Acylation of Lignin with Different Acylating Agents by Mechanical Activation-Assisted Solid Phase Synthesis: Preparation and Properties

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
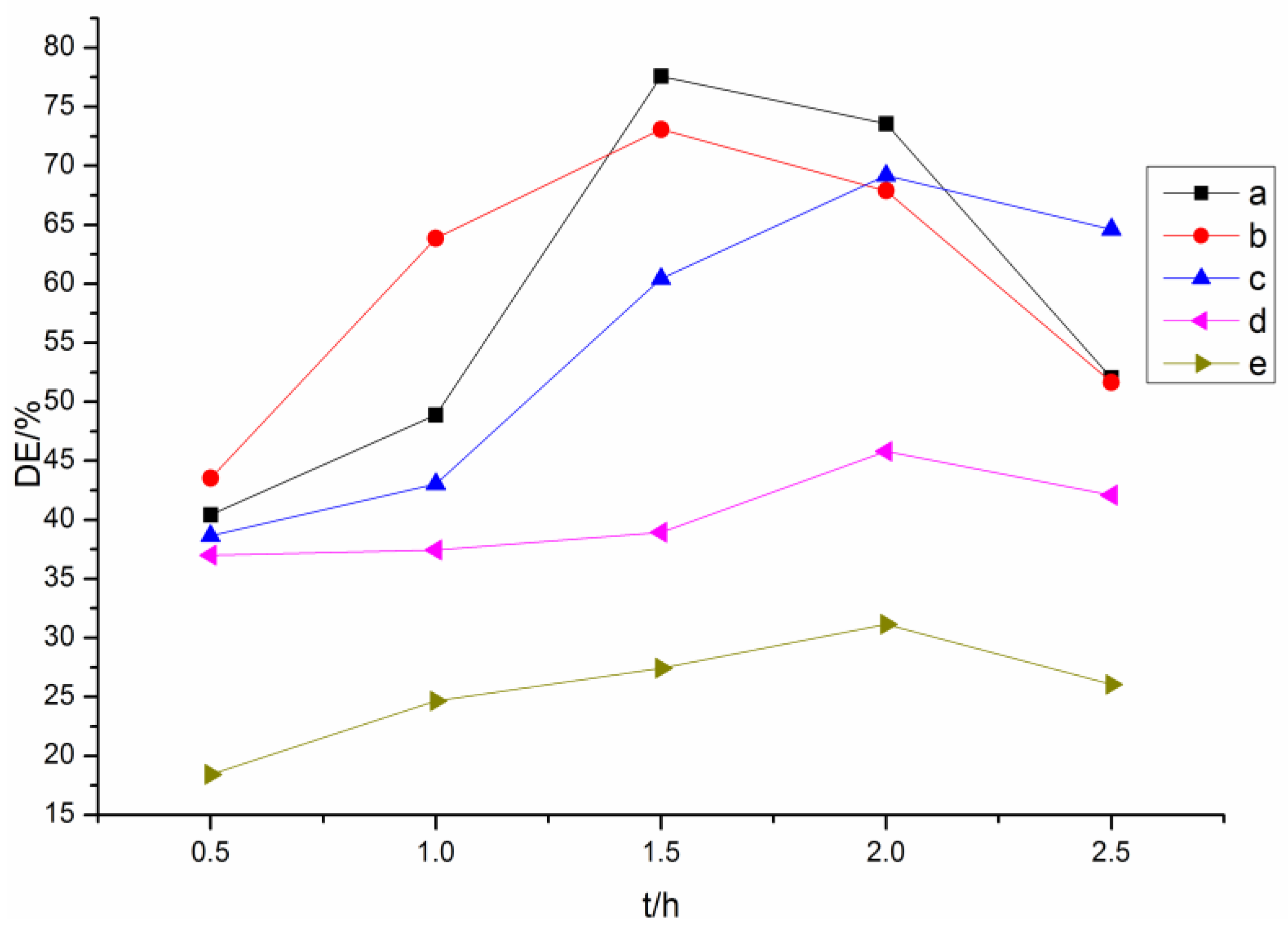
2.2. Preparation of Acylated Lignins
2.3. Characterization
3. Results and Discussion
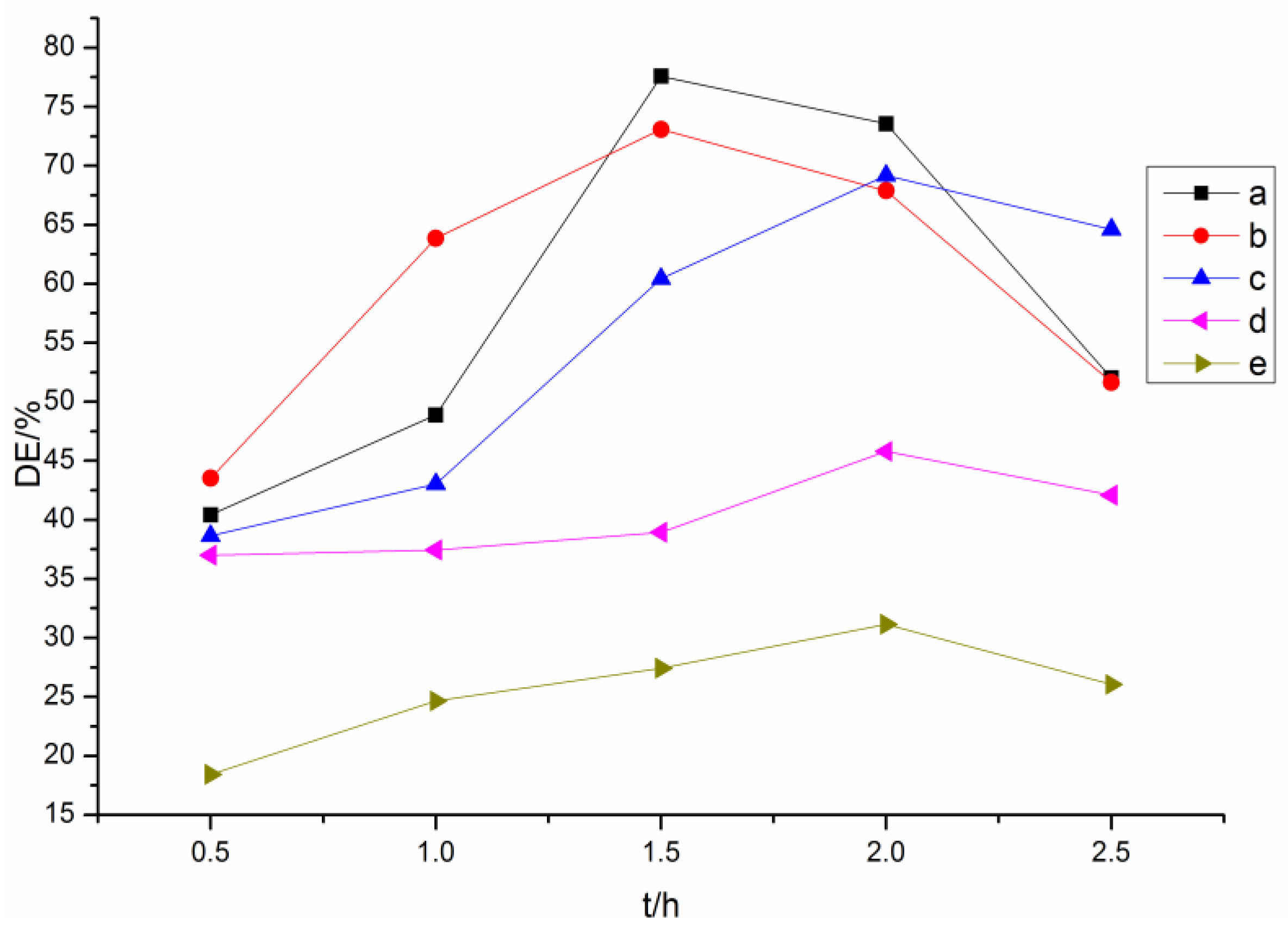
3.1. Effect of Acylating Agent on the Esterification of Lignin
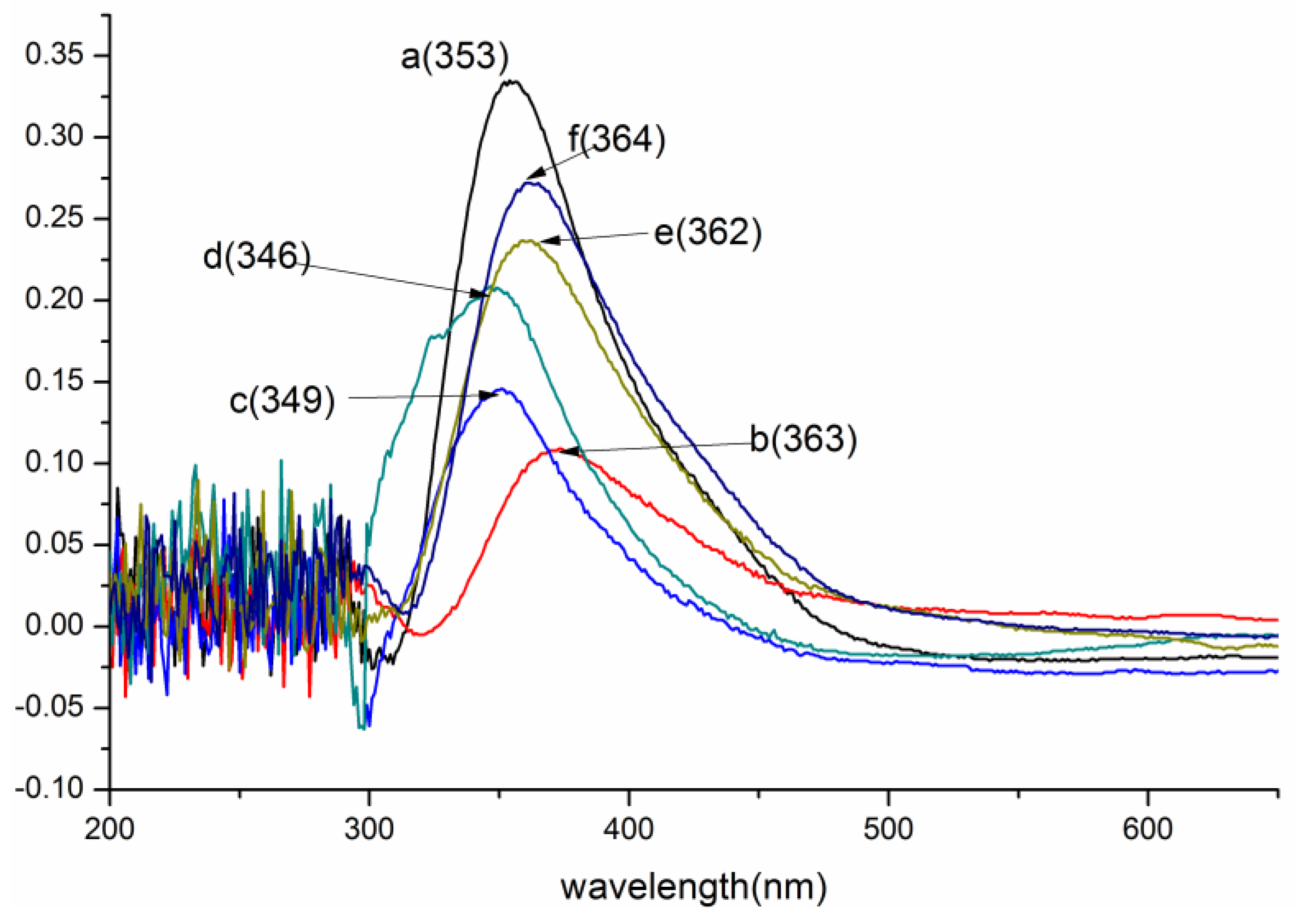
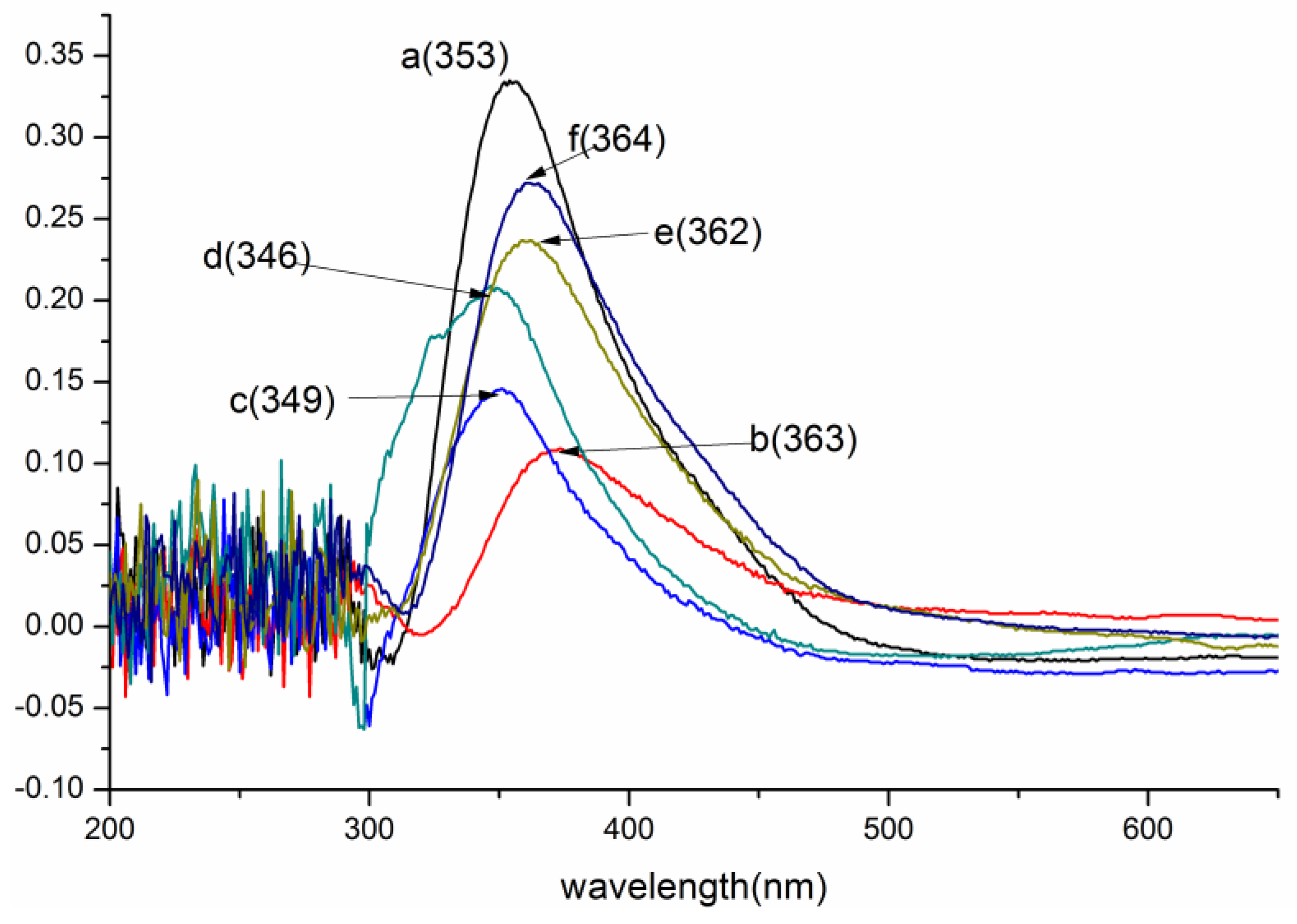
3.1.1. UV/Vis Analysis
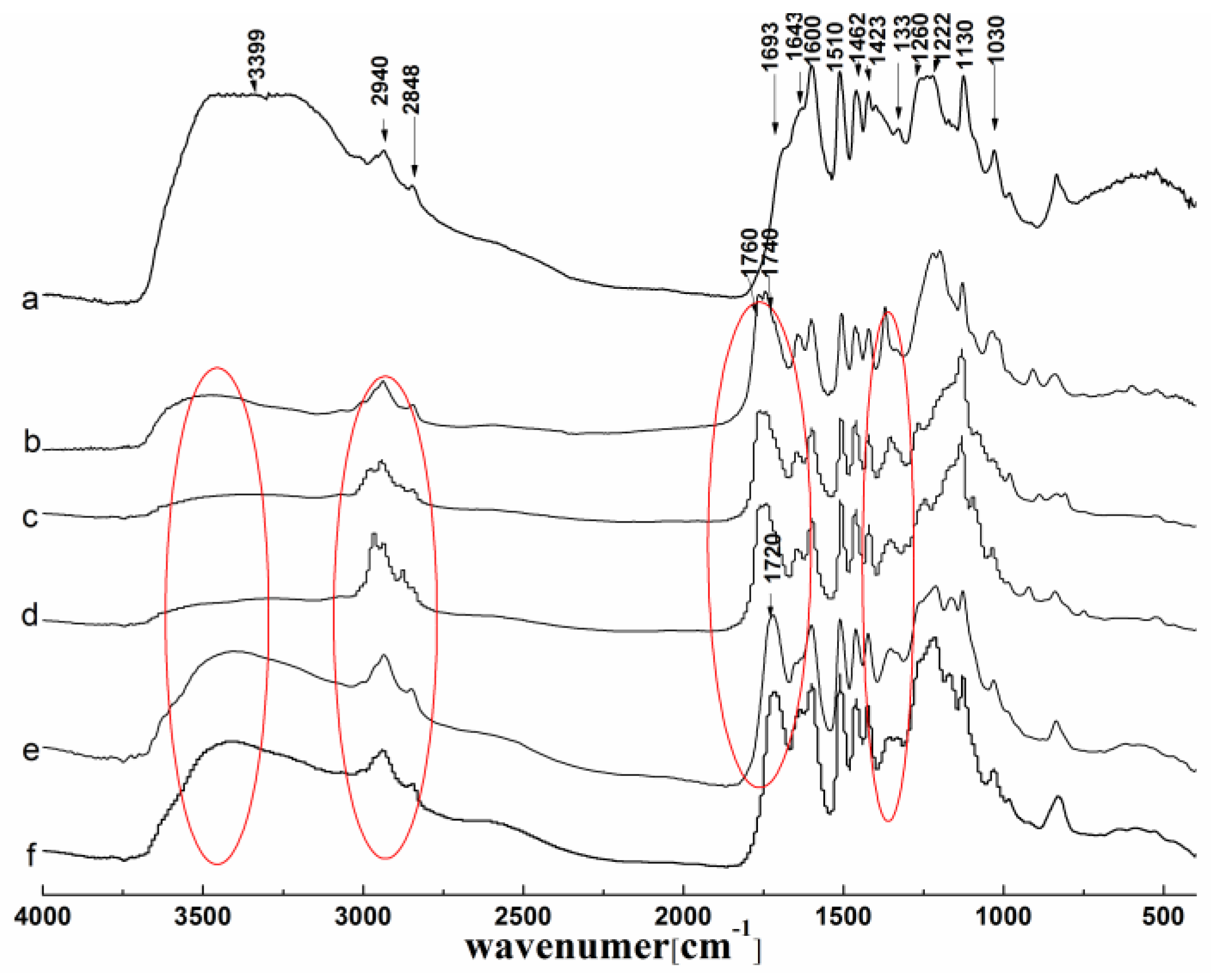
3.1.2. FTIR Analysis
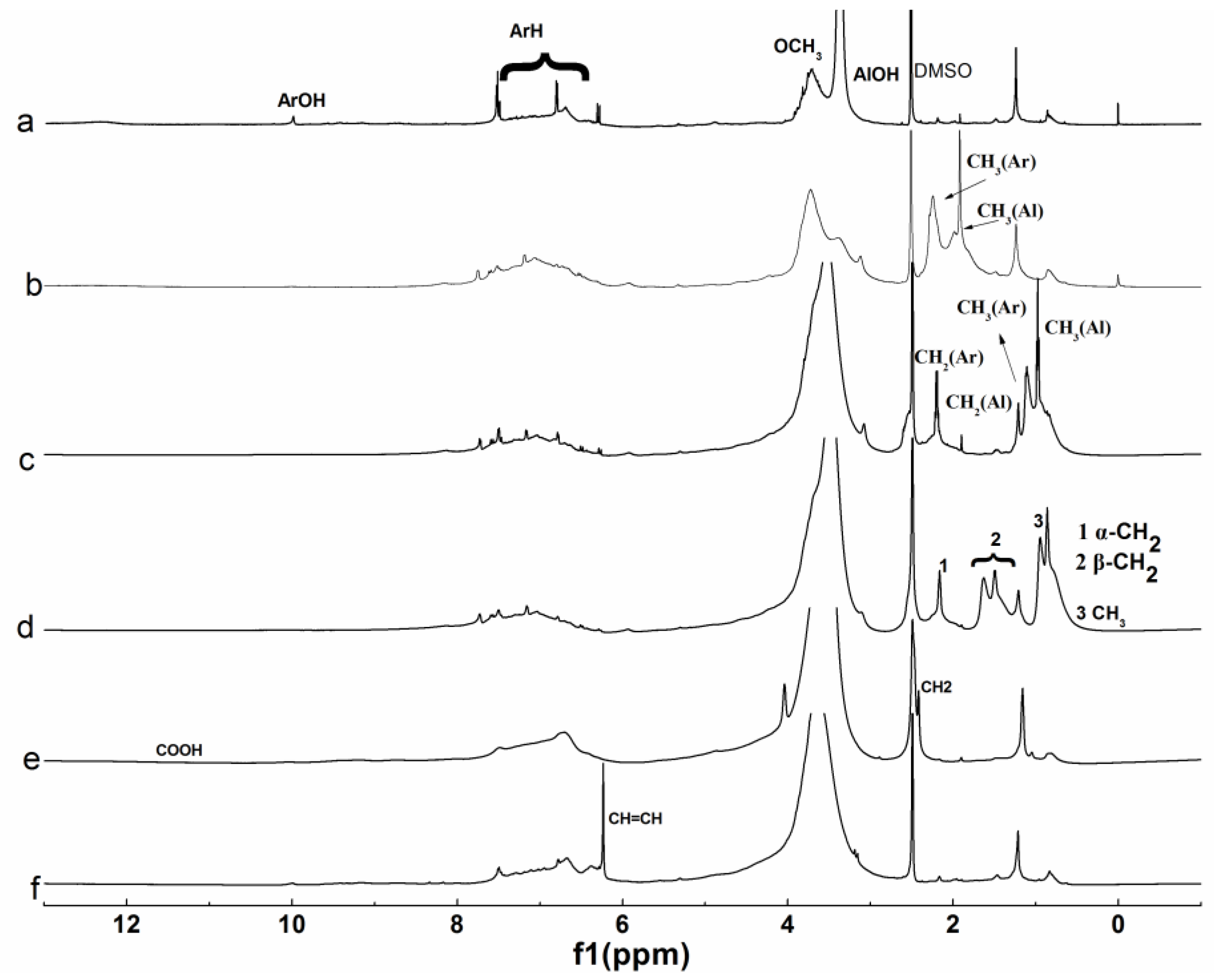
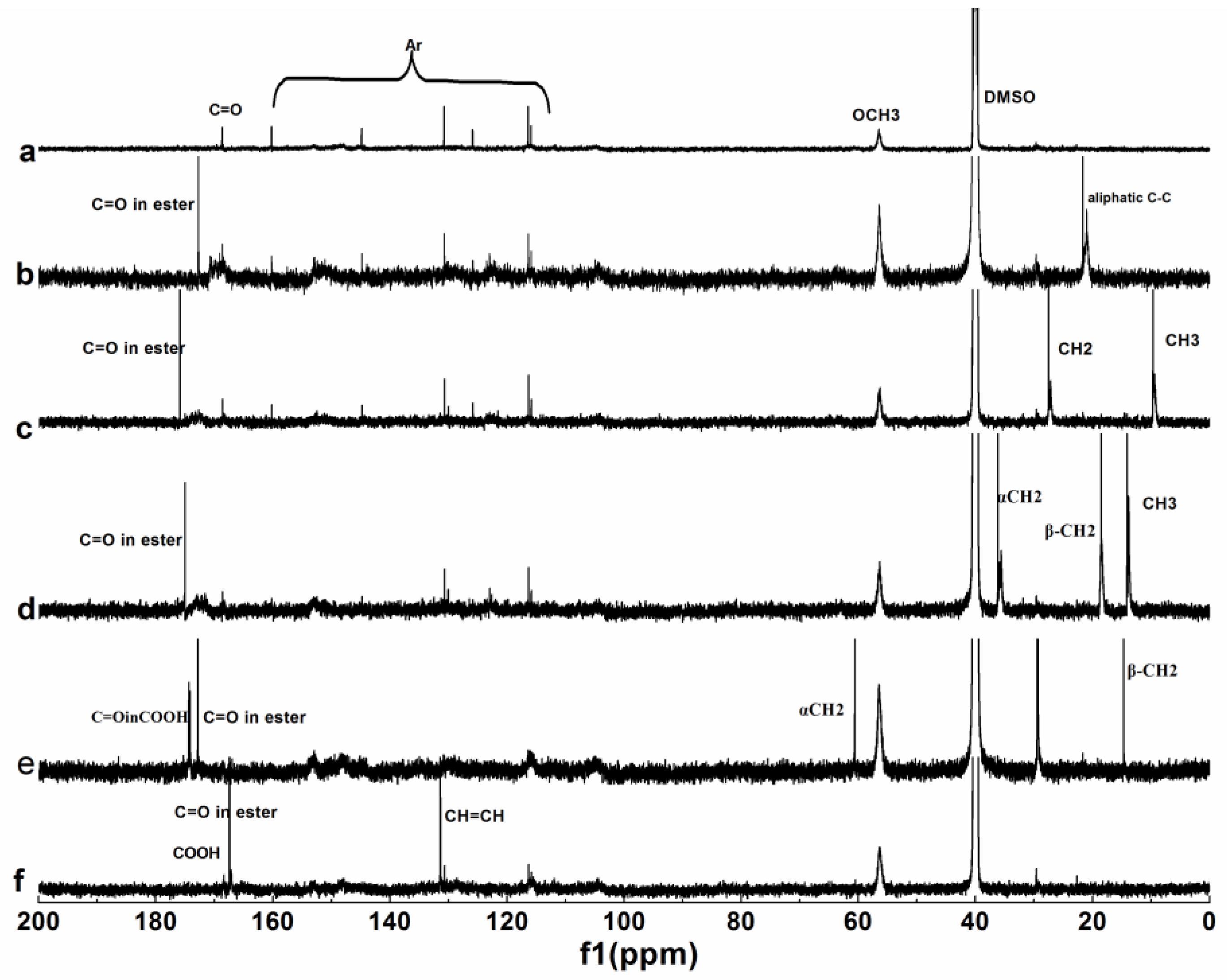
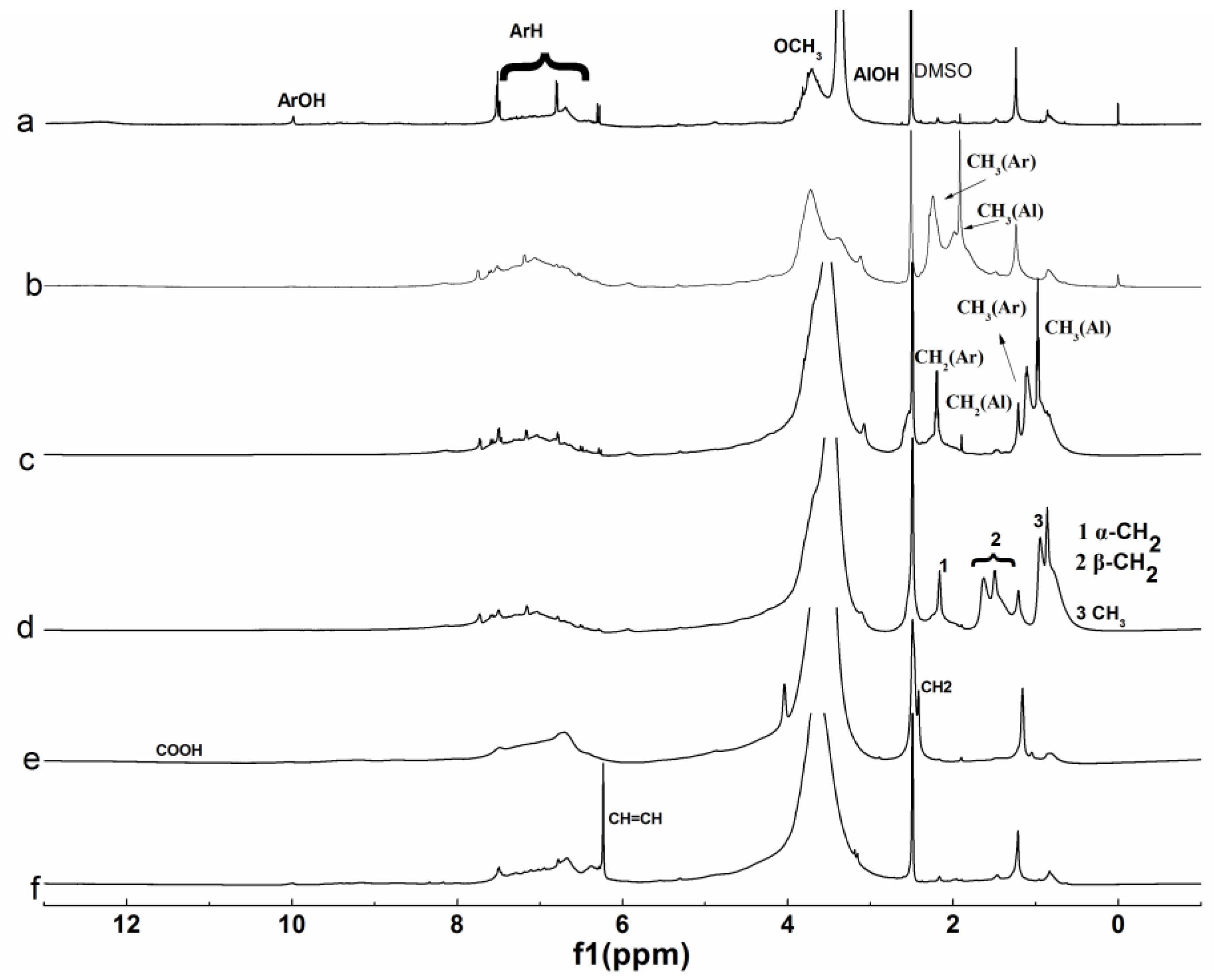
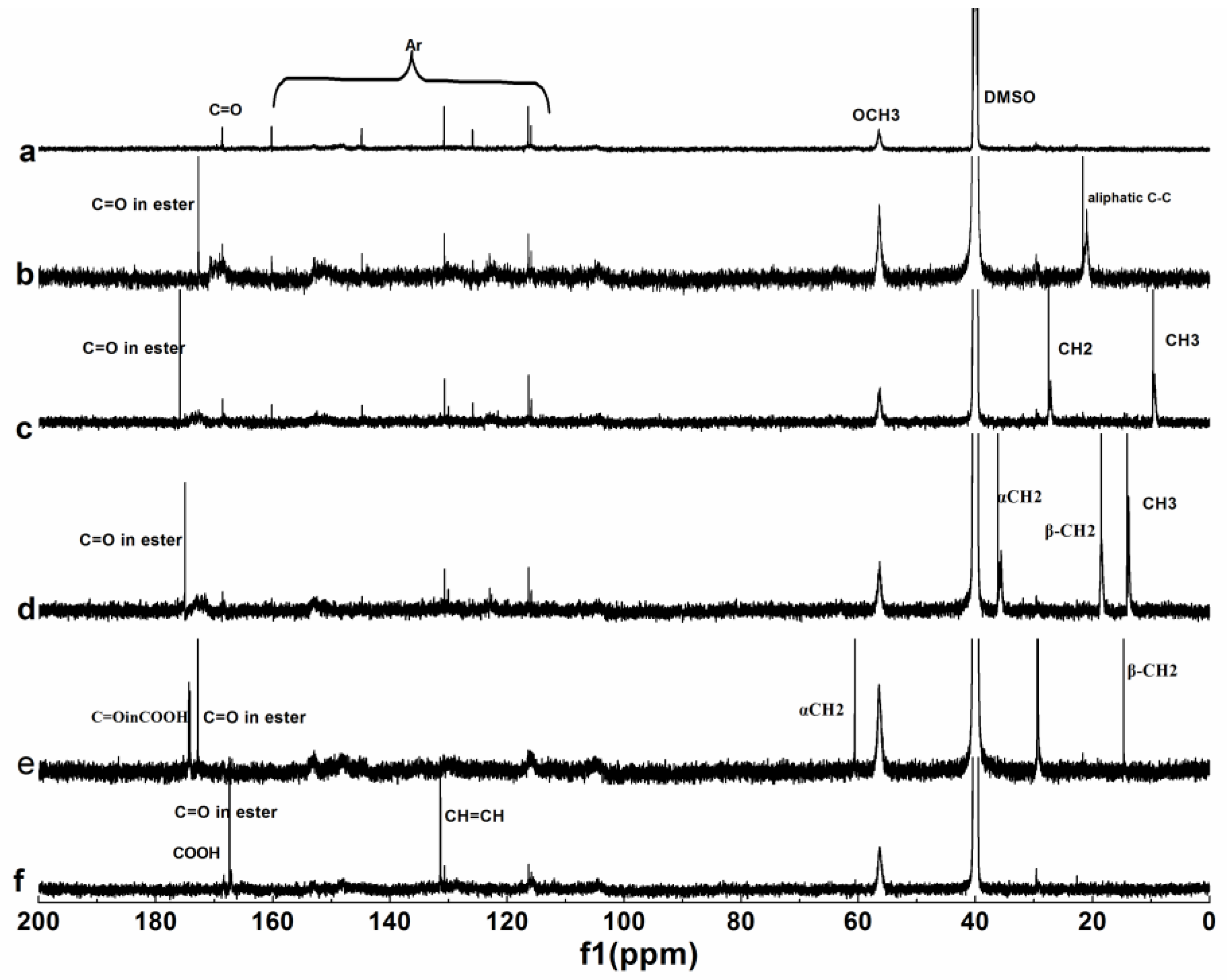
3.1.3. NMR Analysis
3.2. Effect of Acylating Agent on Morphology and Thermal Properties of the Products
3.2.1. SEM Analysis
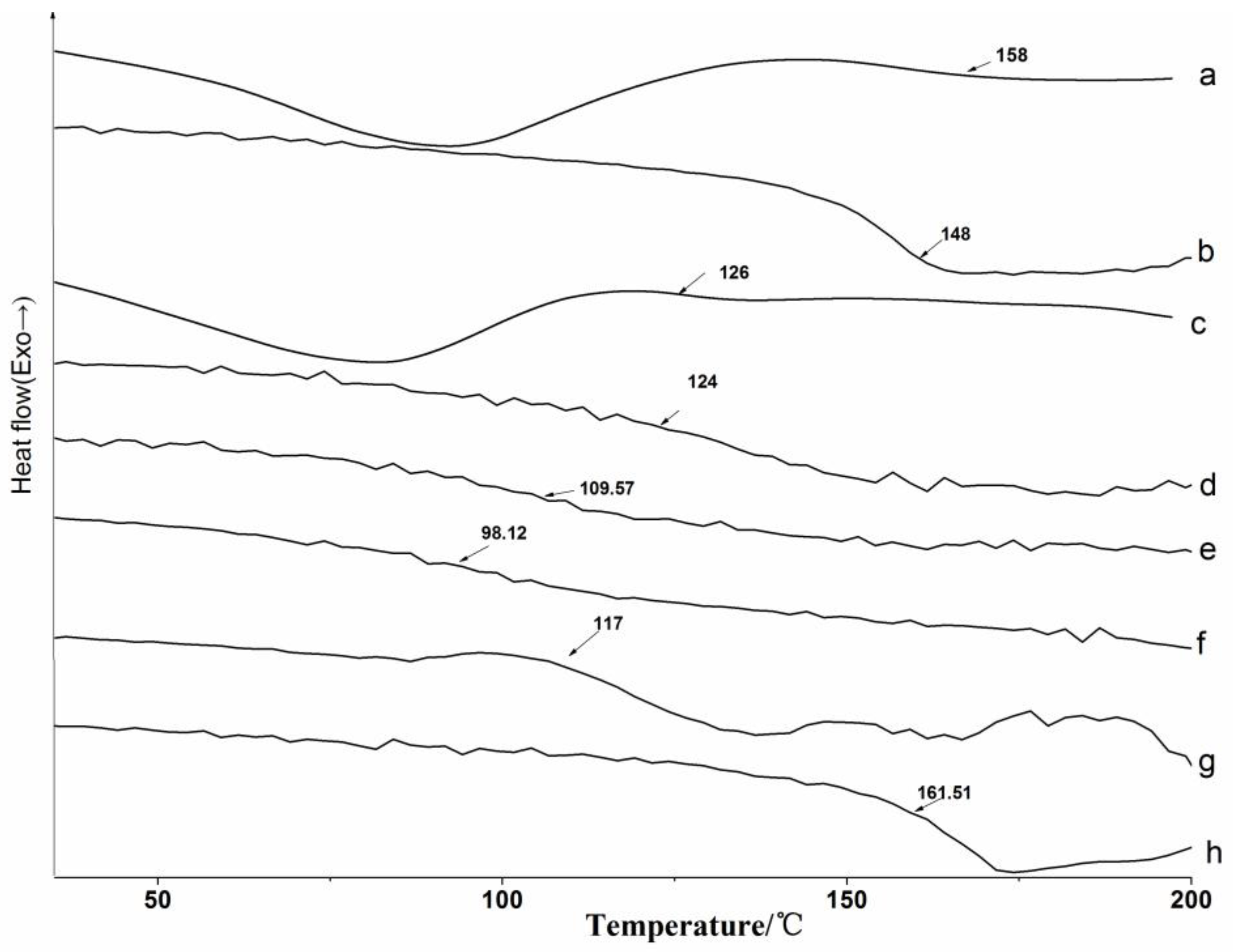
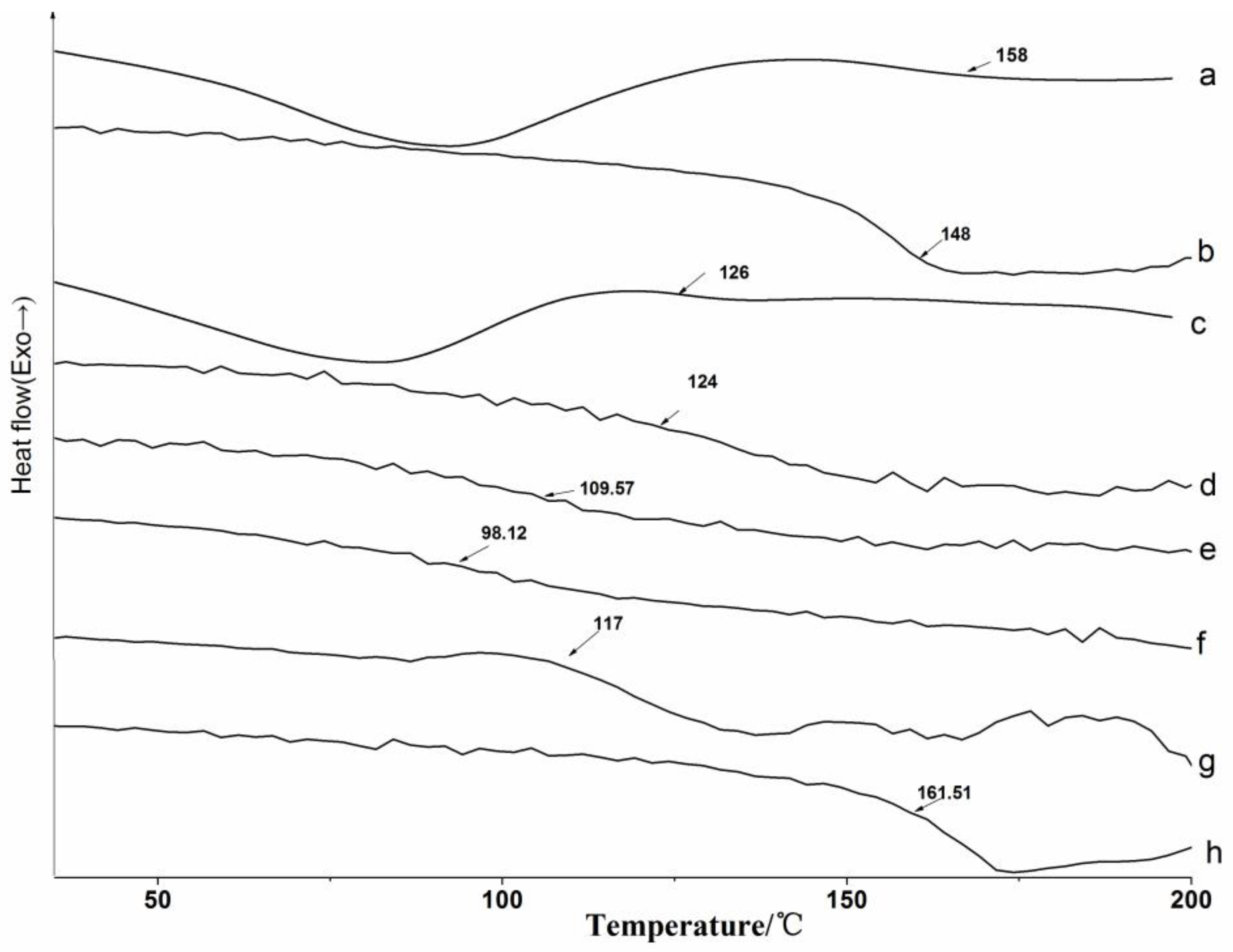
3.2.2. DSC Analysis
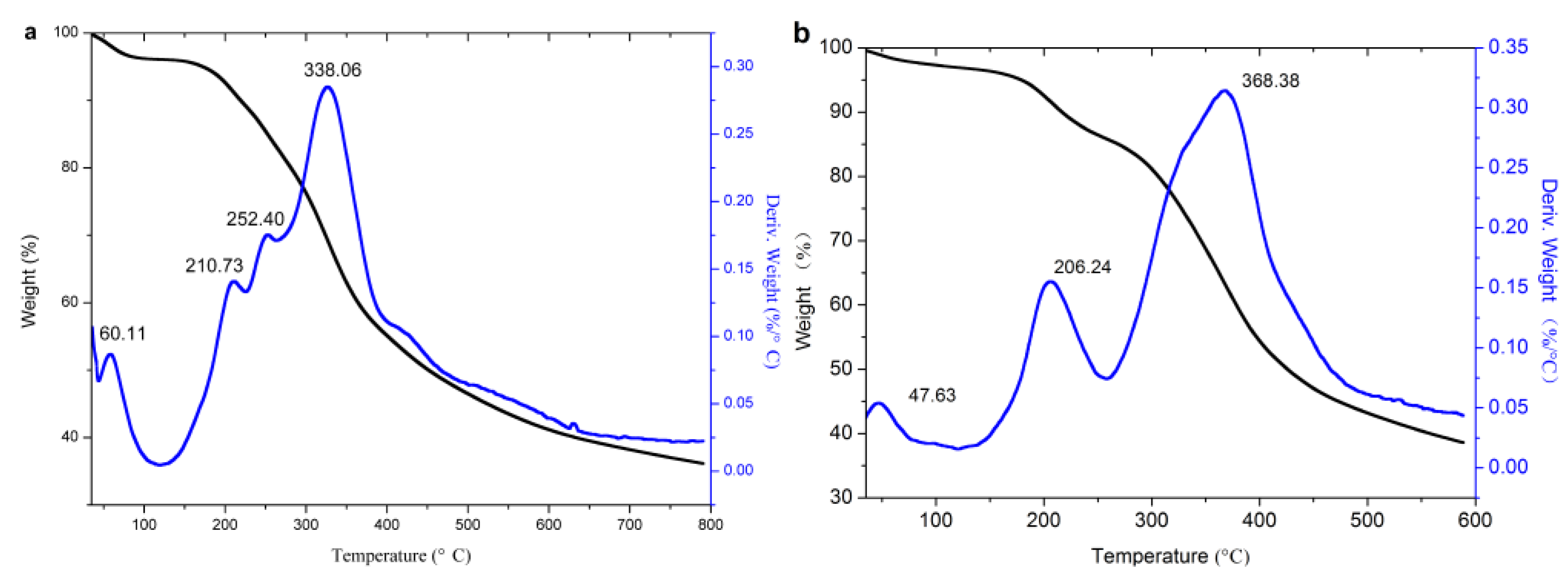
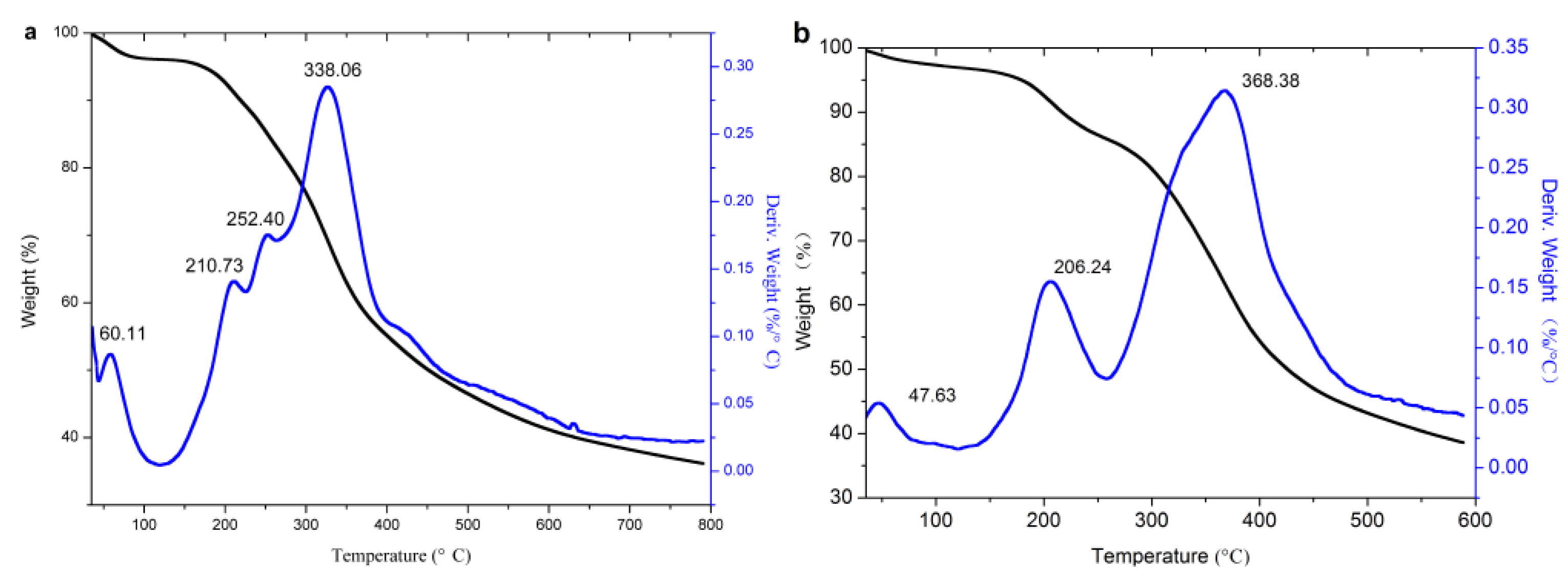
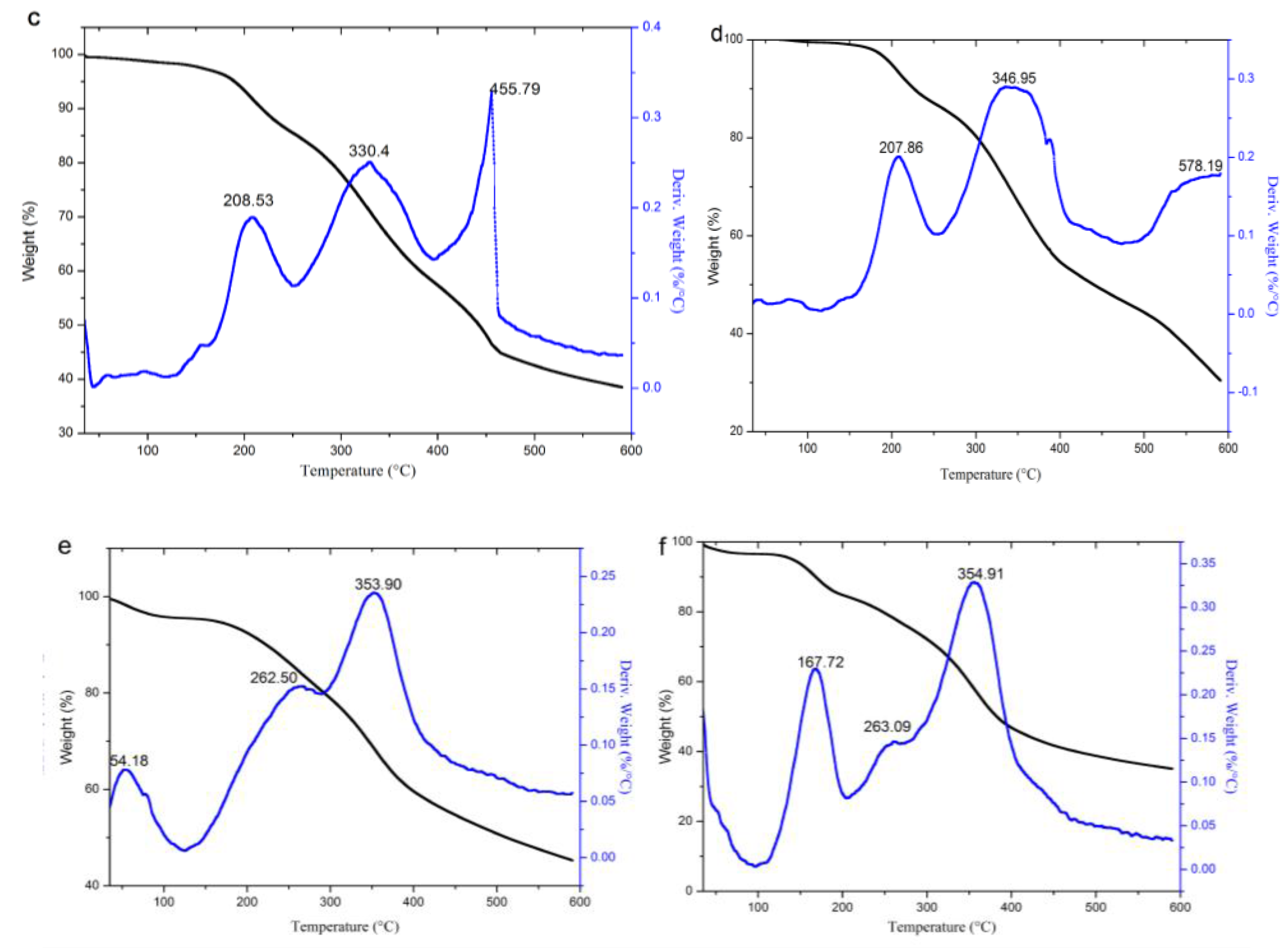
3.2.3. TG Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, S.; Yu, Y.; Di, M. Green Modification of Corn Stalk Lignin and Preparation of Environmentally Friendly Lignin-Based Wood Adhesive. Polymers 2018, 10, 631. [Google Scholar] [CrossRef]
- Zhao, G.; Ni, H.; Ren, S.; Fang, G. Correlation between Solubility Parameters and Properties of Alkali Lignin/PVA Composites. Polymers 2018, 10, 290. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, J.; Shen, D.; Xue, J.; Guan, S.; Gu, S.; Luo, K. Catalytic Oxidation of Lignin in Solvent Systems for Production of Renewable Chemicals: A Review. Polymers 2017, 9, 240. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Kai, D.; Tan, M.J.; Chee, P.L.; Chua, Y.K.; Yap, Y.L.; Loh, X.J. Towards lignin-based functional materials in a sustainable world. Green Chem. 2016, 18, 1175–1200. [Google Scholar] [CrossRef]
- Maldhure, A.V.; Chaudhari, A.R.; Ekhe, J.D. Thermal and structural studies of polypropylene blended with esterified industrial waste lignin. J. Therm. Anal. Calorim. 2011, 103, 625–632. [Google Scholar] [CrossRef]
- Thielemans, W.; Wool, R.P. Lignin esters for use in unsaturated thermosets: lignin modification and solubility modeling. Biomacromolecules 2005, 6, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Cao, J.; McDonald, A.G. Cross-linking of technical lignin via esterification and thermally initiated free radical reaction. Ind. Crops. Prod. 2018, 121, 169–179. [Google Scholar] [CrossRef]
- Kaewtatip, K.; Thongmee, J. Effect of kraft lignin and esterified lignin on the properties of thermoplastic starch. Mater. Des. 2013, 49, 701–704. [Google Scholar] [CrossRef]
- Nevárez, L.A.M.; Casarrubias, L.B.; Celzard, A.; Fierro, V.; Torres, V.; Davila, A.C.; Lubian, J.R.T.; Sánchez, G.G.; Sánchez, A.G.G. Biopolymer-based nanocomposites: effect of lignin acetylation in cellulose triacetate films. Sci. Technol. Adv. Mater. 2011, 12, 45006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordobil, O.; Robles, E.; Egüés, I.; Labidi, J. Lignin-ester derivatives as novel thermoplastic materials. RSC Adv. 2016, 6, 86909–86917. [Google Scholar] [CrossRef]
- Dehne, L.; Vila Babarro, C.; Saake, B.; Schwarz, K.U. Influence of lignin source and esterification on properties of lignin-polyethylene blends. Ind. Crops Prod. 2016, 86, 320–328. [Google Scholar] [CrossRef]
- Nadji, H.; Bedard, Y.; Benaboura, A.; Rodrigue, D.; Stevanovic, T.; Riedl, B. Value-added derivatives of soda lignin from alfa grass (Stipa tenacissima). I. Modification and characterization. J. Appl. Polym. Sci. 2010, 115, 1546–1554. [Google Scholar] [CrossRef]
- Cachet, N.; Camy, S.; Benjelloun-Mlayah, B.; Condoret, J.; Delmas, M. Esterification of organosolv lignin under supercritical conditions. Ind. Crops. Prod. 2014, 58, 287–297. [Google Scholar] [CrossRef]
- Gifford, A.P.; Westland, J.; Neogi, A.N.; Ragan, K.D. Low Tg Lignin Mixed Esters. U.S. Patent Application No. 20100152428A1, 17 June 2010. [Google Scholar]
- Monteil-Rivera, F.; Paquet, L. Solvent-free catalyst-free microwave-assisted acylation of lignin. Ind. Crops. Prod. 2015, 65, 446–453. [Google Scholar] [CrossRef]
- Wang, H.; Chen, W.; Zhang, X.; Wei, Y.; Zhang, A.; Liu, S.; Wang, X.; Liu, C. Structural Changes of Bagasse dusring the Homogeneous Esterification with Maleic Anhydride in Ionic Liquid 1-Allyl-3-methylimidazolium Chloride. Polymers 2018, 10, 433. [Google Scholar] [CrossRef]
- Bridson, J.H.; van de Pas, D.J.; Fernyhough, A. Succinylation of three different lignins by reactive extrusion. J. Appl. Polym. Sci. 2013, 128, 4355–4360. [Google Scholar] [CrossRef]
- Xiao, B.; Sun, X.F.; Sun, R. The chemical modification of lignins with succinic anhydride in aqueous systems. Polym. Degrad. Stab. 2001, 71, 223–231. [Google Scholar] [CrossRef]
- Luo, S.; Cao, J.; McDonald, A.G. Esterification of industrial lignin and its effect on the resulting poly(3-hydroxybutyrate-co-3-hydroxyvalerate) or polypropylene blends. Ind. Crops. Prod. 2017, 97, 281–291. [Google Scholar] [CrossRef]
- Fox, S.C.; McDonald, A.G. Chemical and thermal characterization of three industrial lignins and their corresponding lignin esters. BioResources 2010, 5, 990–1009. [Google Scholar]
- Ashrafi, H.; Emadi, R.; Zamani Foroushani, R. Synthesis and characterization of mullite–zirconia nanostructured composite by combined mechanical activation and reaction sintering. Adv. Powder. Technol. 2015, 26, 1452–1457. [Google Scholar] [CrossRef]
- Pourghahramani, P.; Akhgar, B.N. Influence of mechanical activation on the reactivity of natural pyrite in lead (II) removal from aqueous solutions. J. Ind. Eng. Chem. 2015, 25, 131–137. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, N.; Zhang, Y.; Hu, H.; Luo, Y. Effect of mechanical activation pretreatment on the properties of sugarcane bagasse/poly(vinyl chloride) composites. Compos. Part A 2012, 43, 114–120. [Google Scholar] [CrossRef]
- Huang, Z.; Liang, X.; Hu, H.; Gao, L.; Chen, Y.; Tong, Z. Influence of mechanical activation on the graft copolymerization of sugarcane bagasse and acrylic acid. Polym. Degrad. Stab. 2009, 94, 1737–1745. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Hu, H.; Huang, Z.; Yang, M.; Chen, D.; Huang, K.; Huang, A.; Qin, X.; Feng, Z. Effect of mechanical activation on structure changes and reactivity in further chemical modification of lignin. Int. J. Biol. Macromol. 2016, 91, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gan, T.; Hu, H.; Huang, Z.; Huang, A.; Zhu, Y.; Feng, Z.; Yang, M. A Green Technology for the Preparation of High Fatty Acid Starch Esters: Solid-Phase Synthesis of Starch Laurate Assisted by Mechanical Activation with Stirring Ball Mill as Reactor. Ind. Eng. Chem. Res. 2014, 53, 2114–2120. [Google Scholar] [CrossRef]
- Zhang, Y.; Gan, T.; Luo, Y.; Zhao, X.; Hu, H.; Huang, Z.; Huang, A.; Qin, X. A green and efficient method for preparing acetylated cassava stillage residue and the production of all-plant fibre composites. Compos. Sci. Technol. 2014, 102, 139–144. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, Z.; Zhang, Y.; Yang, M.; Chen, D.; Huang, K.; Hu, H.; Huang, A.; Qin, X.; Feng, Z. Efficient solid-phase synthesis of acetylated lignin and a comparison of the properties of different modified lignins. J. Appl. Polym. Sci. 2016, 44276. [Google Scholar] [CrossRef]
- Hu, H.; Li, H.; Zhang, Y.; Chen, Y.; Huang, Z.; Huang, A.; Zhu, Y.; Qin, X.; Lin, B. Green mechanical activation-assisted solid phase synthesis of cellulose esters using a co-reactant: effect of chain length of fatty acids on reaction efficiency and structure properties of products. RSC Adv. 2015, 5, 20656–20662. [Google Scholar] [CrossRef]
- Huang, Z.; Xie, X.; Chen, Y.; Lu, J.; Tong, Z. Ball-milling treatment effect on physicochemical properties and features for cassava and maize starches. CR. Chim. 2008, 11, 73–79. [Google Scholar] [CrossRef]
- Lin, S.Y.; Dence, C.W. Methods in Lignin Chemistry; Springer Series in Wood Science; Springer: Berlin, Germany, 1992; pp. 217–232. [Google Scholar]
- Cybulska, I.; Brudecki, G.; Rosentrater, K.; Julson, J.L.; Lei, H. Comparative study of organosolv lignin extracted from prairie cordgrass, switchgrass and corn stover. Bioresour. Technol. 2012, 118, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Fang, J.M.; Goodwin, A.; Lawther, J.M.; Bolton, A.J. Fractionation and characterization of ball-milled and enzyme lignins from abaca fibre. J. Sci. Food Agric. 1999, 79, 1091–1098. [Google Scholar] [CrossRef]
- Gouveia, S.; Otero, L.; Fernández-Costas, C.; Filgueira, D.; Sanromán, Á.; Moldes, D. Green Binder Based on Enzymatically Polymerized Eucalypt Kraft Lignin for Fiberboard Manufacturing: A Preliminary Study. Polymers 2018, 10, 642. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin-a review. Cell. Chem. Technol. 2010, 44, 353–363. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | First Stage (30–125 °C) | Second Stage (125–268 °C) | Temperature of 5% Weight Loss(°C) | Third Stage (268–600 °C) | Temperature of Max Weight Loss (°C) | |||
|---|---|---|---|---|---|---|---|---|
| Peak (°C) | Weight Loss (%) | Peak (°C) | Weight Loss (%) | Peak (°C) | Weight Loss (%) | |||
| Original lignin | 60.01 | 3.97 | 210.73, 252.40 | 17.68 | 172.69 | 338.06 | 61.63 | 338.06 |
| Acetylated lignin (1.5 h) | 47.63 | 3.06 | 206.24 | 14.58 | 178.87 | 368.38 | 61.58 | 368.38 |
| Propionylated lignin (1.5 h) | -- | 1.75 | 208.53 | 14.40 | 189.9 | 330.4, 455.79 | 62.53 | 455.79 |
| Butyrated lignin (2 h) | -- | 0.92 | 207.86 | 14.26 | 199.6 | 346.95, 578.19 | 69.53 | 346.95 |
| Succinylated lignin (2 h) | 54.18 | 4.77 | 262.50 | 16.27 | 157.21 | 353.9 | 61.47 | 353.9 |
| Maleylated lignin (2 h) | 35 | 4.51 | 167.72, 263.09 | 23.03 | 140.3 | 354.91 | 54.7 | 354.91 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Zhang, Y.; Yang, M.; Huang, Z.; Hu, H.; Huang, A.; Feng, Z. Acylation of Lignin with Different Acylating Agents by Mechanical Activation-Assisted Solid Phase Synthesis: Preparation and Properties. Polymers 2018, 10, 907. https://doi.org/10.3390/polym10080907
Zhao X, Zhang Y, Yang M, Huang Z, Hu H, Huang A, Feng Z. Acylation of Lignin with Different Acylating Agents by Mechanical Activation-Assisted Solid Phase Synthesis: Preparation and Properties. Polymers. 2018; 10(8):907. https://doi.org/10.3390/polym10080907
Chicago/Turabian StyleZhao, Xiaohong, Yanjuan Zhang, Mei Yang, Zuqiang Huang, Huayu Hu, Aimin Huang, and Zhenfei Feng. 2018. "Acylation of Lignin with Different Acylating Agents by Mechanical Activation-Assisted Solid Phase Synthesis: Preparation and Properties" Polymers 10, no. 8: 907. https://doi.org/10.3390/polym10080907