Compatibility Evaluation of Non-Woven Sheet Composite of Silk Fibroin and Polyurethane in the Wet State

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Production of Silk Fibroin Sponge
2.2. Fabrication of SF/PU Composite Non-Woven Sheet
2.3. Confirmation of Fabrication and Morphological Evaluation of SF/PU Composite Non-Woven Sheet by SEM Observation
2.4. High-Resolution Solid-State 13C NMR Measurement
2.5. Mechanical Property Test
3. Results
3.1. Morphology of SF/PU Composite Non-Woven Sheets
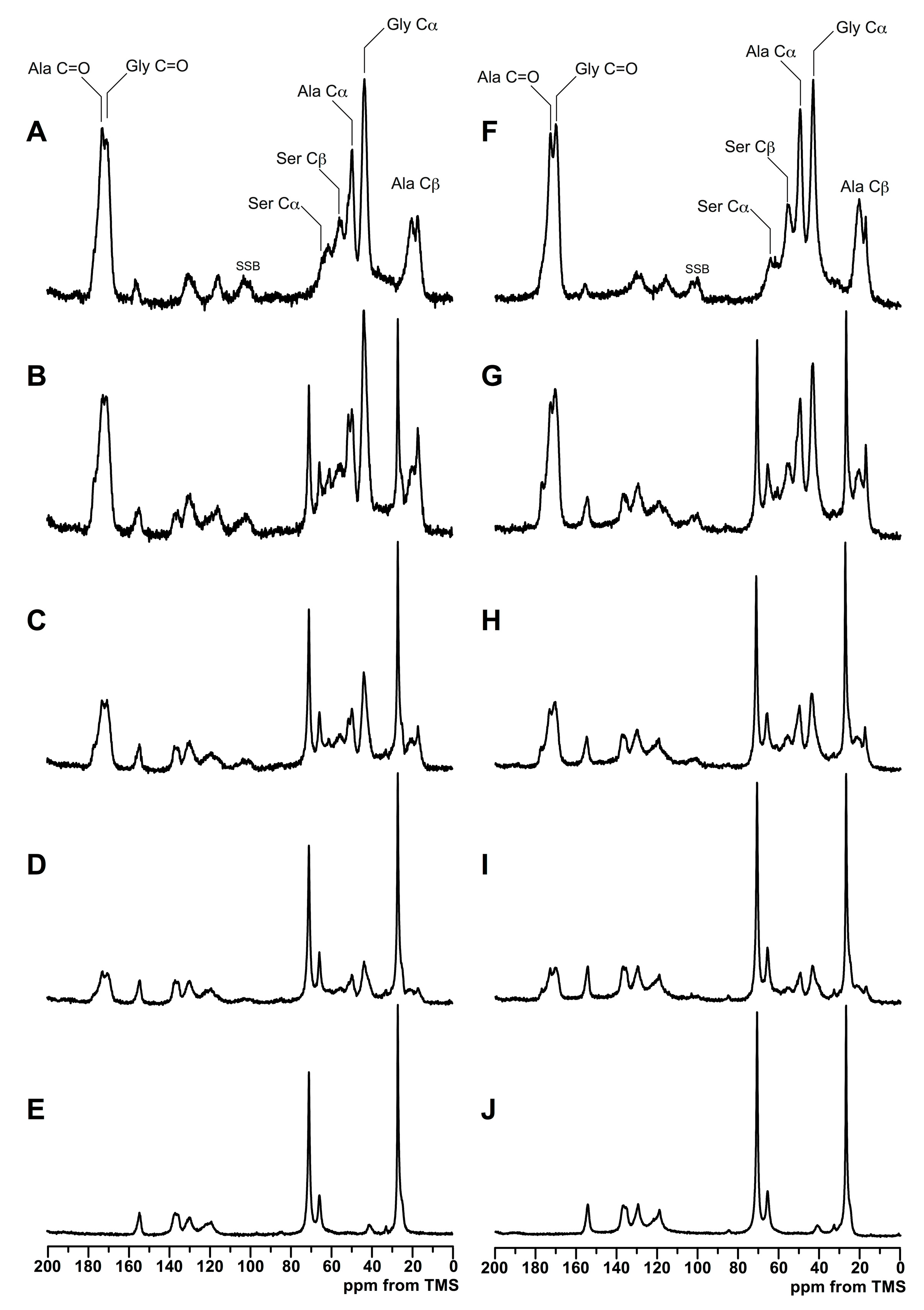
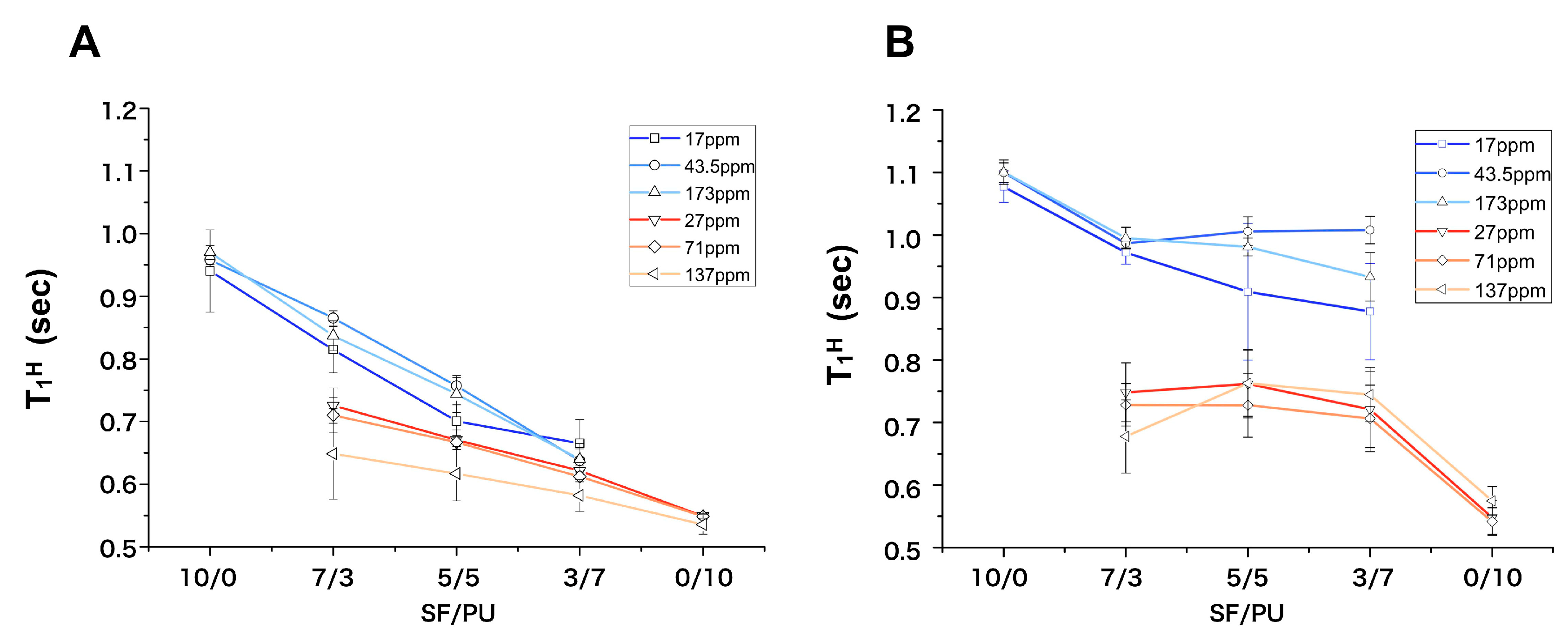
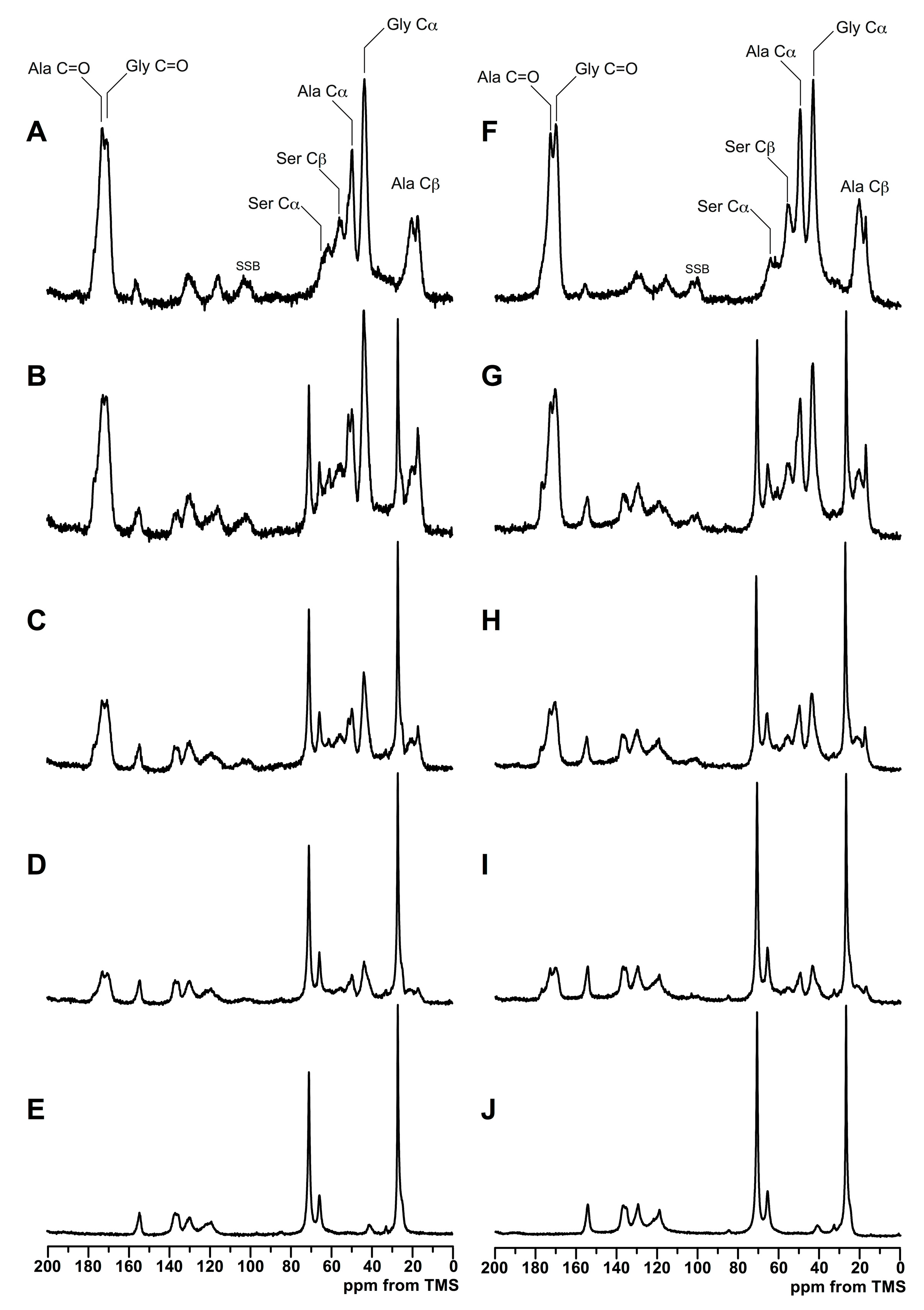
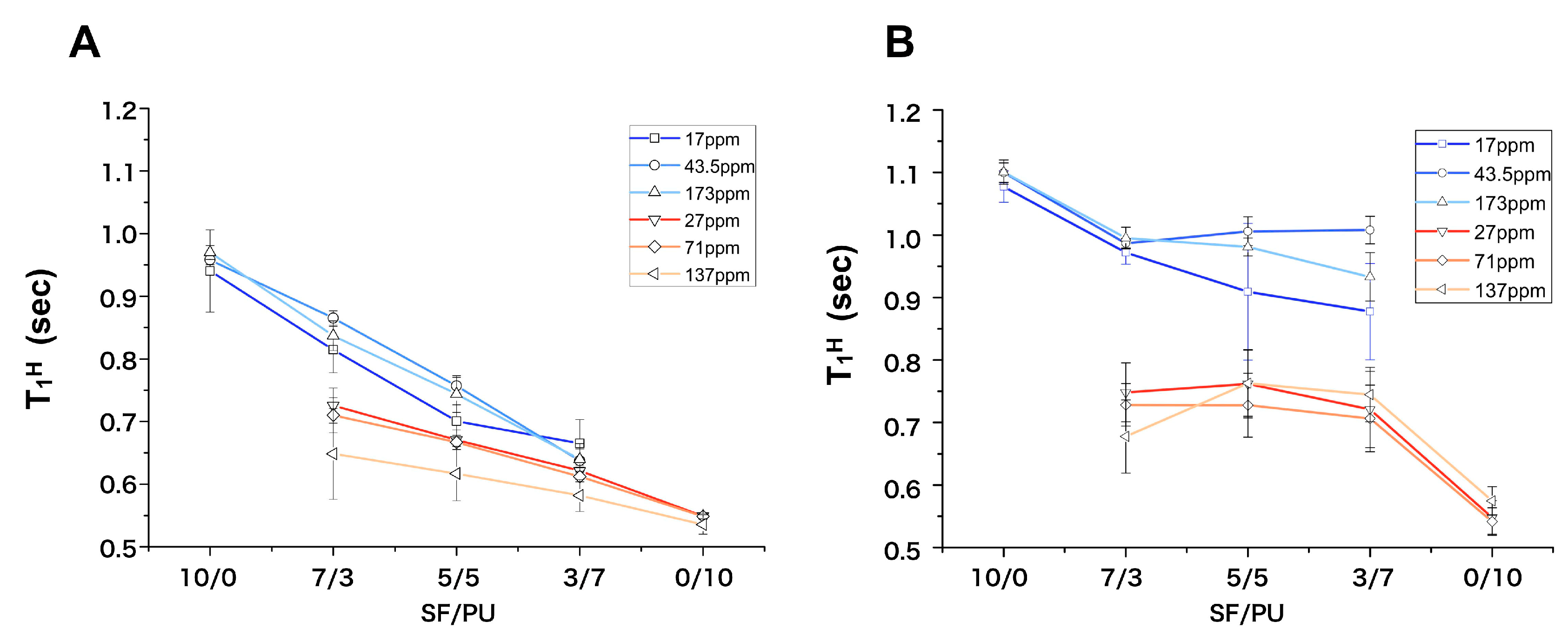
3.2. Structure Analysis by Solid State NMR Measurement
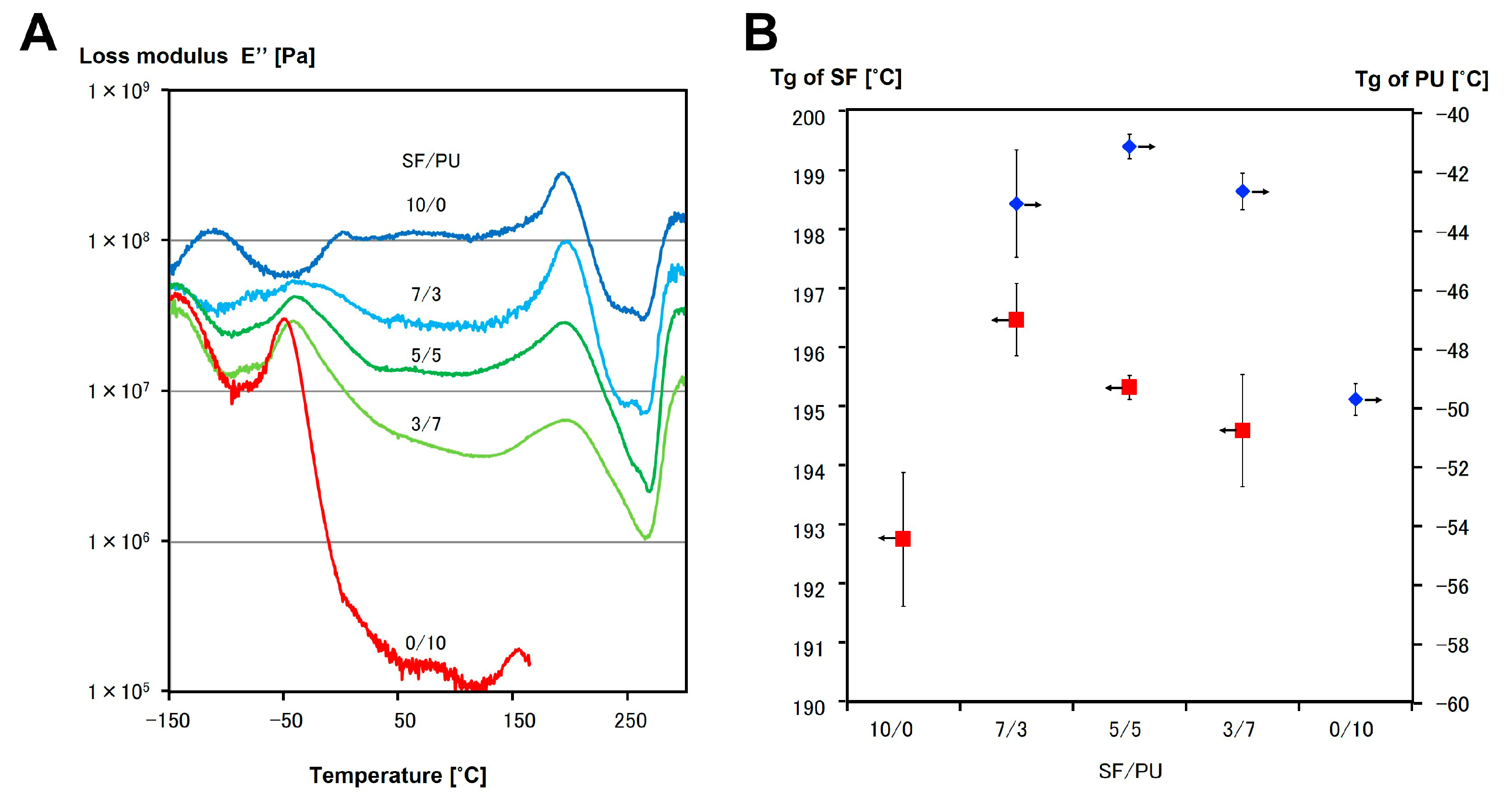
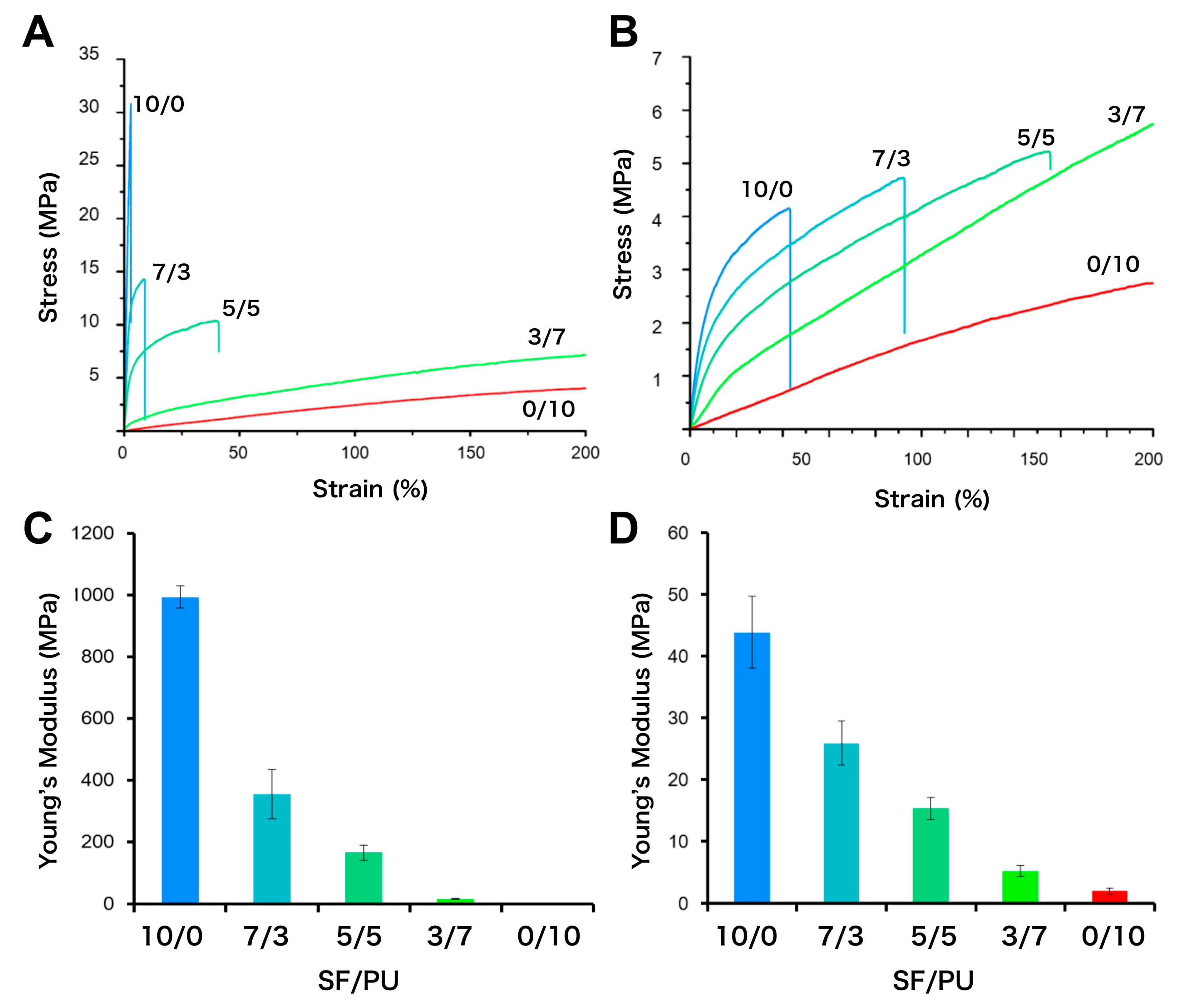
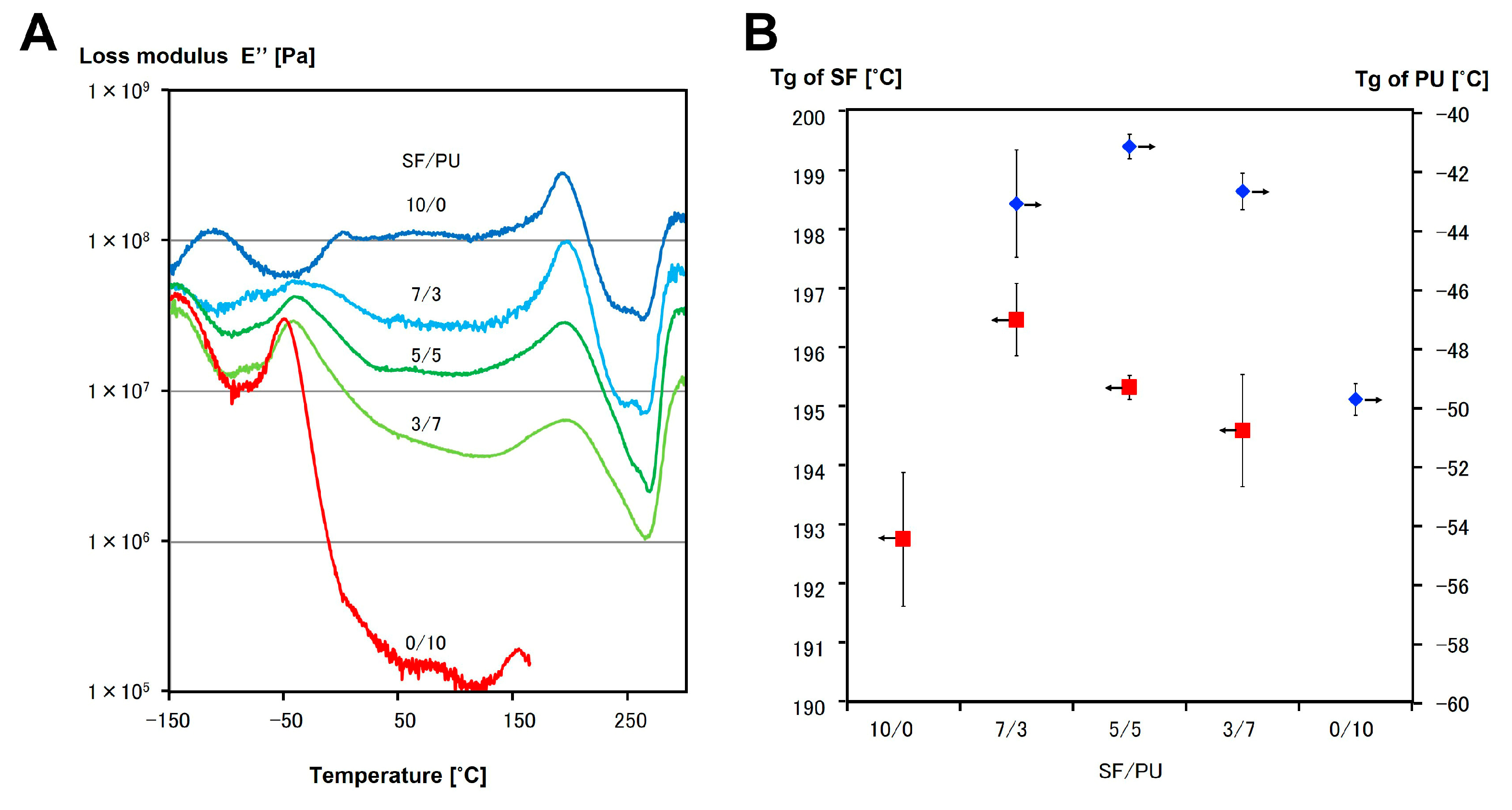
3.3. Mechanical Properties of SF/PU Composite Non-Woven Sheets
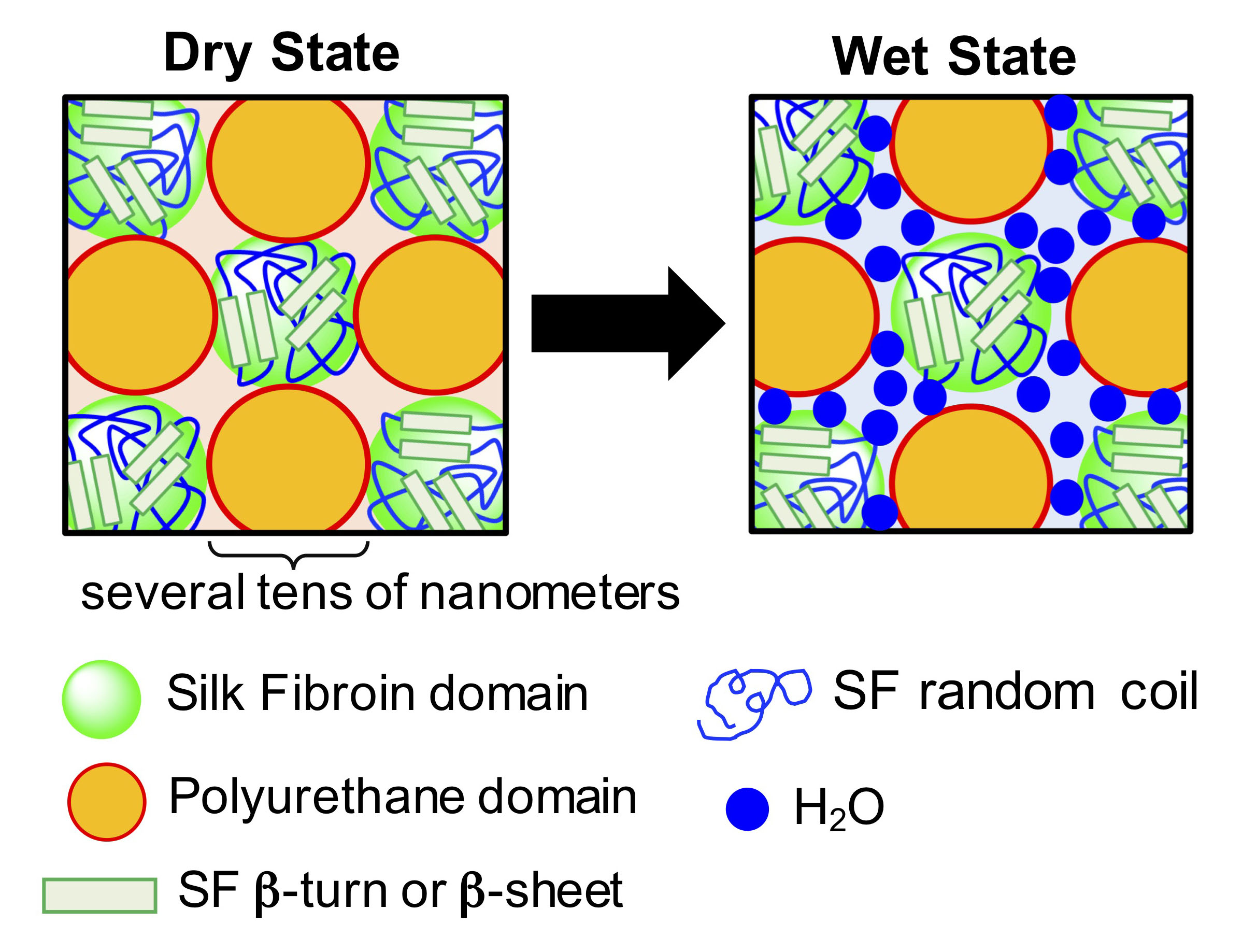
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Scott, T.A.; Towle, H.J.; Assad, D.A.; Nicoll, B.K. Comparison of bioabsorbable laminar bone membrane and non-resorbable ePTFE membrane in mandibular furcations. J. Periodontol. 1997, 68, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Okano, T.; Sakai, H.; Karikusa, F.; Sawasaki, Y.; Sakurai, Y. Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Die Makromol. Chem. Rapid Commun. 1990, 11, 571–576. [Google Scholar] [CrossRef]
- Masumoto, H.; Ikuno, T.; Takeda, M.; Fukushima, H.; Marui, A.; Katayama, S.; Shimizu, T.; Ikeda, T.; Okano, T.; Sakata, R.; et al. Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci. Rep. 2014, 4, 6716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rudym, D.D.; Walsh, A.; Abrahamsen, L.; Kim, H.J.; Kim, H.S.; Kirker-Head, C.; Kaplan, D.L. In vivo degradation of three-dimensional silk fibroin scaffolds. Biomaterials 2008, 29, 3415–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vepari, C.; Kaplan, D.L. Silk as a biomaterial. Prog. Polym. Sci. 2007, 32, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Z.; Confalonieri, F.; Medina, N.; Zivanovic, Y.; Esnault, C.; Yang, T.; Jacquet, M.; Janin, J.; Duguet, M.; Perasso, R.; et al. Fine organization of Bombyx mori fibroin heavy chain gene. Nucleic Acids Res. 2000, 28, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- Panilaitis, B.; Altman, G.H.; Chen, J.; Jin, H.J.; Karageorgiou, V.; Kaplan, D.L. Macrophage responses to silk. Biomaterials 2003, 24, 3079–3085. [Google Scholar] [CrossRef]
- Santin, M.; Motta, A.; Freddi, G.; Cannas, M. In vitro evaluation of the inflammatory potential of the silk fibroin. J. Biomed. Mater. Res. 1999, 46, 382–389. [Google Scholar] [CrossRef]
- Plaza, G.R.; Corsini, P.; Marsano, E.; Pérez-Rigueiro, J.; Elices, M.; Riekel, C.; Vendrely, C.; Guinea, G.V. Correlation Between Processing Conditions, Microstructure and Mechanical Behavior in Regenerated Silkworm Silk Fibers. J. Polym. Sci. B Polym. Phys. 2011, 50, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Shmelev, K.; Sun, L.; Gil, E.-S.; Park, S.-H.; Cebe, P.; Kaplan, D.L. Regulation of silk material structure by temperature-controlled water vapor annealing. Biomacromolecules 2011, 12, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Talano, H.; Hayashi, K.; Taenaka, Y.; Takaichi, S.; Umezu, M.; Nakamura, T.; Iwata, H.; Nakatani, T.; Tanaka, T.; et al. The blood interface with segmented polyurethanws: “Multilayered protein passivation mechanism”. ASAIO J. 1984, 30, 353–358. [Google Scholar]
- Chiarini, A.; Petrini, P.; Bozzini, S.; dal Pra, I.; Armato, U. Silk fibroin/poly(carbonate)-urethane as a substrate for cell growth: In vitro interactions with human cells. Biomaterials 2003, 24, 789–799. [Google Scholar] [CrossRef]
- Park, H.S.; Gong, M.-S.S.; Park, J.-H.H.; Moon, S.I.; Wall, I.B.; Kim, H.-W.W.; Lee, J.H.; Knowles, J.C. Silk fibroin-polyurethane blends: Physical properties and effect of silk fibroin content on viscoelasticity, biocompatibility and myoblast differentiation. Acta Biomater. 2013, 9, 8962–8971. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Chung, O.H.; Park, J.S. Coaxial electrospun poly(lactic acid)/chitosan (core/shell) composite nanofibers and their antibacterial activity. Carbohydr. Polym. 2011, 86, 1799–1806. [Google Scholar] [CrossRef]
- Heiny, M.; Shastri, V.P. Nanofibers of elastin and hydrophilic segmented polyurethane solution blends show enhanced mechanical properties through intermolecular protein-polymer H bonding. Biomacromolecules 2016, 17, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, R.; Venugopal, J.R.; Sundarrajan, S.; Mukherjee, S.; Ramakrishna, S. Poly(glycerol sebacate)/gelatin core/shell fibrous structure for regeneration of myocardial infarction. Tissue Eng. A 2011, 17, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Zhang, J.; Thomson, J.A.; Turng, L.S. Fabrication and characterization of electrospun thermoplastic polyurethane/fibroin small-diameter vascular grafts for vascular tissue engineering. Int. Polym. Process. 2016, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
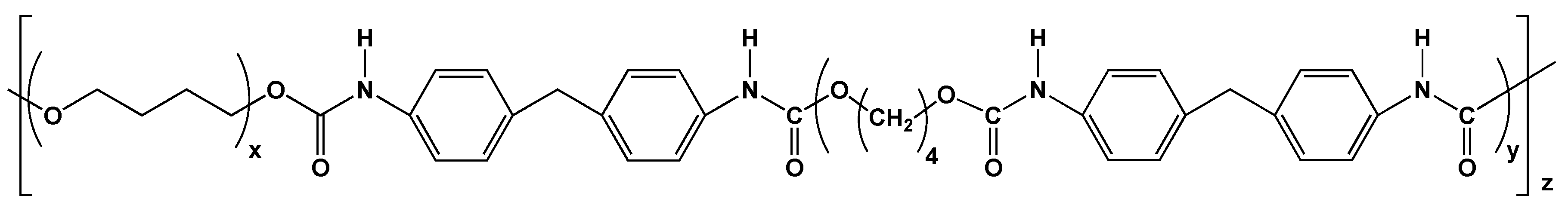
- Nakazawa, C.T.; Higuchi, A.; Asano, A.; Kameda, T.; Aytemiz, D.; Nakazawa, Y. Solid-state NMR studies for the development of non-woven biomaterials based on silk fibroin and polyurethane. Polym. J. 2017, 49, 583–586. [Google Scholar] [CrossRef]
- Shimada, K.; Higuchi, A.; Kubo, R.; Murakami, T.; Nakazawa, Y.; Tanaka, R. The effect of a silk fibroin/polyurethane blend patch on rat vessels. Organogenesis 2017, 13, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chantawong, P.; Tanaka, T.; Uemura, A.; Shimada, K.; Higuchi, A.; Tajiri, H.; Sakura, K.; Murakami, T.; Nakazawa, Y.; Tanaka, R. Silk fibroin-Pellethane® cardiovascular patches: Effect of silk fibroin concentration on vascular remodeling in rat model. J. Mater. Sci. Mater. Med. 2017, 28, 191. [Google Scholar] [CrossRef] [PubMed]
- Shimada, R.; Konishi, H.; Ozawa, H.; Katsumata, T.; Tanaka, R.; Nakazawa, Y.; Nemoto, S. Development of a new surgical sheet containing both silk fibroin and thermoplastic polyurethane for cardiovascular surgery. Surg. Today 2017, 48, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.W.; Paul, D.R. Polymer blends and alloys-a review of selected considerations. Polym. Eng. Sci. 1981, 21, 985–996. [Google Scholar] [CrossRef]
- Fukuda, Y.; Aytemiz, D.; Higuchi, A.; Ichida, Y.; Asakura, T.; Kameda, T.; Nakazawa, Y. Relationship between structure and physical strength of silk fibroin nanofiber sheet depending on insolubilization treatment. J. Appl. Polym. Sci. 2017, 134, 45560. [Google Scholar] [CrossRef]
- Bennett, A.E.; Rienstra, C.M.; Auger, M.; Lakshmi, K.V.; Griffin, R.G. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995, 103, 6951–6958. [Google Scholar] [CrossRef]
- Asano, A. Polymer Blends. In Modern Magnetic Resonance; Graham, W.A., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 623–627. [Google Scholar]
- Plaza, G.R.; Corsini, P.; Pérez-Rigueiro, J.; Marsano, E.; Guinea, G.V.; Elices, M. Effect of water onBombyx mori regenerated silk fibers and its application in modifying their mechanical properties. J. Appl. Polym. Sci. 2008, 109, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Asakura, T.; Saotome, T.; Aytemiz, D.; Shimokawatoko, H.; Yagi, T.; Fukayama, T.; Ozai, Y.; Tanaka, R. Characterization of silk sponge in the wet state using 13C solid state NMR for development of a porous silk vascular graft with small diameter. RSC Adv. 2014, 4, 4427–4434. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SF/PU | 10/0 | 7/3 | 5/5 | 3/7 | 0/10 |
|---|---|---|---|---|---|
| Fiber diameter (µm) | 1.2 ± 1.1 | 0.9 ± 0.6 | 0.9 ± 0.4 | 0.7 ± 0.2 | 1.2 ± 0.4 |
| Chemical Shifts of Ala Cβ Peak in SF (Structure Which Was Assigned to) | Dry State (%) | Wet State (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| SF/PU Ratio | SF/PU Ratio | |||||||
| 10/0 | 7/3 | 5/5 | 3/7 | 10/0 | 7/3 | 5/5 | 3/7 | |
| 16.6 ppm (random coil and repeated β-turn) | 47 | 52 | 42 | 35 | 32 | 36 | 38 | 27 |
| 19.6 ppm (β-sheet) | 27 | 36 | 42 | 34 | 57 | 52 | 34 | 63 |
| 21.9 ppm (β-sheet) | 26 | 12 | 17 | 31 | 12 | 12 | 28 | 10 |
| SF/PU | 10/0 | 7/3 | 5/5 | 3/7 | 0/10 |
|---|---|---|---|---|---|
| Water content (%) | 73 ± 1.7 | 71 ± 1.4 | 70 ± 0.7 | 65 ± 0.5 | 16 ± 6.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aytemiz, D.; Fukuda, Y.; Higuchi, A.; Asano, A.; Nakazawa, C.T.; Kameda, T.; Yoshioka, T.; Nakazawa, Y. Compatibility Evaluation of Non-Woven Sheet Composite of Silk Fibroin and Polyurethane in the Wet State. Polymers 2018, 10, 874. https://doi.org/10.3390/polym10080874
Aytemiz D, Fukuda Y, Higuchi A, Asano A, Nakazawa CT, Kameda T, Yoshioka T, Nakazawa Y. Compatibility Evaluation of Non-Woven Sheet Composite of Silk Fibroin and Polyurethane in the Wet State. Polymers. 2018; 10(8):874. https://doi.org/10.3390/polym10080874
Chicago/Turabian StyleAytemiz, Derya, Yasuhiro Fukuda, Akira Higuchi, Atsushi Asano, Chikako T. Nakazawa, Tsunenori Kameda, Taiyo Yoshioka, and Yasumoto Nakazawa. 2018. "Compatibility Evaluation of Non-Woven Sheet Composite of Silk Fibroin and Polyurethane in the Wet State" Polymers 10, no. 8: 874. https://doi.org/10.3390/polym10080874