consideration, when working with membrane proteins and polymeric membranes, is the extent of reconstitution, a determination of which has not been done to date. It would therefore be beneficial to quantify how many membrane proteins are inserted in a membrane compared to an initial condition. And not least, it would be advantageous to determine whether there are other block copolymers that allow insertion of membrane proteins, and the rationale behind the molecular composition and membrane protein reconstitution.
6. Conclusion
Favorable interactions between biomolecules, such as enzymes, peptides or membrane proteins, and different types of polymer membranes, cannot be taken for granted even though there are many reported combinations and possible applications. This is an emerging field; all of the reported systems, even if at the model stage, are of interest, both from the fundamental point of view of bio-synthetic interactions and when considering them as more stable and robust systems for study in technological and medical applications. In order to form a stable system, the binding that results from the combination of a synthetic membrane and a biomolecule should be strong enough to avoid non-specific detachment of the biomolecule, therefore covalent bonding is generally preferred. However, a decrease in the activity of the biological partner is usually difficult to avoid, and various conditions should be screened in order to obtain a balance between strong coupling and a structurally and functionally preserved biological molecule. In this respect, the manner in which natural molecular recognition interactions occur represents a new way to obtain very specific binding patterns, while not disturbing the biological entity, as reported for the coupling of biotin-streptavidin, antigen-to-antibody, or DNA hybridization. For insertion of transmembrane biomolecules, both the chemical nature and the length of the polymer blocks have an influence, even if this is not yet fully understood. The difference in thickness and the chemical nature of copolymers compared to lipids requires different approaches when developing bio-synthetic membranes in order to obtain an efficient system. This explains why large hydrophobic polymer domains induce a decrease in the activity of the reconstituted protein, or even prohibit the biopore from reconstituting. In certain cases, the reconstitution of membrane proteins in amphiphilic copolymer membranes can be achieved by self-assembly, which indicates that these structures are favored. But in this regard, the combination of biological entities and polymeric membranes should not be viewed as if LEGO™ blocks—which can snap together easily—were being assembled; the situation is more like a puzzle, where the right parts need to be found in order to fit together.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomolecule | Polymer | Type of interaction | Reference |
|---|---|---|---|
| GOx | PoPD | Physical adsorption | [19] |
| Ribonuclease A | PAA | Covalently bonded, complexing | [20] |
| GOx | Poly(n-methyl pyrrole) | Electochemical adsorption | [21] |
| Poly phenol oxidase | PEDT | Physical adsorption | [22] |
| Streptavidin | PMOXA-PDMS-PMOXA | Complexation | [23] |
| RFP, GFP, MBP | Polybutadiene-block-poly(ethylene oxide) | Complexation | [24] |
| GOx | PAN/PANI-4 | Electrochemical immobilized | [25] |
| Oxidases | Poly 1,2-diaminobenzen | Physical adsoption | [26] |
| IgG | NyM-g-GMA | Covalently bonded | [27] |
| DNA | APTES-Glutaraldehyde | Covalently bonded | [28] |
| Gramicidin | PMOXA7-PDMS60-PMOXA7 | Membrane insertion | [29] |
| Alamethicin | PMOXA12–PDMS54–PMOXA12 | Membrane insertion | [30] |
| Alamethicin | PEO-PEE | Membrane insertion | [31] |
| Alamethicin | PMOXA16-PDMS74-PMOXA16 | Membrane insertion | [9] |
| OmpF | PMOXA-PDMS-PMOXA | Membrane insertion | [23,32] |
| LamB | PMOXA-PDMS-PMOXA | Membrane insertion | [33] |
| Tsx | PMOXA-PDMS-PMOXA | Membrane insertion | [34] |
| FhuA (wild type and genetically engineered variants) | PMOXA-PDMS-PMOXA | Membrane insertion | [12,35] |
| AqpZ | PMOXA-PDMS-PMOXA | Membrane insertion | [36] |
| Complex I | PMOXA-PDMS-PMOXA | Membrane insertion | [37] |
| F0F1-ATP synthase | PEtOz-PDMS-PEtOz | Membrane insertion | [38] |
| Bacteriodopsin | PEtOz-PDMS-PEtOz | Membrane insertion | [38] |
Appendix
| APTES | Aminopropyltrietoxysilan |
| NyM-g-GMA | Nylon-grafted-glycidyl methacrylate |
| PAA | Poly(acrylic acid) |
| PAN | Polyacrylonitrile |
| PANI | Polyaniline |
| PDMS | Poly(dimethyl siloxane) |
| PEDT | poly(3,4-ethylenedioxythiophene |
| PEE | Poly(ether esters) |
| PEO | Poly(ethylene oxid) |
| PEtOz | Poly(2-ethyl-2-oxazoline) |
| P(HEMA-g-GMA) | Poly(hydroxyethylmethacrylate)-grafted-poly(glycidyl-methacrylate) |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLLA | Poly(l-lactic acid) |
| PMMA | Poly(methyl methacrylate) |
| PMOXA | Poly(2-methyl-2-oxazoline) |
| PoPD | Poly(ortho-phenylenediamine) |
| PS | Polystyrene |
| Si-g-PAA | Silicon-grafted-poly(acrylic acid) |
References
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta-Rev. Biomembr. 2000, 1469, 159–195. [Google Scholar]
- Simons, K.; Vaz, W.L.C. Model systems, lipid rafts, and cell membranes. Ann. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar]
- Seddon, A.M.; Curnow, P.; Booth, J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 105–117. [Google Scholar]
- Khandelia, H.; Ipsen, J.H.; Mouritsen, O.G. The impact of peptides on lipid membranes. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 1528–1536. [Google Scholar]
- Hagan, C.L.; Kim, S.; Kahne, D. Reconstitution of outer membrane protein assembly from purified components. Science 2010, 328, 890–892. [Google Scholar]
- Ahuja, T.; Mir, I.A.; Kumar, D.; Rajesh. Biomolecular immobilization on conducting polymers for biosensing applications. Biomaterials 2007, 28, 791–805. [Google Scholar]
- Krishna, O.D.; Kiick, K.L. Protein- and peptide-modified synthetic polymeric biomaterials. Biopolymers 2010, 94, 32–48. [Google Scholar]
- Mecke, A.; Dittrich, C.; Meier, W. Biomimetic membranes designed from amphiphilic block copolymers. Soft Matter 2006, 2, 751–759. [Google Scholar]
- Kita-Tokarczyk, K.; Grumelard, J.; Haefele, T.; Meier, W. Block copolymer vesicles—using concepts from polymer chemistry to mimic biomembranes. Polymer 2005, 46, 3540–3563. [Google Scholar]
- Discher, D.E.; Ahmed, F. Polymersomes. Ann. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar]
- Du, J.Z.; O'Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar]
- Onaca, O.; Enea, R.; Hughes, D.W.; Meier, W. Stimuli-responsive polymersomes as nanocarriers for drug and gene delivery. Macromol. Biosci. 2009, 9, 129–139. [Google Scholar]
- Antonietti, M.; Forster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar]
- Herrero-Vanrell, R.; Rincón, A.C.; Alonso, M.; Molina-Martinez, I.T.; Rodriguez-Cabello, J.C. Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. J. Control. Release 2005, 102, 113–122. [Google Scholar]
- Discher, B.M.; Eisenberg, A. Cross-linked polymersome membranes: Vesicles with broadly adjustable properties. J. Phys. Chem. B 2002, 106, 2848–2854. [Google Scholar]
- Palivan, C.G.; Vebert, C.; Axthelm, F.; Meier, W. Responsive self-assembled nanostructures. Nanotechnol. Biol. Med. Methods Dev. Appl. 2007, 32, 1–16. [Google Scholar]
- Kita-Tokarczyk, K.; Meier, W. Biomimetic block copolymer membranes. Chimia 2008, 62, 820–825. [Google Scholar]
- Ho, D.; Chu, B.; Schmidt, J.J.; Brooks, E.K.; Montemagno, C.D. Hybrid protein-polymer biomimetic membranes. IEEE Trans. Nanotechnol. 2004, 3, 256–263. [Google Scholar]
- Rothwell, S.A.; Killoran, S.J.; O'Neill, R.D. Enzyme immobilization strategies and electropolymerization conditions to control sensitivity and selectivity parameters of a polymer-enzyme composite glucose biosensor. Sensors 2010, 10, 6439–6462. [Google Scholar]
- Cullen, S.; Liu, X.; Mandel, I.C.; Himpsel, F.J.; Gopulan, P. Polymeric brushes as functional templates for immobilizing ribonuclease A: Study of binding kinetics and activity. Langmuir 2008, 24, 913–920. [Google Scholar]
- Barlett, N.; Tebbutt, P.; Tyrrell, C.H. Electrochemical immobilization of enzymes. 3. Immobilization of glucose oxidase in thin films of electrochemically polymerized phenols. Anal. Chem. 1992, 64, 138–142. [Google Scholar]
- Vedrine, C.; Fabiano, S.; Tran-Minh, C. Amperometric tyrosinase based biosensor using an electrogenerated polythiophene film as an entrapment support. Talanta 2003, 59, 535–544. [Google Scholar]
- Grzelakowski, M.; Onaca, O.; Rigler, P.; Kumar, M.; Meier, W. Immobilized protein-polymer nanoreactors. Small 2009, 5, 2545–2548. [Google Scholar]
- Nehring, R.; Palivan, C.G.; Moreno-Flores, S.; Mantion, A.; Tanner, P.; Toca-Herrera, J.L.; Thünemann, A.; Meier, W. Protein decorated membranes by specific molecular interactions. Soft Matter 2010, 6, 2815–2824. [Google Scholar]
- Bayramoglu, G.; Metin, A.Ü.; Altintas, B.; Arica, M.Y. Reversible immobilization of glucose oxidase on polyaniline grafted polyacrylonitrile conductive composite membrane. Bioresource Technol. 2010, 101, 6881–6887. [Google Scholar]
- Curulli, A.; Valentini, F.; Orlanduci, S.; Terranova, M.L.; Palleschi, G. Pt based enzyme electrode probes assembled with Prussian Blue and conducting polymer nanostructures. Biosens. Bioelectron. 2004, 20, 1223–1232. [Google Scholar]
- Jackeray, R.; Jain, S.; Chattopadhyay, S.; Yadau, M.; Shrivastav, T.G.; Sing, H. Surface modification of nylon membrane by glycidyl methacrylate graft copolymerization for antibody immobilization. J. Appl. Polym. Sci. 2010, 116, 1700–1709. [Google Scholar]
- Razumovitch, J.; de França, K.; Kehl, F.; Wiki, M.; Meier, W.; Vebert, C. Optimal hybridization efficiency upon immobilization of oligonucleotide double helices. J. Phys. Chem. B 2009, 113, 8383–8390. [Google Scholar]
- Gonzalez-Perez, A.; Stibius, K.B.; Vissing, T.; Nielsen, C.H.; Mouritsen, O.G. Biomimetic triblock copolymer membrane arrays: A stable template for functional membrane proteins. Langmuir 2009, 25, 10447–10450. [Google Scholar]
- Wong, D.; Jeon, T.J.; Schmidt, J. Single molecule measurements of channel proteins incorporated into biomimetic polymer membranes. Nanotechnology 2006, 17, 3710–3717. [Google Scholar]
- Vijayan, K.; Discher, D.E.; Lal, J.; Janmey, P.; Goulian, M. Interactions of membrane-active peptides with thick, neutral, nonzwitterionic bilayers. J. Phys. Chem. B 2005, 109, 14356–14364. [Google Scholar]
- Nardin, C.; Thoeni, S.; Winterhalter, M.; Meier, W. Nanoreactors based on (polymerized) ABA-triblock copolymer vesicles. Chem. Commun. 2000, 15, 1433–1434. [Google Scholar]
- Graff, A.; Sauer, M.; van Gelder, P.; Meier, W. Virus-assisted loading of polymer nanocontainer. Proc. Natl. Acad. Sci. USA 2002, 99, 5064–5068. [Google Scholar]
- Ranquin, A.; Versées, W.; Meier, W.; Steyaert, J.; van Gelder, P. Therapeutic nanoreactors: Combining chemistry and biology in a novel triblock copolymer drug delivery system. Nano Lett. 2005, 5, 2220–2224. [Google Scholar]
- Nallani, M.; Benito, S.; Onaca, O.; Graff, A.; Lindemann, M.; Winterhalter, M.; Meier, W.; Schwaneberg, U. A nanocompartment system (synthosome) designed for biotechnological applications. J. Biotechnol. 2006, 123, 50–59. [Google Scholar]
- Kumar, M.; Grezlakowski, M.; Zilles, J.; Clark, M.; Meier, W. Highly permeable polymeric membranes based on the incorporation of the functional water channel protein Aquaporin Z. Proc. Natl. Acad. Sci. USA 2007, 104, 20719–20724. [Google Scholar]
- Graff, A.; Fraysse-Ailhas, C.; Palivan, C.G.; Grzelakowski, M.; Friedrich, T.; Vebert, C.; Gescheidt, G.; Meier, W. Amphiphilic copolymer membranes promote NADH: Ubiquinone oxidoreductase activity: towards an electron-transfer nanodevice. Macromol. Chem. Phys. 2010, 211, 229–238. [Google Scholar]
- Choi, H.J.; Montemagno, C.D. Artificial organelle: ATP synthesis from cellular mimetic polymersomes. Nano Lett. 2005, 5, 2538–2542. [Google Scholar]
- Delaittre, G.; Reynhount, I.C.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Cascade Reactions in an all-enzyme nanoreactor. Chem.-A Eur. J. 2009, 15, 12600–12603. [Google Scholar]
- Bayramoglu, G.; Arica, M.Y. Immobilization of laccase onto poly(glycidylmethacrylate) brush grafted poly(hydroxyethylmethacrylate) films: Enzymatic oxidation of phenolic compounds. Mater. Sci. Eng. C-Mater. Biol. Appl. 2009, 29, 1990–1997. [Google Scholar]
- Teles, F.R.R.; Fonseca, L.R. Applications of polymers for biomolecule immobilization in electrochemical biosensors. Mater. Sci. Eng. C-Biomim. Supramol. Syst. 2008, 28, 1530–1543. [Google Scholar]
- Dirks, A.J.T.; van Berkel, S.S; Hatzakis, N.S.; Opsteen, J.A.; van Delft, F.L.; Cornelissen, J.J.L.M.; Rowan, A.E.; van Hest, J.C.M.; Rutjes, F.P.J.T.; Nolte, R.J.M. Preparation of biohybrid amphiphiles via the copper catalysed Huisgen [3 + 2] dipolar cycloaddition reaction. Chem. Commun. 2005, 33, 4172–4174. [Google Scholar]
- Nehring, R.; Palivan, C.G.; Casse, O.; Tanner, P.; Tüxen, J.; Meier, W. Amphiphilic diblock copolymers for molecular recognition: metal-nitrilotriacetic acid functionalized vesicles. Langmuir 2009, 25, 1122–1130. [Google Scholar]
- Stengel, G.; Gill, J.P.; Sandin, P.; Wilhemsson, L.M.; Albinosson, B.; Nordén, B.; Millar, D. Conformational dynamics of DNA polymerase probed with a novel fluorescent DNA base analogue. Biochemistry 2007, 46, 12289–12297. [Google Scholar]
- Burns, S.A.; Hard, R.; Hicks, W.L., Jr.; Cohan, P.; Sigurfson, L.; Gardella, J.A., Jr. Determining the protein drug release characteristics and cell adhesion to a PLLA or PLGA biodegradable polymer membrane. J. Biomed. Mater. Res. A 2010, 94A, 27–37. [Google Scholar]
- Liu, X.H.; Ma, X. Polymeric scaffolds for bone tissue engineering. Ann. Biomed. Eng. 2004, 32, 477–486. [Google Scholar]
- Andersen, O.S. Ion movement through gramicidin A channels. Single-channel measurements at very high potentials. Biophys. J. 1983, 41, 119–133. [Google Scholar]
- Lei, S.B.; Tero, R.; Misawa, N.; Yamamura, S.; Wan, L.J.; Urisu, T. AFM characterization of gramicidin-A in tethered lipid membrane on silicon surface. Chem. Phys. Lett. 2006, 429, 244–249. [Google Scholar]
- Urry, D.W.; Goodall, M.C.; Glickson, J.D.; Mayers, D.F. The gramicidin A transmembrane channel: Characteristics of head-to-head dimerized π(L,D) helices. Proc. Natl. Acad. Sci. USA 1971, 68, 1907–1911. [Google Scholar]
- Sarges, R.; Witkop, B.; Gramicidin, VII. The Structure of Valine- and Isoleucine-gramicidin B. J. Amer. Chem. Soc. 1965, 87, 2027–2030. [Google Scholar]
- Elliott, J.R.; Needham, D.; Dilger, J.P.; Haydon, D.A. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta 1983, 735, 95–103. [Google Scholar]
- Cafiso, D.S. Alamethicin: A peptide model for voltage gating and protein-membrane interactions. Ann. Rev. Biophys. Biomol. Struct. 1994, 23, 141–165. [Google Scholar]
- Hanke, W.; Boheim, G. The lowest conductance state of the alamethicin pore. Biochim. Biophys. Acta 1980, 596, 456–462. [Google Scholar]
- Haefele, T.; Kita-Tokarczyk, K.; Meier, W. Phase behavior of mixed Langmuir monolayers from amphiphilic block copolymers and an antimicrobial peptide. Langmuir 2006, 22, 1164–1172. [Google Scholar]
- Sansom, M.S. The biophysics of peptide models of ion channels. Prog. Biophys. Mol. Biol. 1991, 55, 139–235. [Google Scholar]
- Salzer, R.; Li, J.; Rautenberg, C.; Friedrich, S.; Habicher, W.D. Integration of ion channel proteins into a polymer matrix—Investigation by the patch-clamp technique. Macromol. Symp. 2001, 164, 239–245. [Google Scholar]
- Dorn, J.; Belegrinou, S.; Kreiter, M.; Sinner, E.-K.; Meier, W. Planar block copolymer membranes by vesicle spreading. Macromol. Biosci. 2011. in press. [Google Scholar]
- Collarini, M.; Amblard, G.; Lazduanski, C.; Pattus, F. Gating processes of channels induced by colicin A, its C-terminal fragment and colicin E1 in planar lipid bilayers. Eur. Biophys. J. Biophys. Lett. 1987, 14, 147–153. [Google Scholar]
- Graff, A.; Winterhalter, M.; Meier, W. Nanoreactors from polymer-stabilized liposomes. Langmuir 2001, 17, 919–923. [Google Scholar]
- Tamm, L.K.; Hong, H.; Liang, B.Y. Folding and assembly of beta-barrel membrane proteins. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 250–263. [Google Scholar]
- Mouritsen, O.G. Self-assembly and organization of lipid-protein membranes. Curr. Opin. Colloid Interface Sci. 1998, 3, 78–87. [Google Scholar]
- Pata, V.; Dan, N. The effect of chain length on protein solubilization in polymer-based vesicles (polymersomes). Biophys. J. 2003, 85, 2111–2118. [Google Scholar]
- Dan, N.; Safran, S. A. Self-Assembly in mixtures of diblock copolymers. Macromolecules 1994, 27, 5766–5772. [Google Scholar]
- Ahmed, F.; Photos, J.; Discher, D.E. Polymersomes as viral capsid mimics. Drug Develop. Res. 2006, 67, 4–14. [Google Scholar]
- Kim, M.S.; Lee, D.S. Biodegradable and pH-sensitive polymersome with tuning permeable membrane for drug delivery carrier. Chem. Commun. 2010, 46, 4481–4483. [Google Scholar]
- Christian, D.A.; Cai, S.; Bowen, D.M.; Kim, Y.; Pajerowska, D.; Discher, D.E. Polymersome carriers: From self-assembly to siRNA and protein therapeutics. Eur. J. Pharm. Biopharm. 2009, 71, 463–474. [Google Scholar]
- Broz, P.; Benito, S.; Saw, C.L.; Burger, P.; Heider, H.; Pfister, M.; Marsch, S.; Meier, M.; Hunziker, P. Toward intelligent nanosize bioreactors: A pH-switchable, channel-equipped, functional polymer nanocontainer. Nano Lett. 2006, 6, 2349–2353. [Google Scholar]
- Axthelm, F.; Casse, O.; Koppenol, W.H.; Nauser, T.; Meier, W.; Palivan, C.G. Antioxidant nanoreactor based on superoxide dismutase encapsulated in superoxide-permeable vesicles. J. Phys. Chem. B 2008, 112, 8211–8217. [Google Scholar]
- Kamphuis, M.M.J.; Johnston, A.P.R.; Such, G.K.; Dam, H.H.; Evans, R.A.; Scott, A.M.; Nice, E.C.; Heath, J.K.; Caruso, F. Targeting of cancer cells using click-functionalized polymer capsules. J. Am. Chem. Soc. 2010, 132, 15881–15883. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baumann, P.; Tanner, P.; Onaca, O.; Palivan, C.G. Bio-Decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics. Polymers 2011, 3, 173-192. https://doi.org/10.3390/polym3010173
Baumann P, Tanner P, Onaca O, Palivan CG. Bio-Decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics. Polymers. 2011; 3(1):173-192. https://doi.org/10.3390/polym3010173
Chicago/Turabian StyleBaumann, Patric, Pascal Tanner, Ozana Onaca, and Cornelia G. Palivan. 2011. "Bio-Decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics" Polymers 3, no. 1: 173-192. https://doi.org/10.3390/polym3010173
APA StyleBaumann, P., Tanner, P., Onaca, O., & Palivan, C. G. (2011). Bio-Decorated Polymer Membranes: A New Approach in Diagnostics and Therapeutics. Polymers, 3(1), 173-192. https://doi.org/10.3390/polym3010173