Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure

 ,
,  ,
,  ,
, 
Abstract
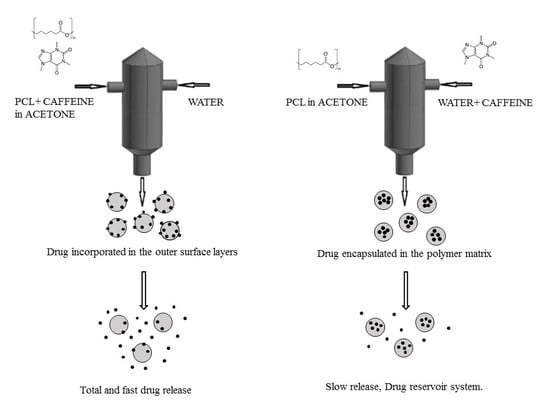
:
1. Introduction
2. Materials and Methods
2.1. Materials
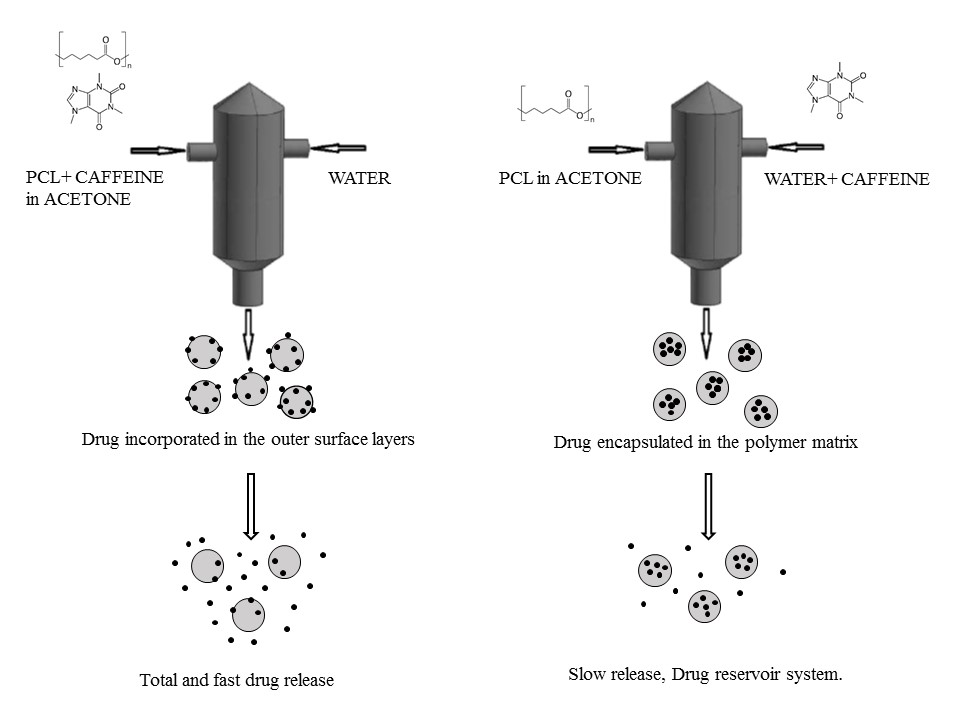
2.2. Nanoparticle Preparation
2.3. Analytical Methods
2.3.1. Nanoparticle Size
2.3.2. Determination of Loading Capacity and Encapsulation Efficiency
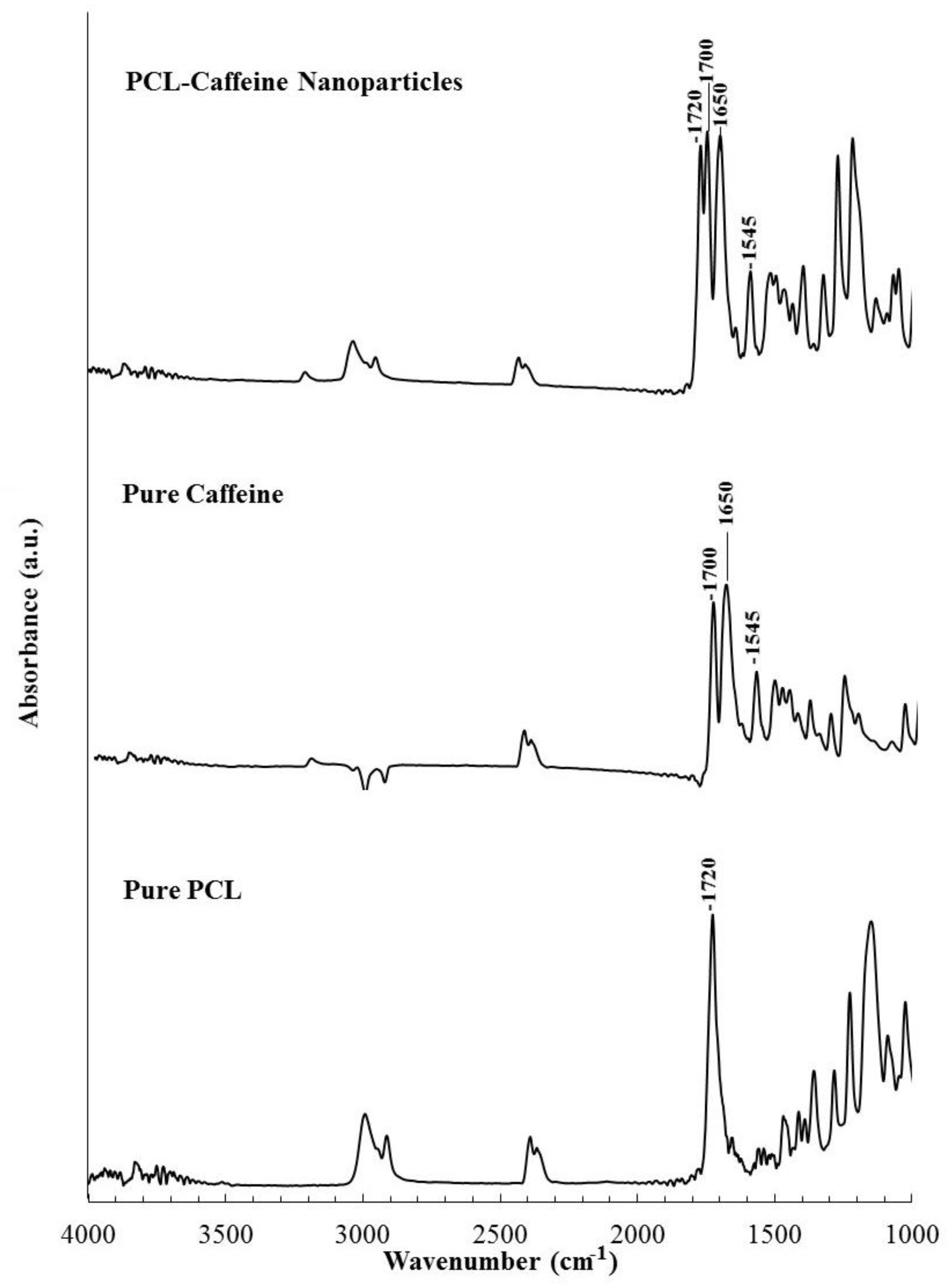
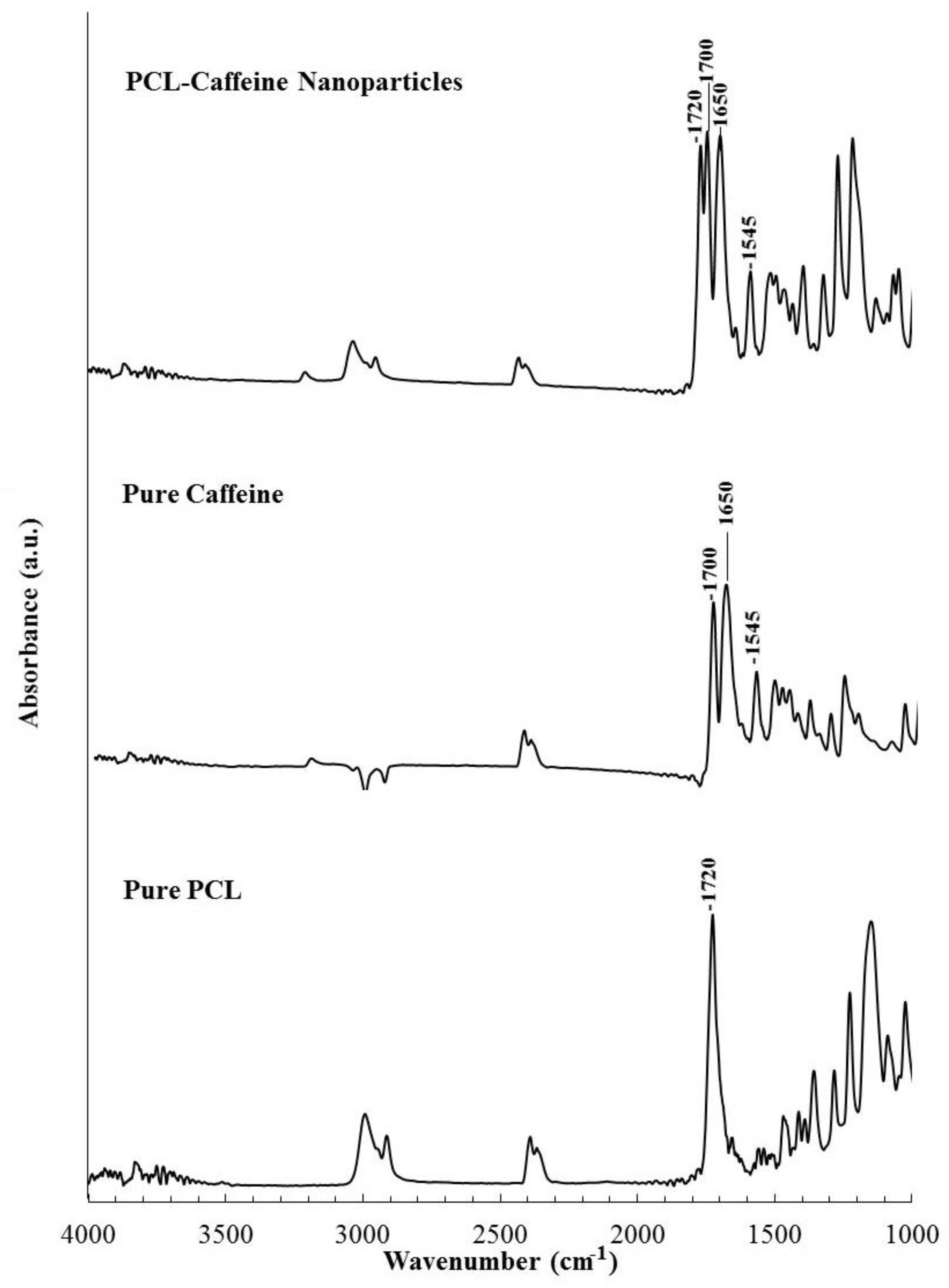
2.3.3. Fourier Transform Infrared Spectroscopy
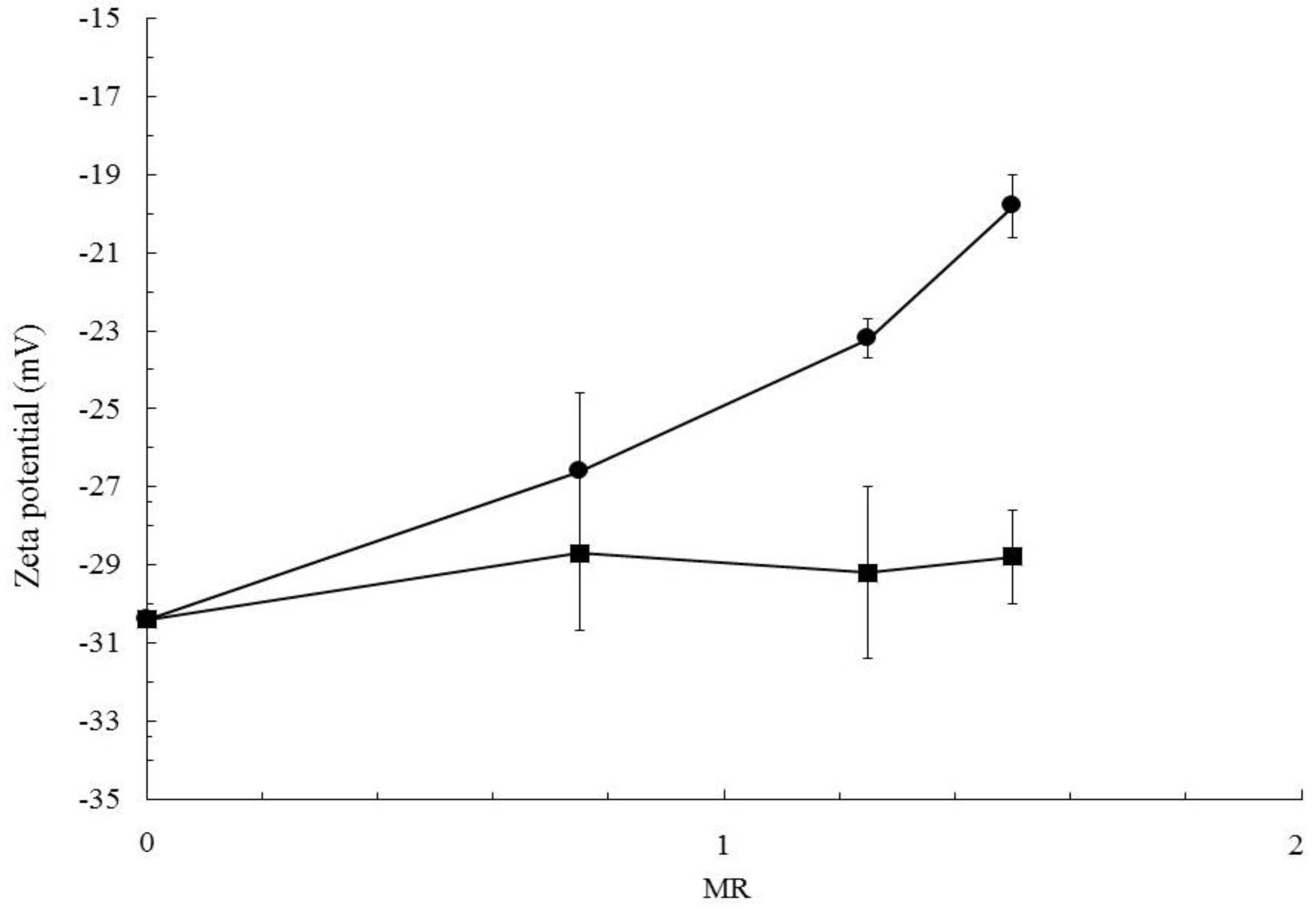
2.3.4. Zeta Potential
2.3.5. X-ray Photoelectron Spectroscopy
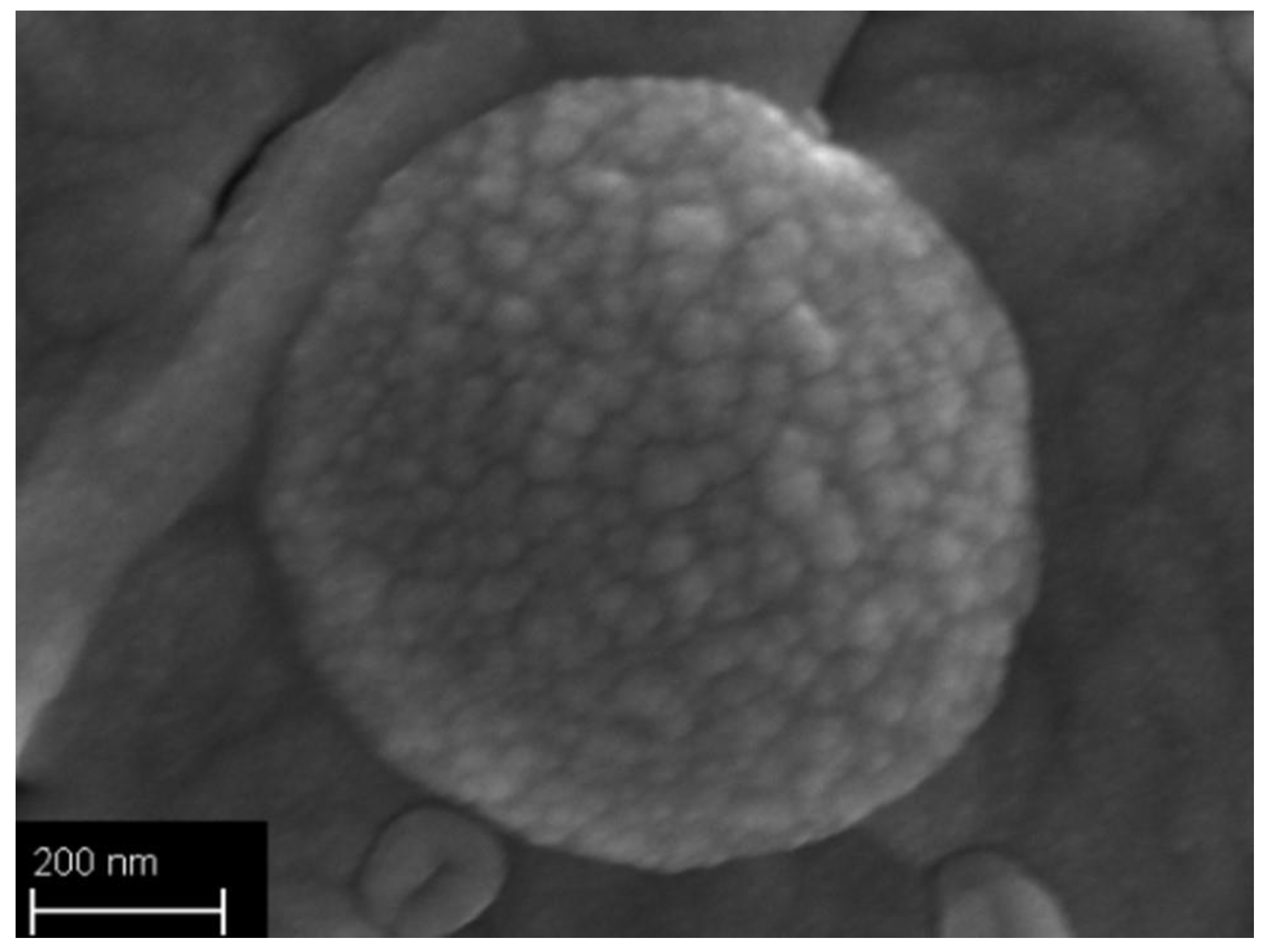
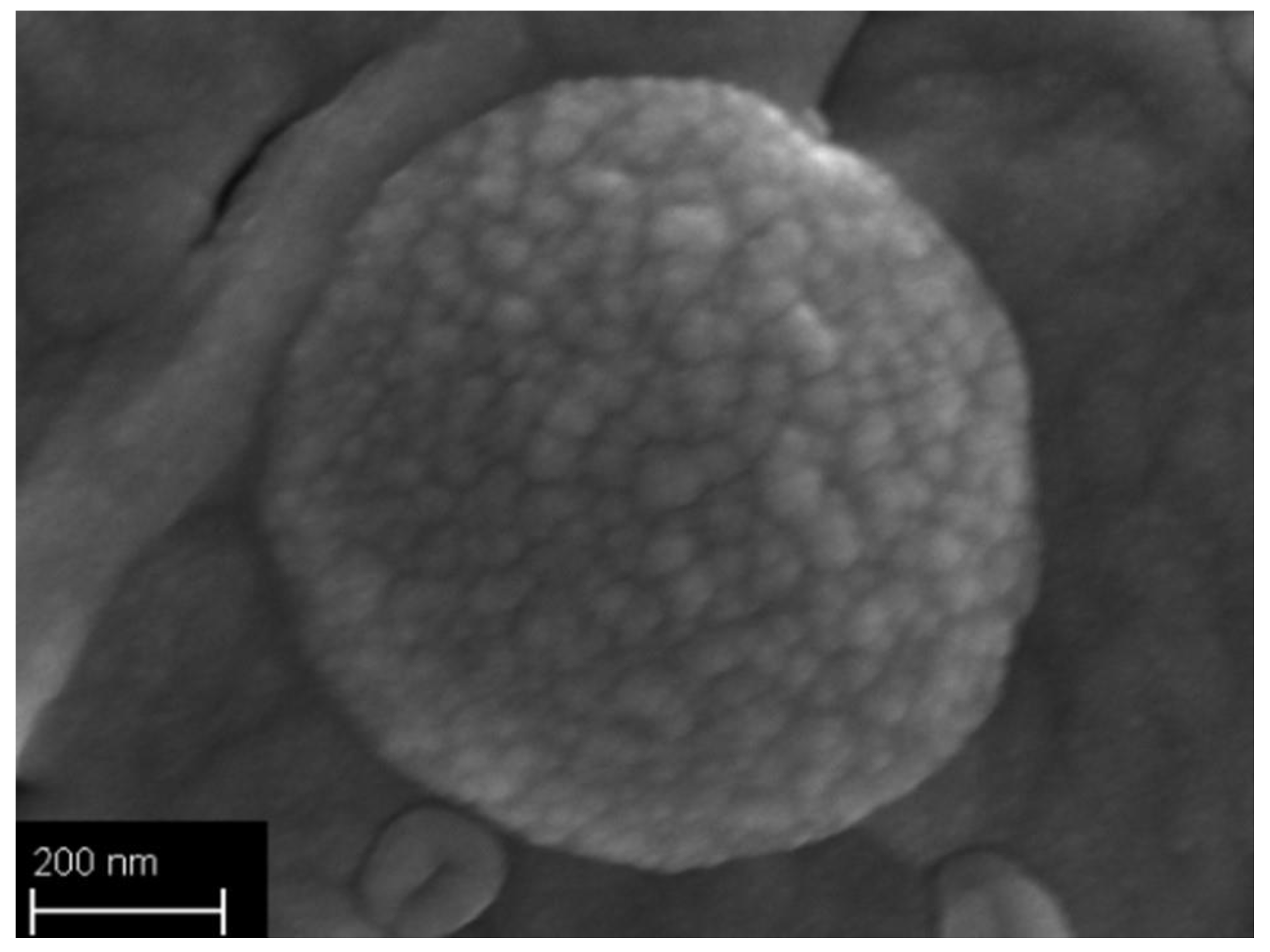
2.3.6. Field Emission Scanning Electron Microscopy
2.3.7. In Vitro Release Test
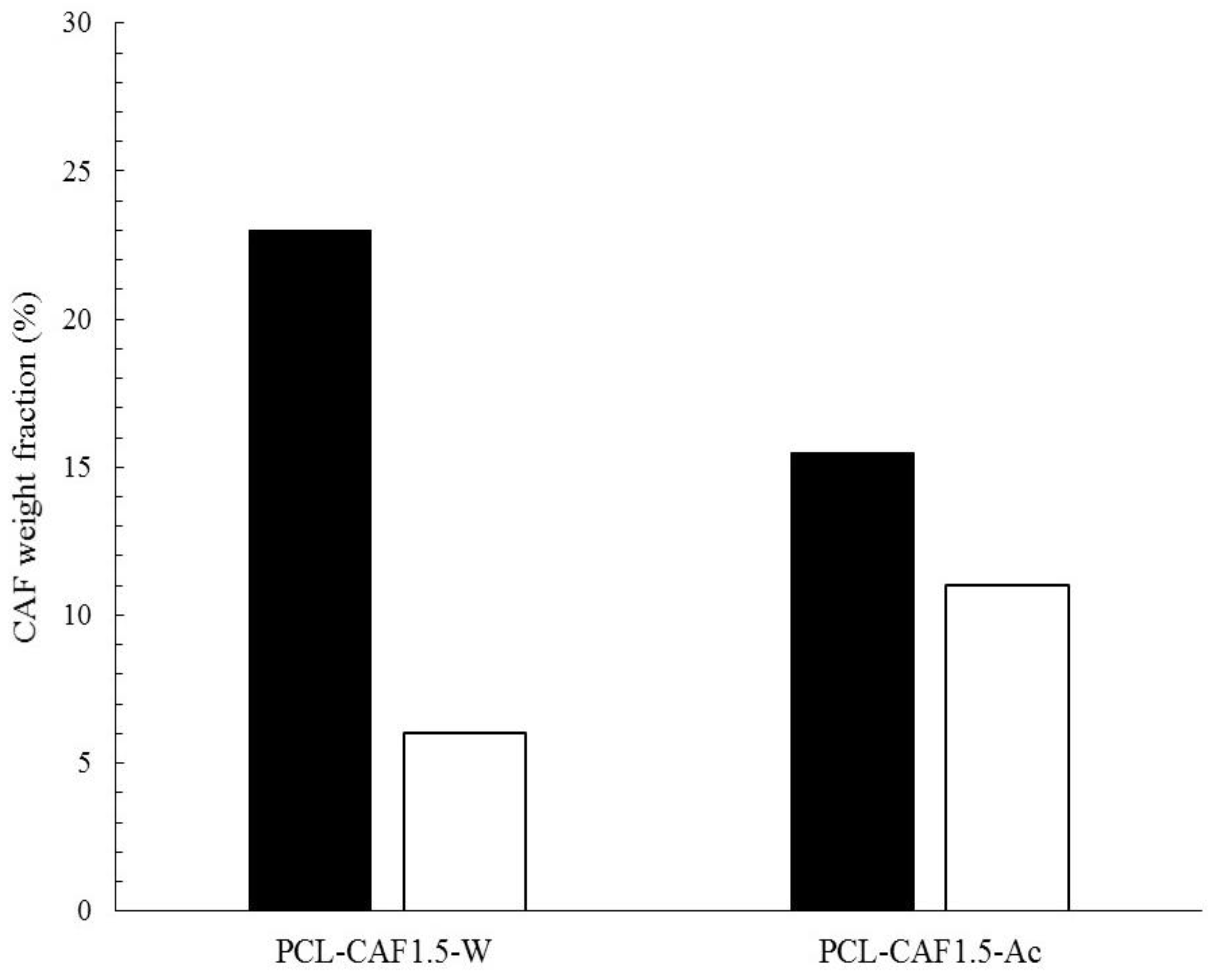
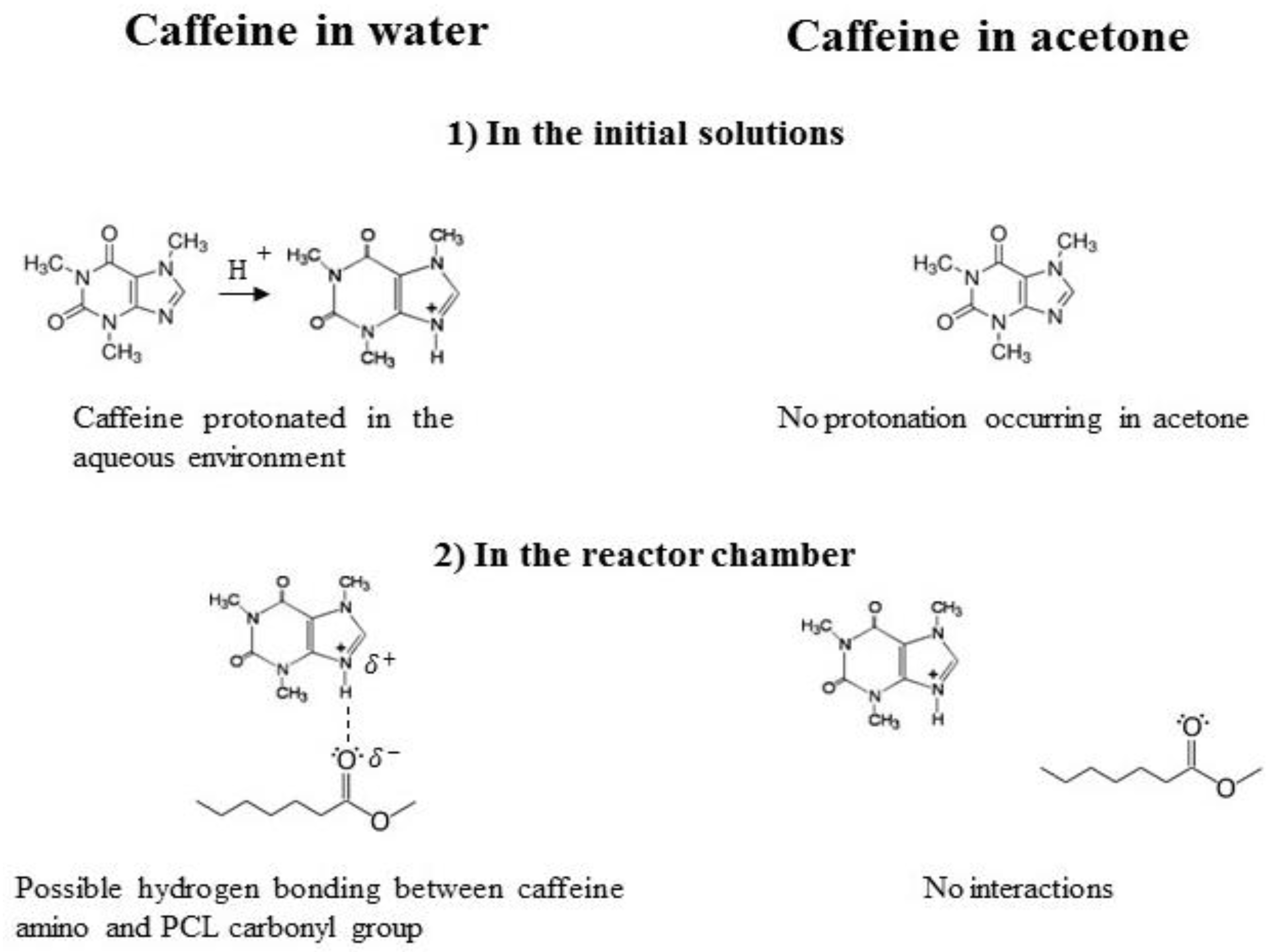
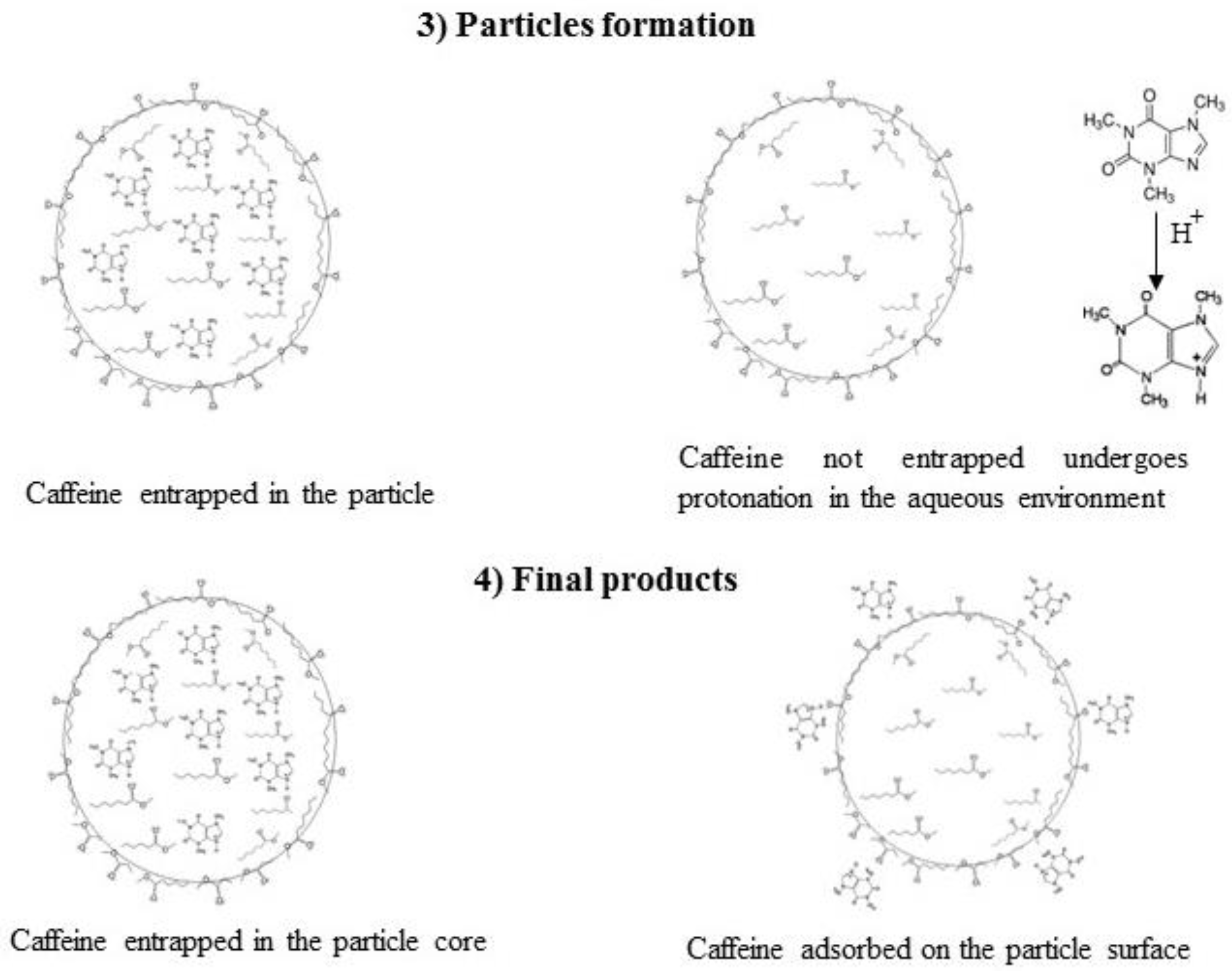
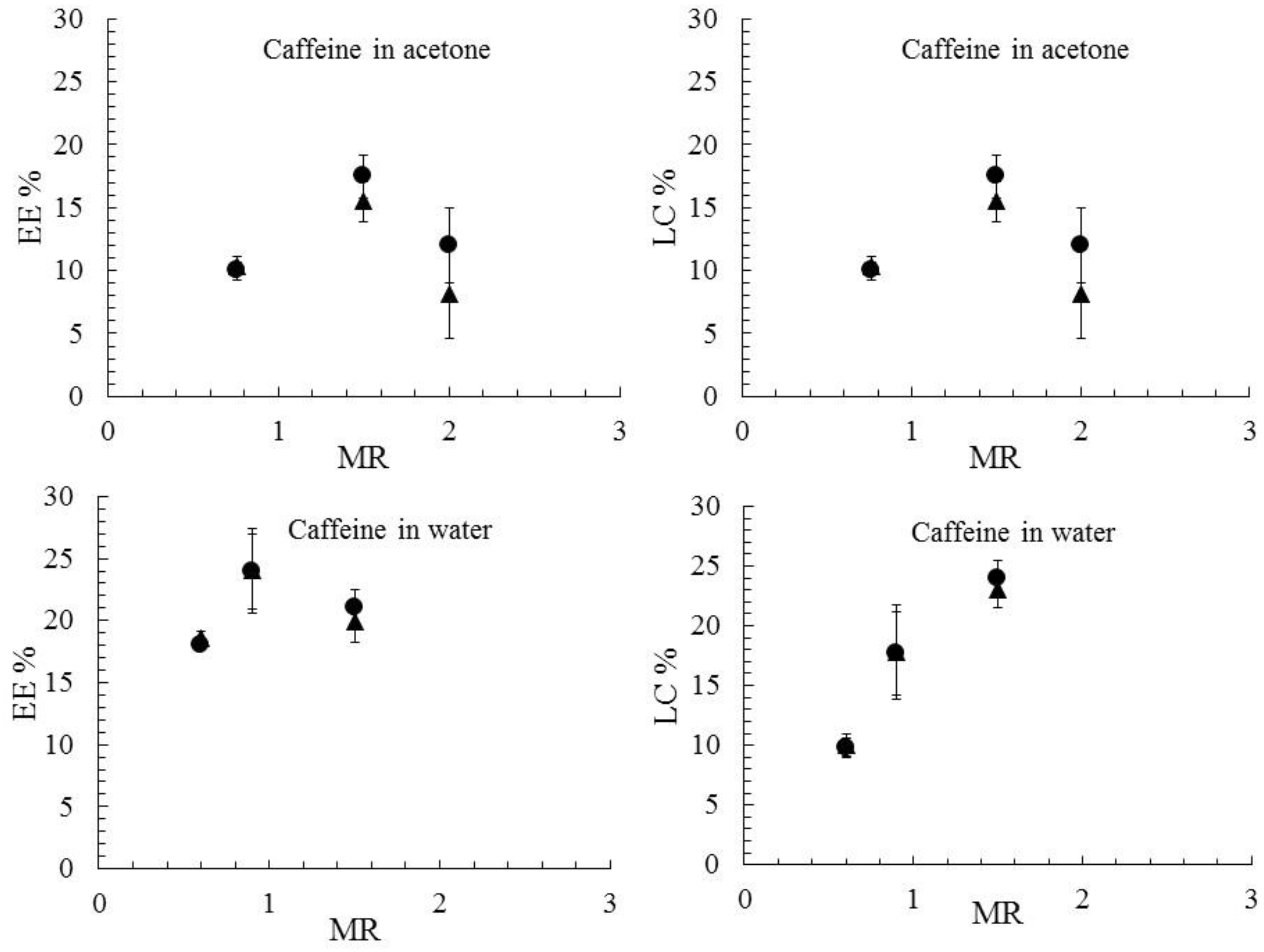
3. Results and Discussions
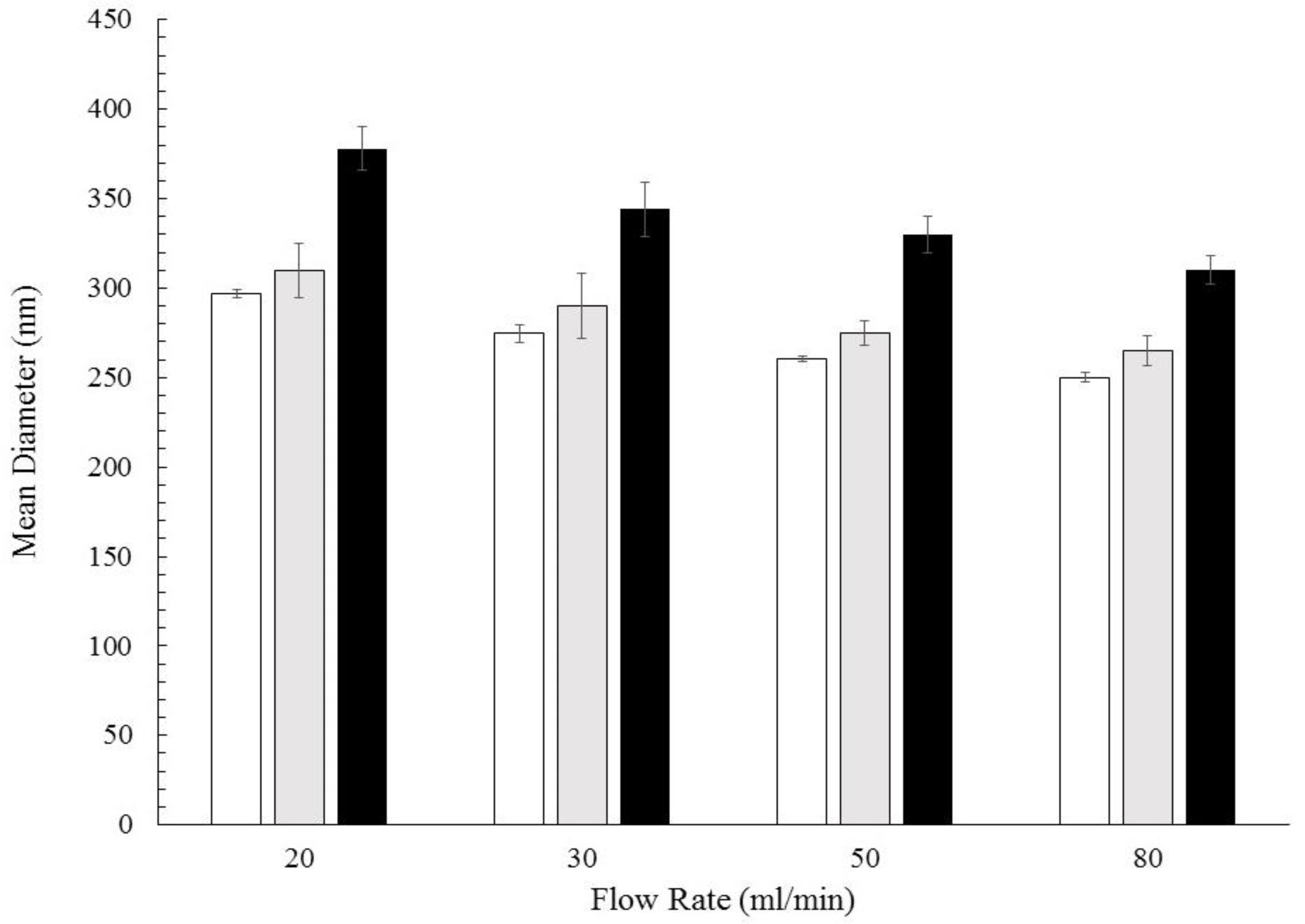
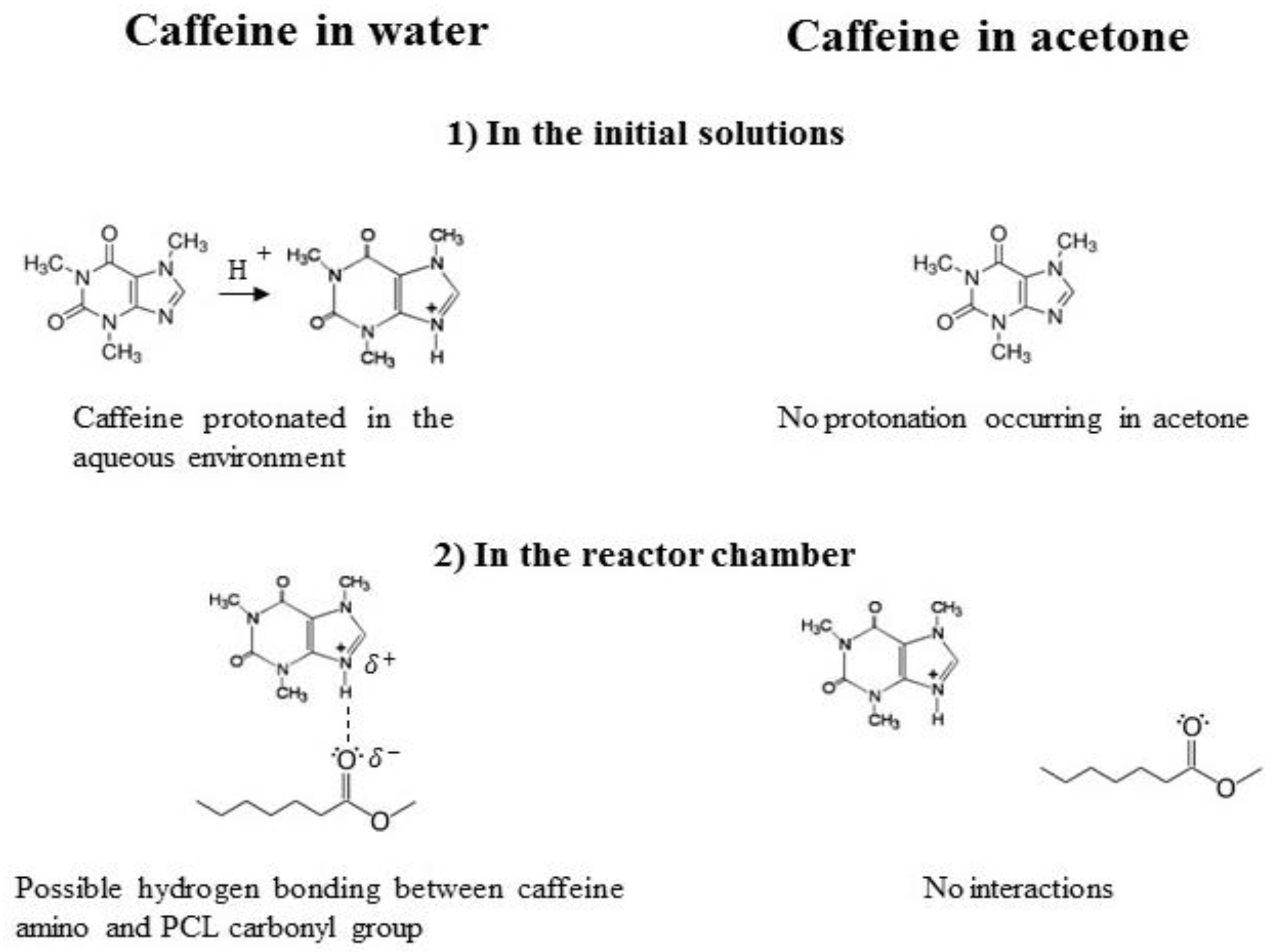
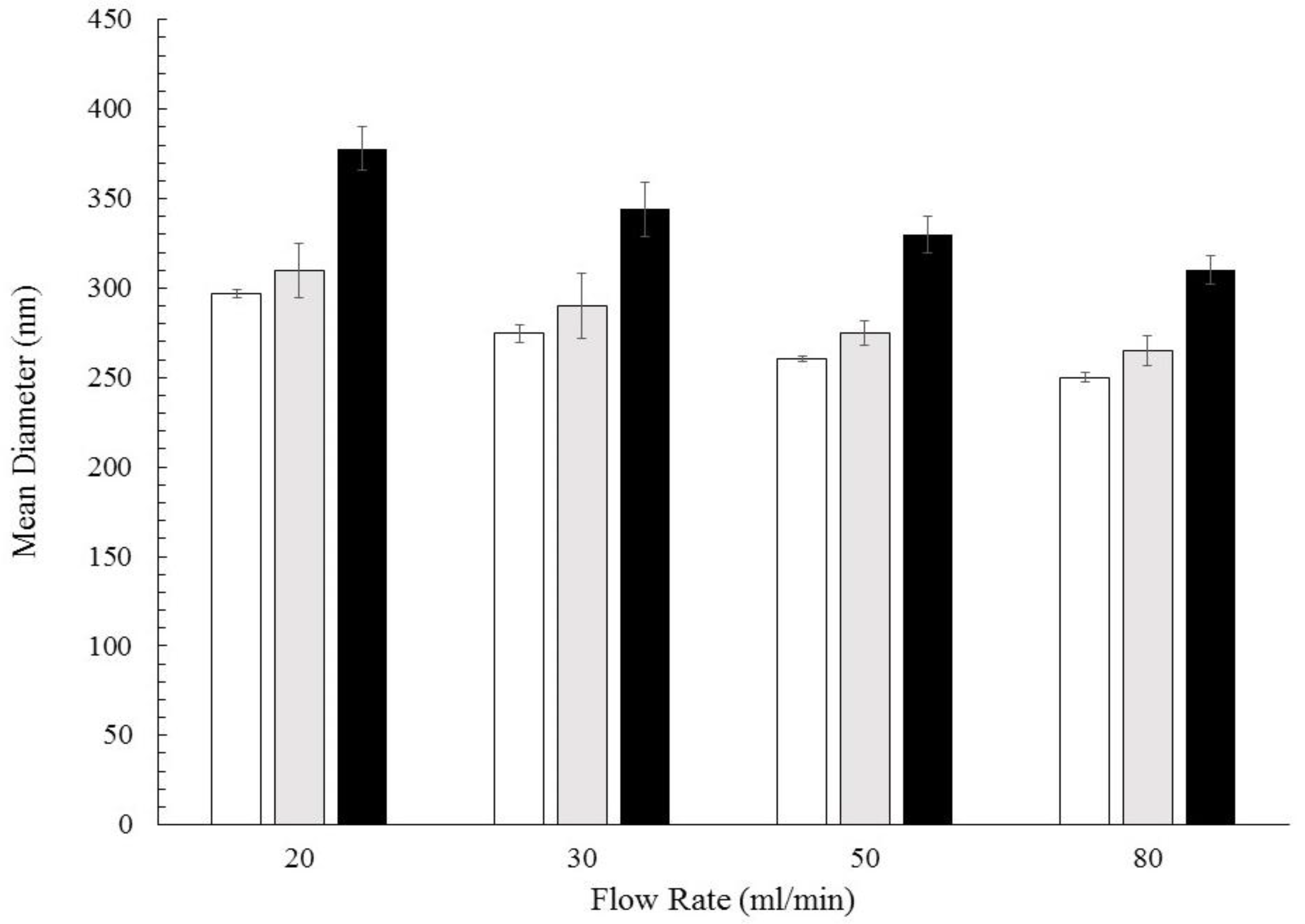
3.1. Formation of PCL-caffeine Nanoparticles
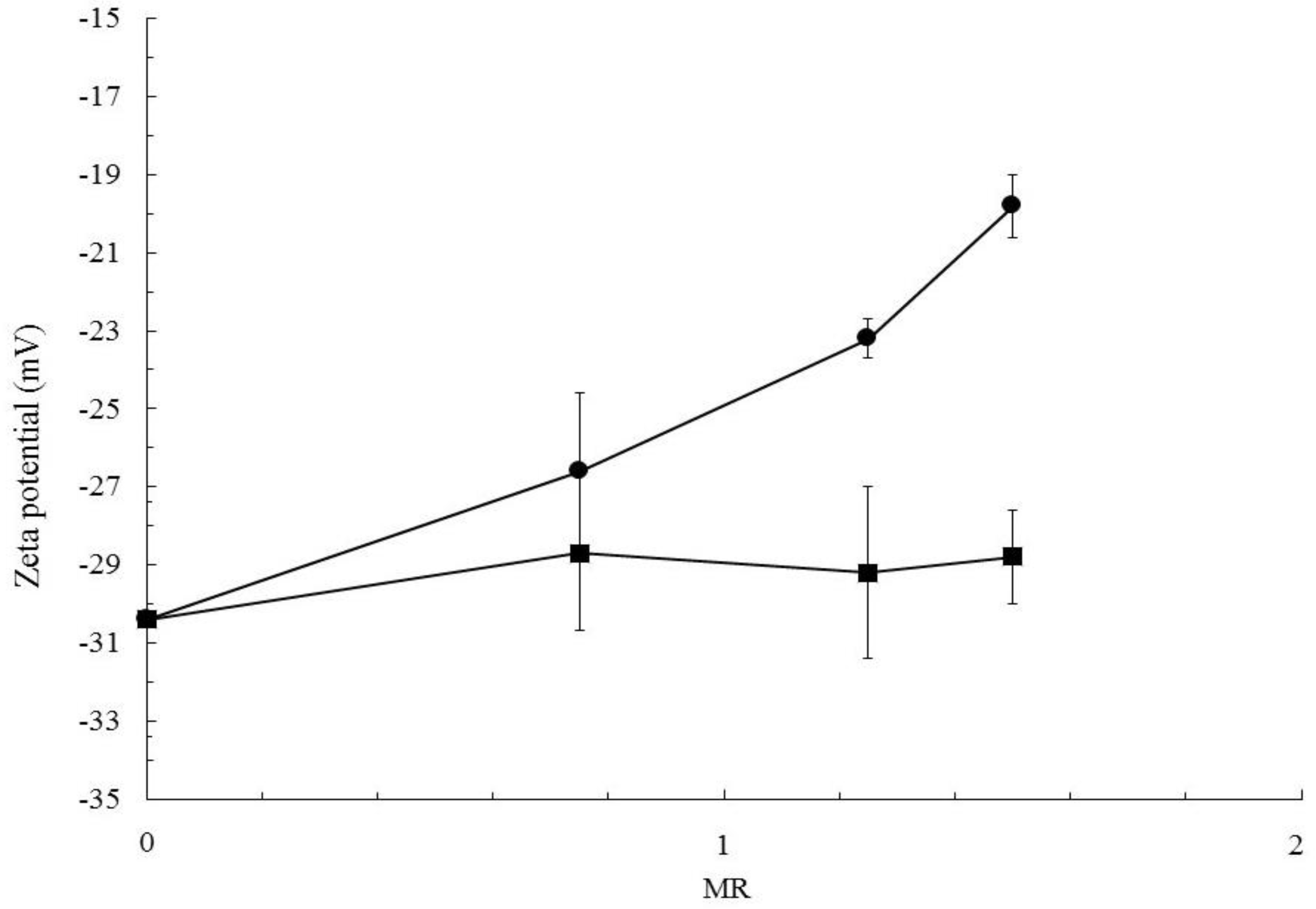
3.2. Particles Structure
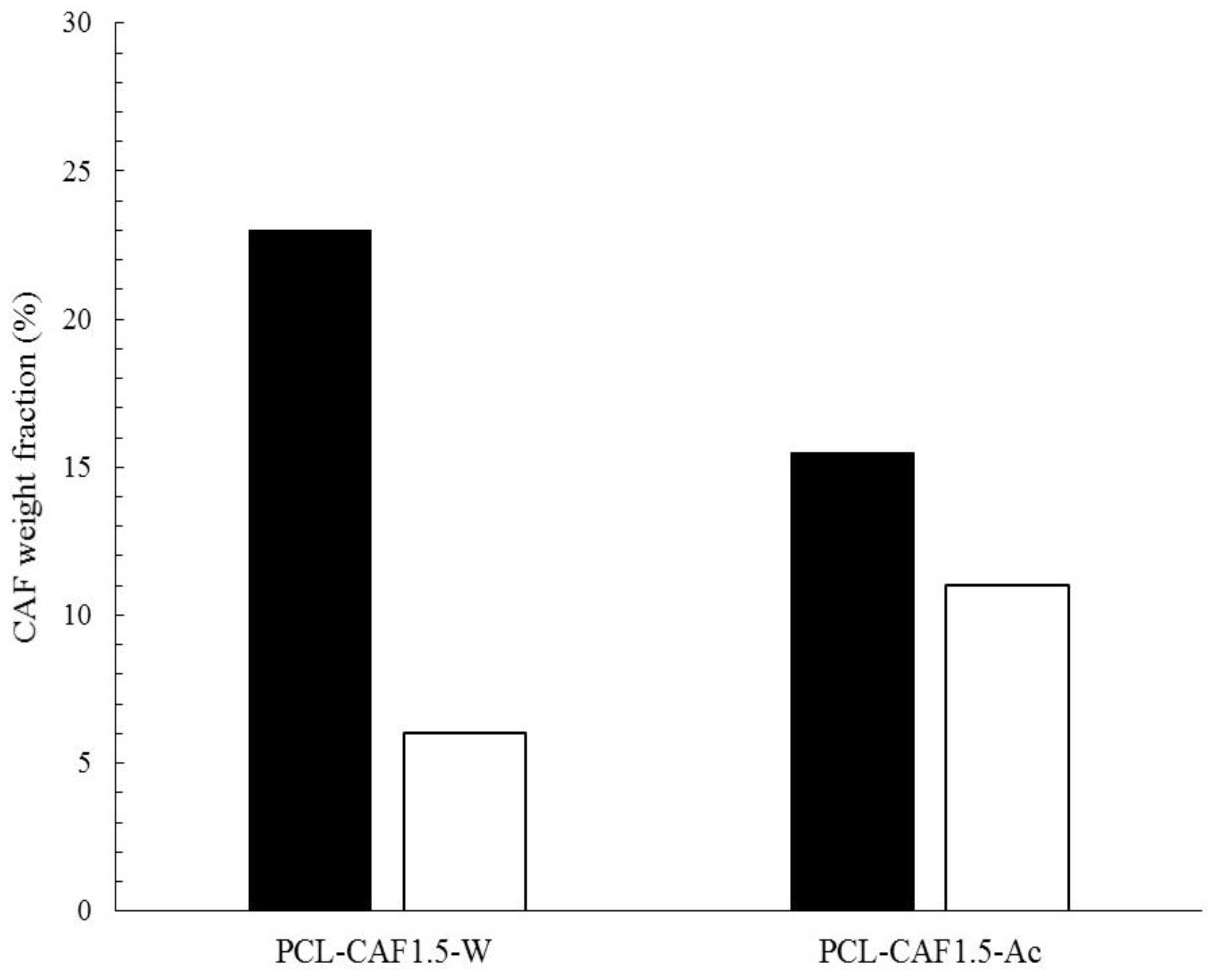
3.3. Drug Encapsulation and Release
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Fay, F.; Hak, S.; Manuel Perez-Aguilar, J.; Sanchez-Gaytan, B.L.; Goode, B.; Duivenvoorden, R.; de Lange Davies, C.; Bjørkøy, A.; Weinstein, H.; et al. Augmenting drug–carrier compatibility improves tumour nanotherapy efficacy. Nat. Commun. 2016, 7, 11221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadjavi, A.; Stura, I.; Prato, M.; Minero, V.G.; Panariti, A.; Rivolta, I.; Gulino, G.R.; Bessone, F.; Giribaldi, G.; Quaglino, E.; et al. ‘in vitro’, ‘In vivo’ and ‘in silico’ investigation of the anticancer effectiveness of oxygen-loaded chitosan-shelled nanodroplets as potential drug vector. Pharm. Res. 2018, 35, 75. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qu, Y.; Wang, K.; Zhang, X.; Zha, J.; Song, T.; Bao, C.; Liu, H.; Wang, Z.; Wang, J.; et al. In vivo nanoparticle-mediated radiopharmaceutical-excited fluorescence molecular imaging. Nat. Commun. 2015, 6, 7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumontel, B.; Canta, M.; Engelke, H.; Chiodoni, A.; Racca, L.; Ancona, A.; Limongi, T.; Canavese, G.; Cauda, V. Enhanced biostability and cellular uptake of zinc oxide nanocrystals shielded with a phospholipid bilayer. J. Mater. Chem. B 2017, 5, 8799–8813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.K.; Ferrari, J.D.; LaMothe, R.A.; Kolte, P.N.; Griset, A.P.; O’Neil, C.; Chan, V.; Browning, E.; Chalishazar, A.; Kuhlman, W.; et al. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat. Nanotechnol. 2016, 11, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Ancona, A.; Dumontel, B.; Garino, N.; Demarco, B.; Chatzitheodoridou, D.; Fazzini, W.; Engelke, H.; Cauda, V. Lipid-coated zinc oxide nanoparticles as innovative ROS-generators for photodynamic therapy in cancer cells. Nanomaterials 2018, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Luo, S.; Du, Z.; Zhou, M.; Li, P.; Fu, Y.; Sun, X.; Huang, Y.; Zhang, Z. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat. Commun. 2017, 8, 878. [Google Scholar] [CrossRef] [PubMed]
- Femminò, S.; Penna, C.; Bessone, F.; Caldera, F.; Dhakar, N.; Cau, D.; Pagliaro, P.; Cavalli, R.; Trotta, F. α-Cyclodextrin and α-Cyclodextrin polymers as oxygen nanocarriers to limit hypoxia/reoxygenation injury: Implications from an in vitro model. Polymers 2018, 10, 211. [Google Scholar] [CrossRef]
- Lamprecht, A. Nanomedicines in gastroenterology and hepatology. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Martínez Rivas, C.J.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Galindo Rodríguez, S.A.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Prud’homme, R.K. Flash nanoprecipitation of organic actives and block copolymers using a confined impinging jets mixer. Aust. J. Chem. 2003, 56, 1021–1024. [Google Scholar] [CrossRef]
- Lince, F.; Marchisio, D.L.; Barresi, A.A. Strategies to control the particle size distribution of poly-ε-caprolactone nanoparticles for pharmaceutical applications. J. Colloid Interface Sci. 2008, 322, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lince, F.; Marchisio, D.L.; Barresi, A.A. A comparative study for nanoparticle production with passive mixers via solvent-displacement: Use of CFD models for optimization and design. Chem. Eng. Process. Process Intensif. 2011, 50, 356–368. [Google Scholar] [CrossRef]
- Johnson, B.K.; Prud’homme, R.K. Chemical processing and micromixing in confined impinging jets. AIChE J. 2003, 49, 2264–2282. [Google Scholar] [CrossRef]
- Han, J.; Zhu, Z.; Qian, H.; Wohl, A.R.; Beaman, C.J.; Hoye, T.R.; Macosko, C.W. A simple confined impingement jets mixer for flash nanoprecipitation. J. Pharm. Sci. 2012, 101, 4018–4023. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.F.; Sun, C.C.; Chow, A.H.L. Assessment of the relative performance of a confined impinging jets mixer and a multi-inlet vortex mixer for curcumin nanoparticle production. Eur. J. Pharm. Biopharm. 2014, 88, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Valente, I.; Celasco, E.; Marchisio, D.L.; Barresi, A.A. Nanoprecipitation in confined impinging jets mixers: Production, characterization and scale-up of pegylated nanospheres and nanocapsules for pharmaceutical use. Chem. Eng. Sci. 2012, 77, 217–227. [Google Scholar] [CrossRef]
- Cheng, J.C.; Vigil, R.D.; Fox, R.O. A competitive aggregation model for flash nanoprecipitation. J. Colloid Interface Sci. 2010, 351, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Lavino, A.D.; Di Pasquale, N.; Carbone, P.; Marchisio, D.L. A novel multiscale model for the simulation of polymer flash nano-precipitation. Chem. Eng. Sci. 2017, 171, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Di Pasquale, N.; Marchisio, D.L.; Barresi, A.A. Model validation for precipitation in solvent-displacement processes. Chem. Eng. Sci. 2012, 84, 671–683. [Google Scholar] [CrossRef]
- Margulis, K.; Magdassi, S.; Lee, H.S.; Macosko, C.W. Formation of curcumin nanoparticles by flash nanoprecipitation from emulsions. J. Colloid Interface Sci. 2014, 434, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Johnstone, S.A.; Tardi, P.G.; Lo, L.; Xie, S.; Shu, Y.; Harasym, T.O.; Harasym, N.L.; Williams, L.; Bermudes, D.; et al. Modulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugates. J. Med. Chem. 2008, 51, 3288–3296. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, M.; Ginart, P.; Gindy, M.E.; Theriault, C.; Chin, K.H.; Soboyejo, W.; Prud’homme, R.K. Generic method of preparing multifunctional fluorescent nanoparticles using flash nanoprecipitation. Adv. Funct. Mater. 2009, 19, 718–725. [Google Scholar] [CrossRef]
- Miladi, K.; Sfar, S.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From particle preparation to in vivo applications. In Polymer Nanoparticles for Nanomedicines; Vauthier, C., Ponchel, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 17–53. ISBN 978-3-319-41419-5. [Google Scholar]
- Tam, Y.T.; To, K.K.W.; Chow, A.H.L. Fabrication of doxorubicin nanoparticles by controlled antisolvent precipitation for enhanced intracellular delivery. Colloids Surf. B Biointerfaces 2016, 139, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Massella, D.; Leone, F.; Peila, R.; Barresi, A.; Ferri, A. Functionalization of cotton fabrics with polycaprolactone nanoparticles for transdermal release of melatonin. J. Funct. Biomater. 2017, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Osorio, O.; Liu, Y.-G.; Scott, E. Facile assembly and loading of theranostic polymersomes via multi-impingement flash nanoprecipitation. J. Controll. Release 2017, 262, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Lane, M.E. Topical and transdermal delivery of caffeine. Int. J. Pharm. 2015, 490, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckman, M.A.; Weil, J.; de Mejia, E.G. Caffeine (1, 3, 7-trimethylxanthine) in foods: A comprehensive review on consumption, functionality, safety, and regulatory matters. J. Food Sci. 2010, 75, R77–R87. [Google Scholar] [CrossRef] [PubMed]
- Harpaz, E.; Tamir, S.; Weinstein, A.; Weinstein, Y. The effect of caffeine on energy balance. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, J.J.; Roberts, H.R. Caffeine consumption. Food Chem. Toxicol. 1996, 34, 119–129. [Google Scholar] [CrossRef]
- Goldstein, E.R.; Ziegenfuss, T.; Kalman, D.; Kreider, R.; Campbell, B.; Wilborn, C.; Taylor, L.; Willoughby, D.; Stout, J.; Graves, B.S.; et al. International society of sports nutrition position stand: Caffeine and performance. J. Int. Soc. Sports Nutr. 2010, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-P.; Lou, Y.-R.; Xie, J.-G.; Peng, Q.-Y.; Zhou, S.; Lin, Y.; Shih, W.J.; Conney, A.H. Caffeine and caffeine sodium benzoate have a sunscreen effect, enhance UVB-induced apoptosis, and inhibit UVB-induced skin carcinogenesis in SKH-1 mice. Carcinogenesis 2007, 28, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.-W.; Hirakawa, S.; Fujii, S.; Kawasumi, M.; Nghiem, P. Protection from photodamage by topical application of caffeine after ultraviolet irradiation. Br. J. Dermatol. 2007, 156, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Roure, R.; Oddos, T.; Rossi, A.; Vial, F.; Bertin, C. Evaluation of the efficacy of a topical cosmetic slimming product combining tetrahydroxypropyl ethylenediamine, caffeine, carnitine, forskolin and retinol, in vitro, ex vivo and in vivo studies: Evaluation of the efficacy of a topical cosmetic slimming. Int. J. Cosmet. Sci. 2011, 33, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, P.; Jordan, S.; Eastwood, J.; Rotstein, J.; Hugenholtz, A.; Feeley, M. Effects of caffeine on human health. Food Addit. Contam. 2003, 20, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, G.; Hattis, D.; Russ, A.; Sonawane, B. Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: Implications for assessing children’s risks from environmental agents. J. Toxicol. Environ. Health A 2004, 67, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Dias, M. Topical delivery of caffeine from some commercial formulations. Int. J. Pharm. 1999, 182, 41–47. [Google Scholar] [CrossRef]
- Budai, L. Liposomes for topical use: A physico-chemical comparison of vesicles prepared from egg or soy lecithin. Sci. Pharm. 2013, 81, 1151–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chorilli, M.; Calixto, G.; Rimério, T.C.; Scarpa, M.V. Caffeine encapsulated in small unilamellar liposomes: Characterization and in vitro release profile. J. Dispers. Sci. Technol. 2013, 34, 1465–1470. [Google Scholar] [CrossRef]
- Tiew, S.X.; Misran, M. Physicochemical properties of acylated low molecular weight chitosans. Int. J. Polym. Mater. Polym. Biomater. 2017, 67, 619–628. [Google Scholar] [CrossRef]
- Yew, H.-C.; Misran, M. Preparation and characterization of pH dependent κ-carrageenan-chitosan nanoparticle as potential slow release delivery carrier. Iran. Polym. J. 2016, 25, 1037–1046. [Google Scholar] [CrossRef]
- Rodrigues, F.; Alves, A.C.; Nunes, C.; Sarmento, B.; Amaral, M.H.; Reis, S.; Oliveira, M.B.P.P. Permeation of topically applied caffeine from a food by-product in cosmetic formulations: Is nanoscale in vitro approach an option? Int. J. Pharm. 2016, 513, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Puglia, C.; Offerta, A.; Tirendi, G.G.; Tarico, M.S.; Curreri, S.; Bonina, F.; Perrotta, R.E. Design of solid lipid nanoparticles for caffeine topical administration. Drug Deliv. 2016, 23, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-C.; Yeh, I.-T.; Niyama, E.; Huang, W.-R.; Ebara, M.; Wu, C.-S. Electrospun poly(ε-caprolactone) nanofibrous mesh for imiquimod delivery in melanoma therapy. Polymers 2018, 10, 231. [Google Scholar] [CrossRef]
- Limongi, T.; Lizzul, L.; Giugni, A.; Tirinato, L.; Pagliari, F.; Tan, H.; Das, G.; Moretti, M.; Marini, M.; Brusatin, G.; et al. Laboratory injection molder for the fabrication of polymeric porous poly-epsilon-caprolactone scaffolds for preliminary mesenchymal stem cells tissue engineering applications. Microelectron. Eng. 2017, 175, 12–16. [Google Scholar] [CrossRef]
- Cesca, F.; Limongi, T.; Accardo, A.; Rocchi, A.; Orlando, M.; Shalabaeva, V.; Di Fabrizio, E.; Benfenati, F. Fabrication of biocompatible free-standing nanopatterned films for primary neuronal cultures. RSC Adv. 2014, 4, 45696–45702. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Azadi, S.; Ashrafi, H.; Azadi, A. Mathematical modeling of drug release from swellable polymeric nanoparticles. J. Appl. Pharm. Sci. 2017, 7, 125–133. [Google Scholar] [CrossRef]
- Saad, W.S.; Prud’homme, R.K. Principles of nanoparticle formation by flash nanoprecipitation. Nano Today 2016, 11, 212–227. [Google Scholar] [CrossRef]
- Ferri, A.; Kumari, N.; Peila, R.; Barresi, A.A. Production of menthol-loaded nanoparticles by solvent displacement. Can. J. Chem. Eng. 2017, 95, 1690–1706. [Google Scholar] [CrossRef]
- Di Pasquale, N.; Marchisio, D.L.; Carbone, P.; Barresi, A.A. Identification of nucleation rate parameters with MD and validation of the CFD model for polymer particle precipitation. Chem. Eng. Res. Des. 2013, 91, 2275–2290. [Google Scholar] [CrossRef]
- Zelenková, T.; Fissore, D.; Marchisio, D.L.; Barresi, A.A. Size control in production and freeze-drying of poly-ε-caprolactone nanoparticles. J. Pharm. Sci. 2014, 103, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Barresi, A.A.; Vanni, M.; Fissore, D.; Zelenková, T. Synthesis and preservation of polymer nanoparticles for pharmaceutical applications. In Handbook of Polymers for Pharmaceutical Technologies; Thakur, V.K., Thakur, M.K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 229–280. ISBN 978-1-119-04141-2. [Google Scholar]
- Celasco, E.; Valente, I.; Marchisio, D.L.; Barresi, A.A. Dynamic light scattering and X-ray photoelectron spectroscopy characterization of PEGylated polymer nanocarriers: Internal structure and surface properties. Langmuir 2014, 30, 8326–8335. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, H.; Tabrizchi, M.; Farrokhpour, H. Protonation of caffeine: A theoretical and experimental study. Chem. Phys. 2013, 415, 222–227. [Google Scholar] [CrossRef]
- Pinkerton, N.M.; Behar, L.; Hadri, K.; Amouroux, B.; Mingotaud, C.; Talham, D.R.; Chassaing, S.; Marty, J.-D. Ionic Flash NanoPrecipitation (iFNP) for the facile, one-step synthesis of inorganic–organic hybrid nanoparticles in water. Nanoscale 2017, 9, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, N.; Marchisio, D.L.; Barresi, A.A.; Carbone, P. Solvent structuring and its effect on the polymer structure and processability: The case of water–acetone poly-ε-caprolactone mixtures. J. Phys. Chem. B 2014, 118, 13258–13267. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.F.; Wan, K.Y.; Cheng, K.K.; Wong, K.W.; Sun, C.C.; Baum, L.; Chow, A.H.L. Development of highly stabilized curcumin nanoparticles by flash nanoprecipitation and lyophilization. Eur. J. Pharm. Biopharm. 2015, 94, 436–449. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
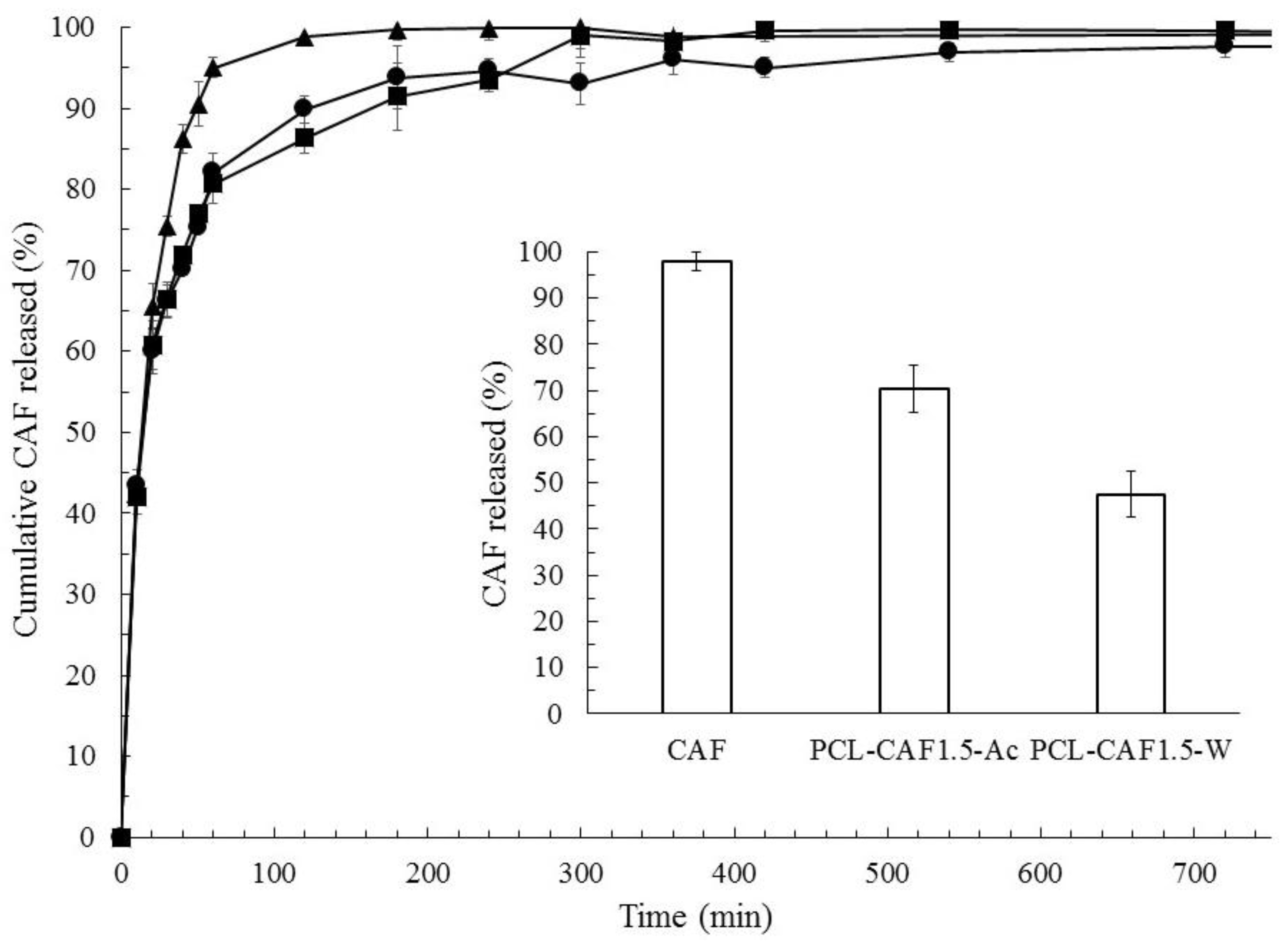{kind=link}
| Sample Label | CPCL (mg/mL) | CCAF (mg/mL) | CAF Solvent | MR |
|---|---|---|---|---|
| PCL-CAF0.76-Ac | 10.0 | 7.6 | Acetone | 0.76 |
| PCL-CAF1.5-Ac | 6.0 | 9.0 | Acetone | 1.50 |
| PCL-CAF2-Ac | 4.5 | 9.0 | Acetone | 2.00 |
| PCL-CAF0.6-W | 15.0 | 9.0 | Water | 0.60 |
| PCL-CAF0.9-W | 10.0 | 9.0 | Water | 0.90 |
| PCL-CAF1.5-W | 6.0 | 9.0 | Water | 1.50 |
| Sample Label | CPCL (mg/mL) | CCAF (mg/mL) | CAF Solvent | MR |
|---|---|---|---|---|
| Pure PCL | 6.0 | 0.0 | - | 0.00 |
| PCL-CAF0.75-Ac | 6.0 | 4.5 | Acetone | 0.75 |
| PCL-CAF1.25-Ac | 6.0 | 7.5 | Acetone | 1.25 |
| PCL-CAF1.5-Ac | 6.0 | 9.0 | Acetone | 1.50 |
| PCL-CAF0.75-W | 6.0 | 4.5 | Water | 0.75 |
| PCL-CAF1.25-W | 6.0 | 7.5 | Water | 1.25 |
| PCL-CAF1.5-W | 6.0 | 9.0 | Water | 1.50 |
| Sample | Zero Order | First Order | Higuchi | Hixon Crowell | Baker Lonsdale |
|---|---|---|---|---|---|
| CAF Control | 0.14 | 0.90 | 0.60 | 0.48 | 0.70 |
| PCL-CAF1.5-Ac | 0.19 | 0.85 | 0.84 | 0.69 | 0.89 |
| PCL-CAF1.5-W | 0.07 | 0.94 | 0.84 | 0.84 | 0.99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massella, D.; Celasco, E.; Salaün, F.; Ferri, A.; Barresi, A.A. Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure. Polymers 2018, 10, 1092. https://doi.org/10.3390/polym10101092
Massella D, Celasco E, Salaün F, Ferri A, Barresi AA. Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure. Polymers. 2018; 10(10):1092. https://doi.org/10.3390/polym10101092
Chicago/Turabian StyleMassella, Daniele, Edvige Celasco, Fabien Salaün, Ada Ferri, and Antonello A. Barresi. 2018. "Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure" Polymers 10, no. 10: 1092. https://doi.org/10.3390/polym10101092