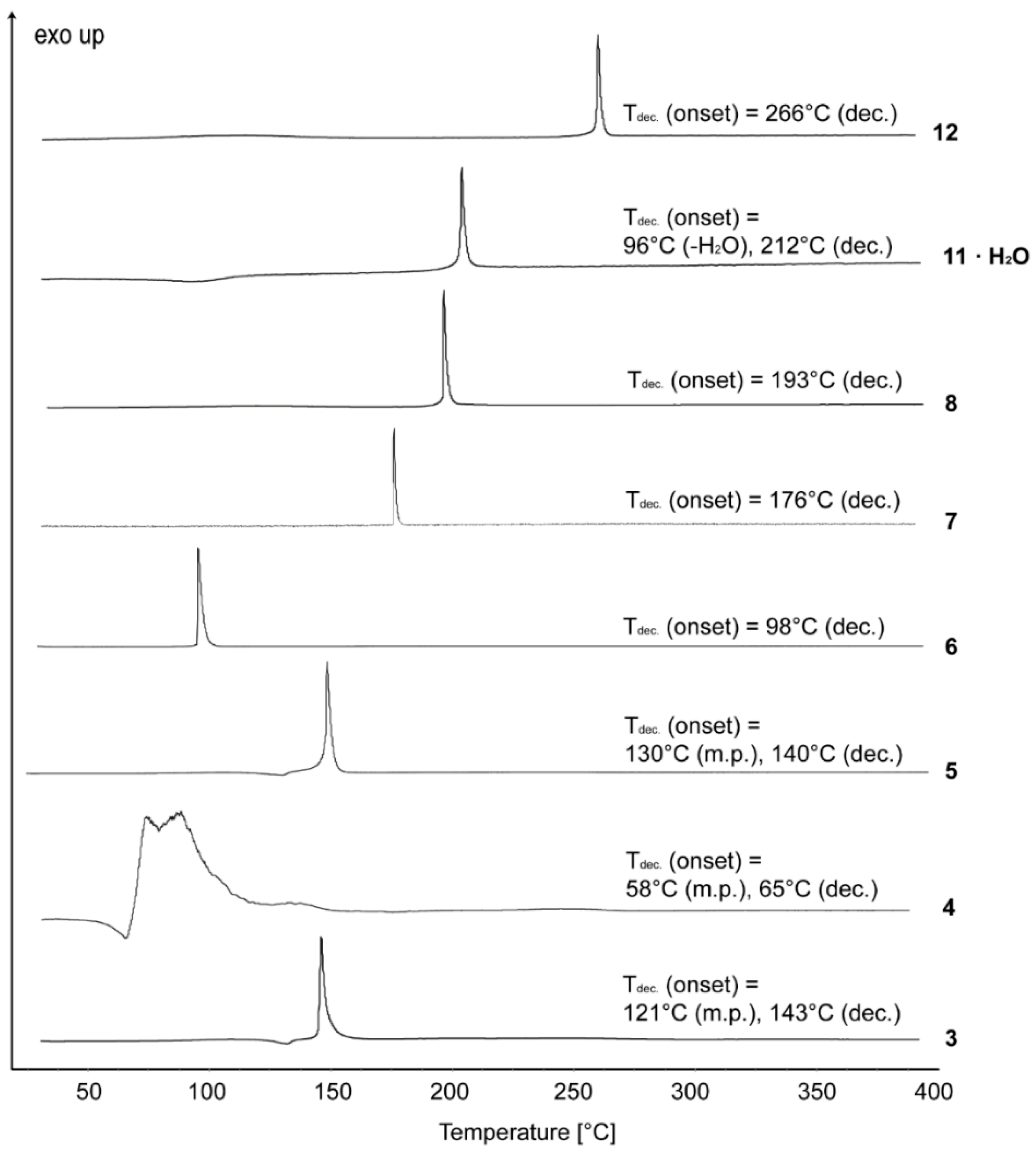
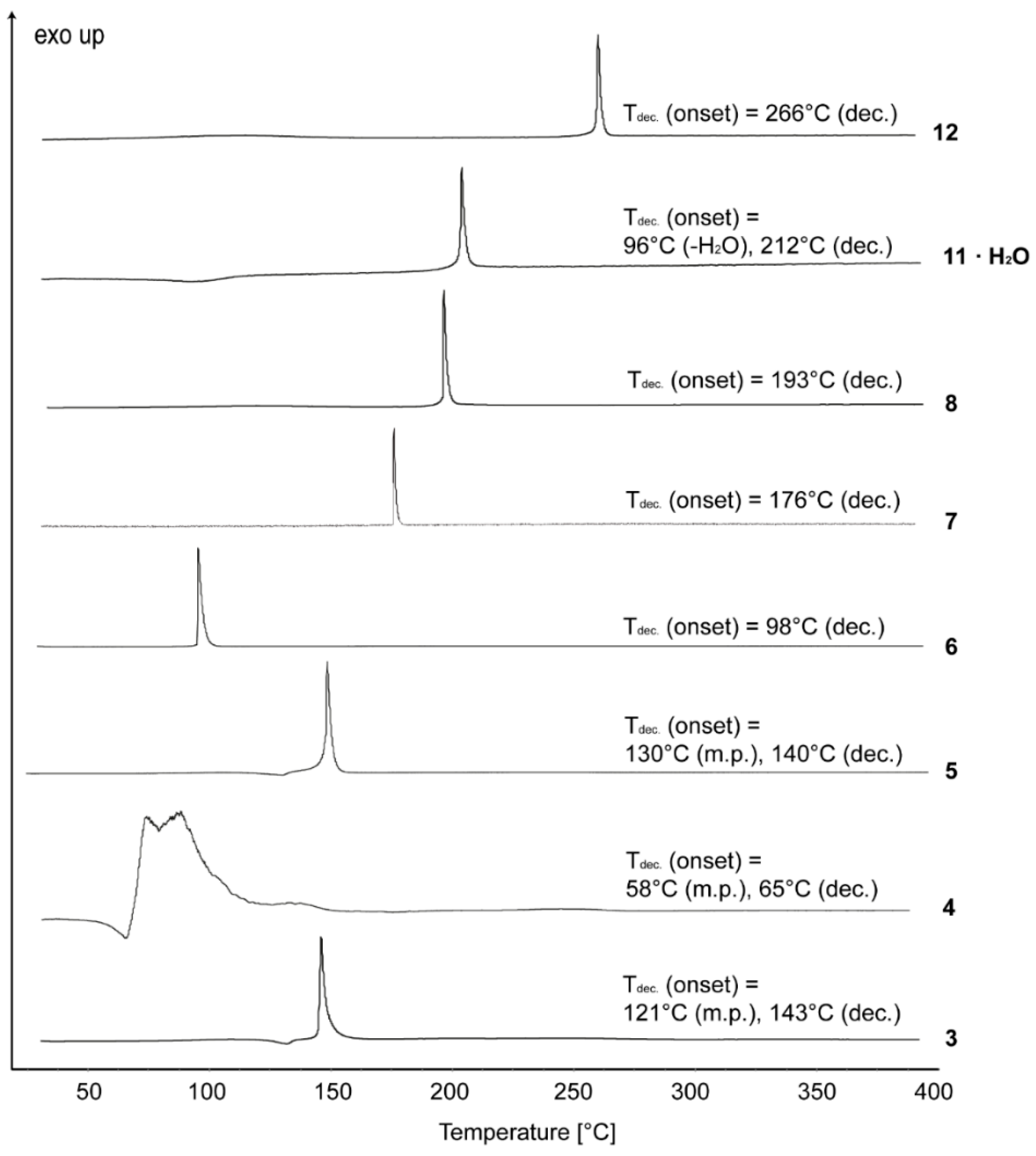
2.2. Crystal Structure
The crystal structures of compounds
3–
6,
8 and
12 were determined.
Tables S1–S2 in the Supporting Information gather selected data and parameters of the X-ray measurements. Once crystals were obtained for each compound, we did not attempt to search for additional polymorphs of each.
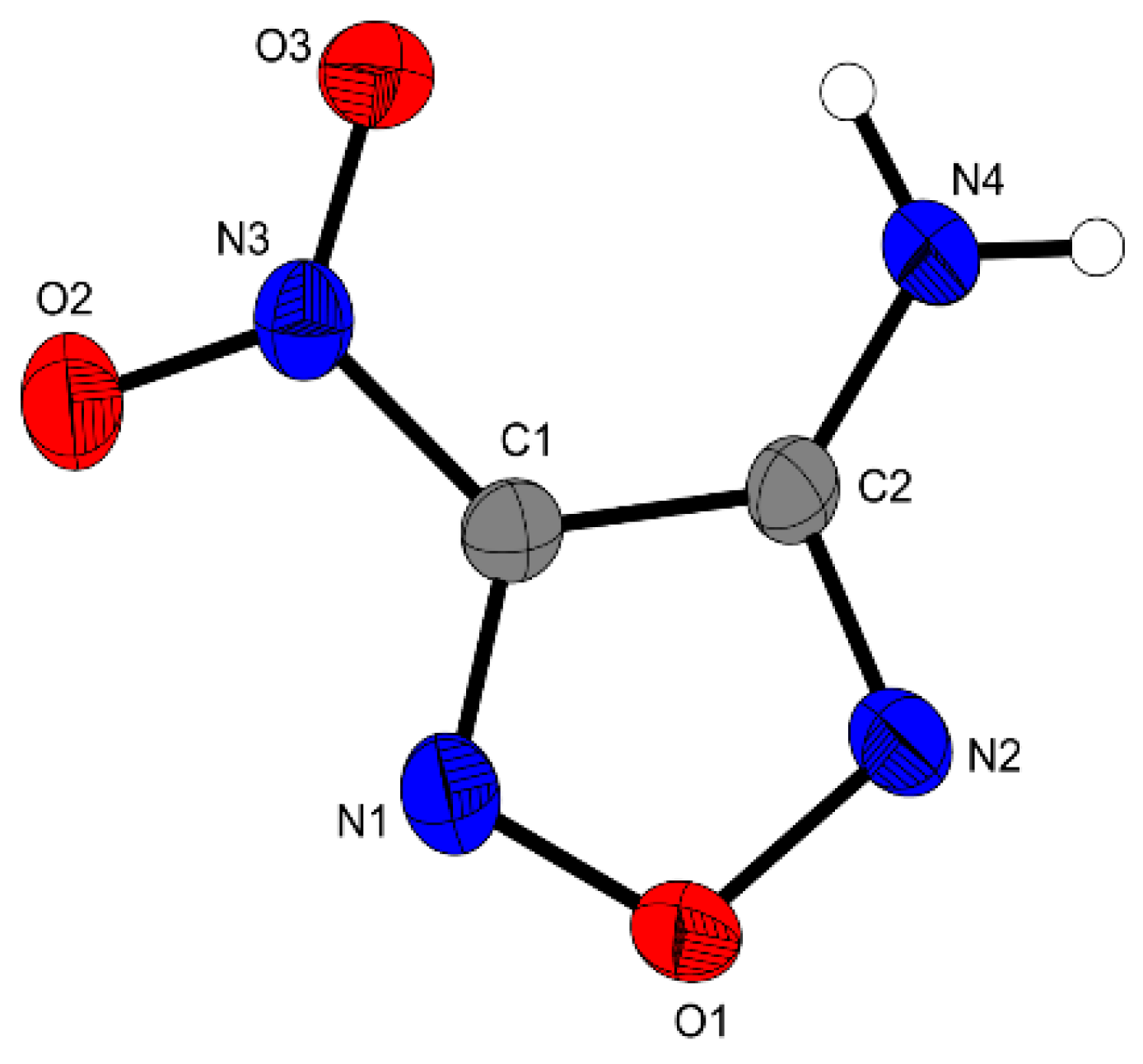
The compound 3-Amino-4-nitraminofurazan
3 crystallizes in the orthorhombic space group
Pna2
1 with a cell volume of 938.26 Å
3 and eight molecular units per unit cell (
Figure 2). The calculated density at 173 K is 1.842 g·cm
−3, which is similar to the previously reported density of 1.84 g·cm
−3 (X-ray analysis at 153 K) [
11]. Compound
3 is planar, only the nitro moiety is slightly tilted against the furazan plane by a torsion angle of O2–N3–C1–C2 = 4.4°. Short hydrogen bonding can be observed between the amino and nitro groups, similar bond distances have been reported for other furazanes in the literature [
6]. A more detailed description of the structure can be found in the literature [
11].
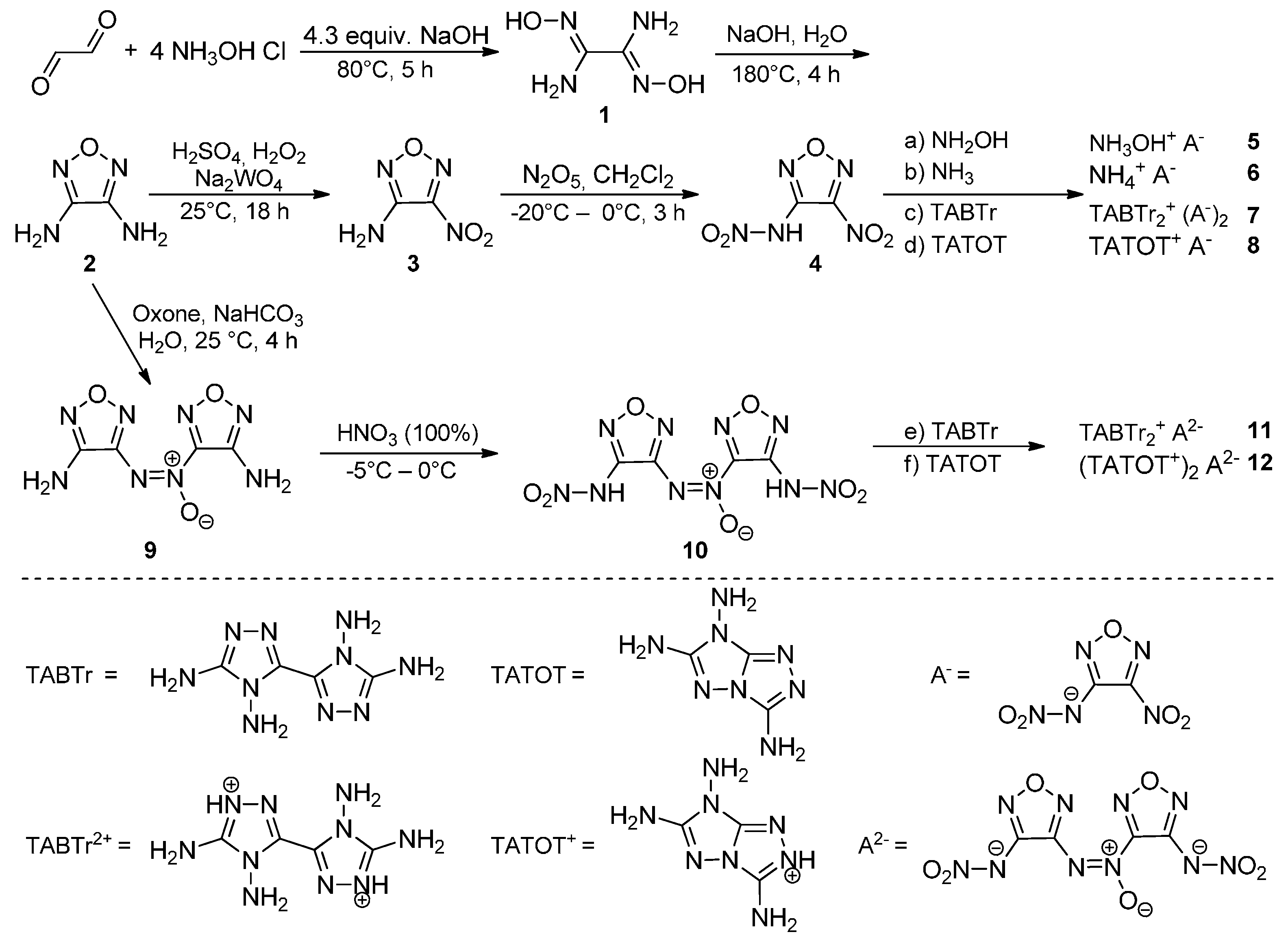
Figure 2.
Molecular unit of 3. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): O1–N1 1.352(4), O1–N2 1.409(5), C1–C2 1.428(6), N4–N2 1.331(7), N3–C1 1.450(5), O2–N3 1.226(5); selected bond angles (°): N1–O1–N2 111.1(3), N1–C1–C2 111.8(3), N3–C1–C2 128.1(4), N4–C2–C1 129.5(4); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H···A), d(D…A), (D−H…A)): N4−H4A…O3: 0.87(5), 2.38(5), 2.919(5), 120(4); N4–H4A…N1 0.87(5), 2.29(2), 3.084(6), 152(5); selected torsion angles (°): N2–O1–N1–C1 0.6(4), O2–N3–C1–C2 4.4(5), O1–N2–C2–N4 179.2(4).
Figure 2.
Molecular unit of 3. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): O1–N1 1.352(4), O1–N2 1.409(5), C1–C2 1.428(6), N4–N2 1.331(7), N3–C1 1.450(5), O2–N3 1.226(5); selected bond angles (°): N1–O1–N2 111.1(3), N1–C1–C2 111.8(3), N3–C1–C2 128.1(4), N4–C2–C1 129.5(4); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H···A), d(D…A), (D−H…A)): N4−H4A…O3: 0.87(5), 2.38(5), 2.919(5), 120(4); N4–H4A…N1 0.87(5), 2.29(2), 3.084(6), 152(5); selected torsion angles (°): N2–O1–N1–C1 0.6(4), O2–N3–C1–C2 4.4(5), O1–N2–C2–N4 179.2(4).
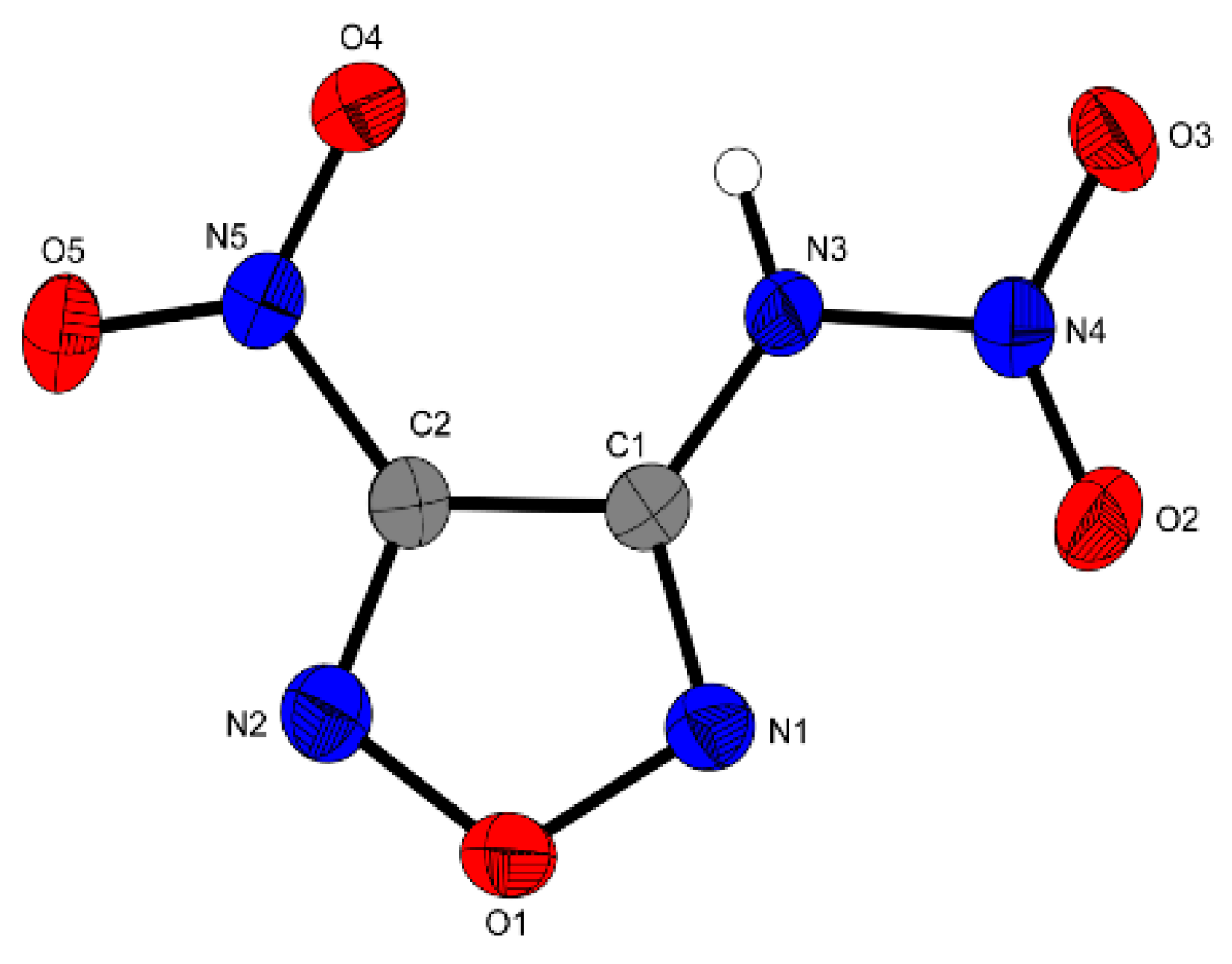
The compound 3-Nitramino-4-nitrofurazan
4 crystallizes in the orthorhombic space group
Pbca with a cell volume of 1177.45 Å
3 and eight molecular units per unit cell. The calculated density at 173 K is 1.975 g·cm
−3, which is comparable to the density reported in the literature: 1.93 g·cm
−3 (gas pycnometer) [
5] and 1.95 g·cm
−3 [
7]. The small discrepancies are likely due to the different temperatures at which the densities were measured. The molecular unit of
4 is displayed in
Figure 3.
The nitro- and nitramino groups in compound
4 are only slightly tilted against the furazan ring as shown by the torsion angles O4–B5–C2–C1 = 4.2°, C1–N3–N4–O2 = 179.4° and C1–N3–N4–O2 = 3.9°. Together with the planar furazan ring (N2–O1–N1–C1 = 0.5°) the complete compound forms an almost planar unit, which might be one of the reasons that can explain the high density of this compound. Another reason might be the hydrogen-bonds of N3−H3…O4 and N3−H3…O3. The bond distances of the furazan ring (C1–C2 = 1.420 Å) and the nitro group (N5–C2 1.444 Å, O5–N5 = 1.215 Å) are similar to those in other nitrofurazanes as compound
3. The bond distances of the nitramino moiety (O2–N4 = 1.210 Å, N3–N4 = 1.370 Å) are also in the same range compared to other nitramino moieties in heterocyclic ring systems [
6,
12].
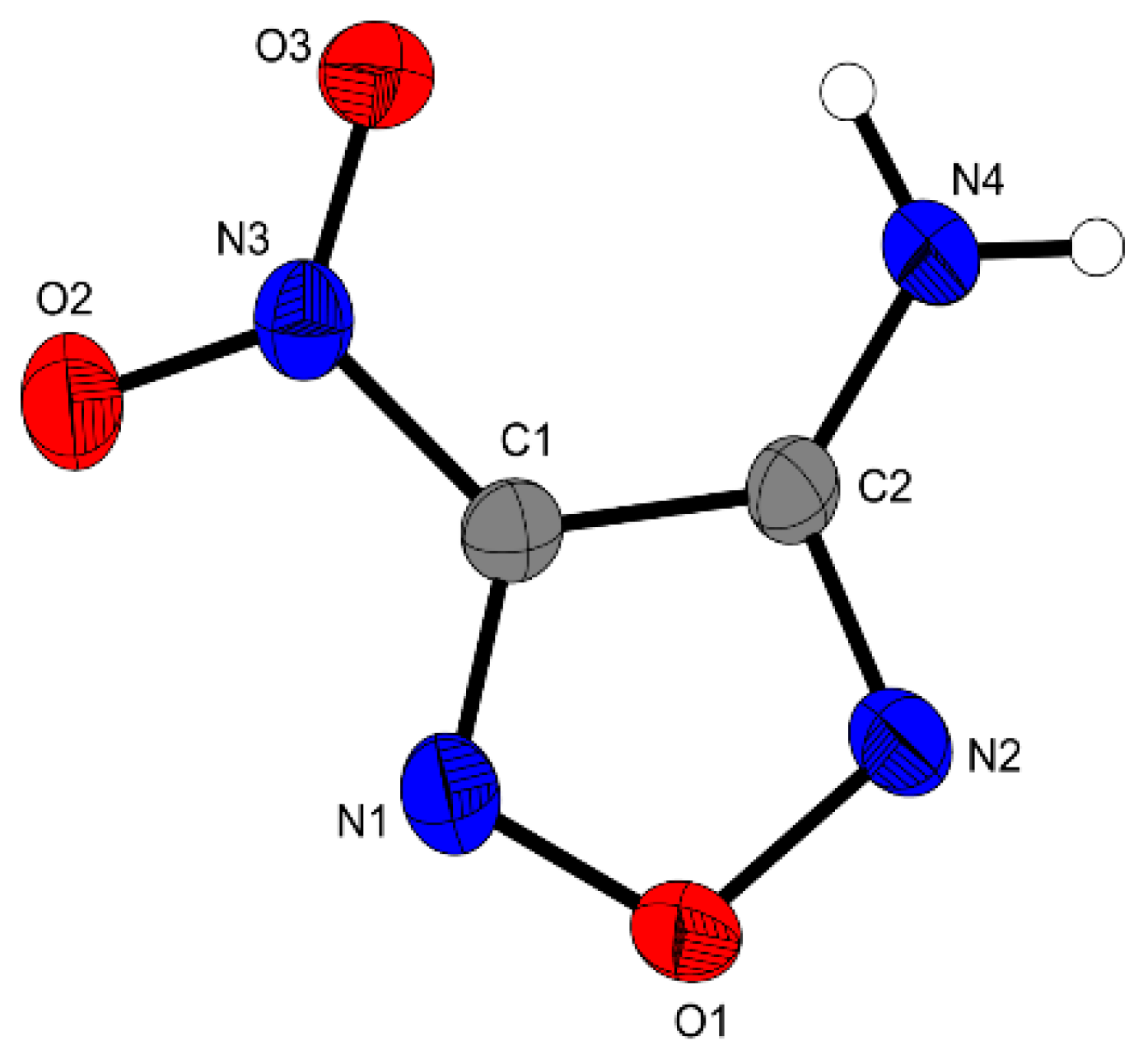
Figure 3.
Molecular unit of 4. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): O2–N4 1.210(2), N3–N4 1.370(2), N3–C1 1.365(2), N5–C2 1.444(2), O5–N5 1.215(2), C1–C2 1.420(3); selected bond angles (°): N2–C2–N5 111.23 (17), N3–C1–C2 107.69(16), N1–O1–N2 112.26(14); selected hydrogen bond distances [Å] and angles [°] (D−H∙∙∙A, d(D−H), d(H…A), d(D…A), (D−H…A)): N3−H3…O4: 0.85(3), 2.29(2), 2.787(2), 111.8(2); N3–H3…O3 0.85(3), 2.21(3), 3.016(2), 159(2); selected torsion angles (°): N2–O1–N1–C1 0.5(2), O4–B5–C2–C1 4.2(3), C1–N3–N4–O2 179.4(2), C1–N3–N4–O2 3.9 (3).
Figure 3.
Molecular unit of 4. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): O2–N4 1.210(2), N3–N4 1.370(2), N3–C1 1.365(2), N5–C2 1.444(2), O5–N5 1.215(2), C1–C2 1.420(3); selected bond angles (°): N2–C2–N5 111.23 (17), N3–C1–C2 107.69(16), N1–O1–N2 112.26(14); selected hydrogen bond distances [Å] and angles [°] (D−H∙∙∙A, d(D−H), d(H…A), d(D…A), (D−H…A)): N3−H3…O4: 0.85(3), 2.29(2), 2.787(2), 111.8(2); N3–H3…O3 0.85(3), 2.21(3), 3.016(2), 159(2); selected torsion angles (°): N2–O1–N1–C1 0.5(2), O4–B5–C2–C1 4.2(3), C1–N3–N4–O2 179.4(2), C1–N3–N4–O2 3.9 (3).
Hydroxylammonium 3-nitramino-4-nitrofurazan
5 crystallizes in the monoclinic space group
Pc with a cell volume of 361.86 Å
3 and two molecular units per unit cell. The calculated density at 173 K is 1.910 g·cm
−3, which is comparable to the reported values in the literature: 1.875 g·cm
−3 (X-ray analysis) [
5] and 1.89 g·cm
−3 [
7]. The slightly higher density values of
5 might be because of the different temperatures at which the densities were measured. The molecular unit of
5 is displayed in
Figure 4.
Similar to neutral compound 4, the furazan unit in 5 is planar (N2–O2–N1–C1 = 1.1°), whereas the nitro- and nitramino groups are slightly tilted against the furazan ring as indicated by the torsion angles of O5–N5–C2–N2 = 5.1° and N4–N3–C1–C2 = 171.7°, C1–N3–N4–O3 = 2.5°. Hydrogen-bonding can for example be observed between the hydrogen atoms of the hydroxylammonium cation and the O4 of the nitramino moiety. The bond distances of the furazan ring (C1–C2 = 1.432 Å), the nitro group (N5–C2 = 1.451 Å, O6–N5 = 1.243 Å) and the nitramino moiety (N3–N4 = 1.307 Å, O4–N4 = 1.280 Å) are in accordance with parent compound 4.
Figure 4.
Molecular unit of 5. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): N5–C2 1.451(8), O6–N5 1.243(6), N3–C1 1.385(7), N3–N4 1.307(2), O4–N4 1.280(6), C1–C2 1.432(8); selected bond angles (°): N1–O2–N2 111.8(4), N2–C2–N5 119.7(5), N1–C1–N3 129.7(5), O3–N4–N3 125.5(5); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N6−H6B…O4: 0.910, 2.440, 2.892(6), 111.00; O1–H4A…O4 0.73(6), 1.94(6), 2.653(6), 169(6); selected torsion angles (°): N2–O2–N1–C1 1.1(6), O5–N5–C2–N2 5.1(8), N4–N3–C1–C2 171.7(5), C1–N3–N4–O3 2.5(8).
Figure 4.
Molecular unit of 5. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): N5–C2 1.451(8), O6–N5 1.243(6), N3–C1 1.385(7), N3–N4 1.307(2), O4–N4 1.280(6), C1–C2 1.432(8); selected bond angles (°): N1–O2–N2 111.8(4), N2–C2–N5 119.7(5), N1–C1–N3 129.7(5), O3–N4–N3 125.5(5); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N6−H6B…O4: 0.910, 2.440, 2.892(6), 111.00; O1–H4A…O4 0.73(6), 1.94(6), 2.653(6), 169(6); selected torsion angles (°): N2–O2–N1–C1 1.1(6), O5–N5–C2–N2 5.1(8), N4–N3–C1–C2 171.7(5), C1–N3–N4–O3 2.5(8).
Ammonium 3-nitramino-4-nitrofurazan
6 crystallizes in the monoclinic space group
P2
1/
n with a cell volume of 704.59 Å
3 and four molecular units per unit cell. The calculated density at 173 K is 1.811 g·cm
−3, which is virtually the same as the reported density in the literature: 1.82 g·cm
−3 [
7], but significantly lower than of hydroxylammonium salt
5. The molecular unit of compound
6 is illustrated in
Figure 5.
Figure 5.
Molecular unit of 6. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.4293(17), N3–C2 1.451(2), O3–N3 1.217(2), N4–C1 1.3684(15), N4–N5 1.3179(15), O4–N4 1.2570(14); selected bond angles (°): N1–O1–N2 111.72(10), N2–C2–N3 119.64(11), N1–C1–N4 132.09(11), O5–N5–N4 124.25(11); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N6−H4A…O3: 0.87(2), 2.51(3), 3.249 (2), 143(2); N6−H4A…O2: 0.87(2), 2.38(3), 3.043 (2), 133.6(18); N4–H4B…O5 0.91(2), 2.09(2), 2.993(2), 176(2); selected torsion angles (°): N2–O1–N1–C1 0.27(14), N5–N4–C1–N1 5.5(2), O2–N3–C2–N2 29.5(2), C1–N4–N5–O5 2.9(2).
Figure 5.
Molecular unit of 6. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.4293(17), N3–C2 1.451(2), O3–N3 1.217(2), N4–C1 1.3684(15), N4–N5 1.3179(15), O4–N4 1.2570(14); selected bond angles (°): N1–O1–N2 111.72(10), N2–C2–N3 119.64(11), N1–C1–N4 132.09(11), O5–N5–N4 124.25(11); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N6−H4A…O3: 0.87(2), 2.51(3), 3.249 (2), 143(2); N6−H4A…O2: 0.87(2), 2.38(3), 3.043 (2), 133.6(18); N4–H4B…O5 0.91(2), 2.09(2), 2.993(2), 176(2); selected torsion angles (°): N2–O1–N1–C1 0.27(14), N5–N4–C1–N1 5.5(2), O2–N3–C2–N2 29.5(2), C1–N4–N5–O5 2.9(2).
As in salt 5 and neutral compound 4, the furazan ring in 6 has a planar structure (N2–O1–N1–C1 = 0.27°). While the nitramino group is only slightly tilted against the furazan plane (N5–N4–C1–N1 = 5.5°, C1–N4–N5–O5 = 2.9°), the nitro group is tilted strongly against the plane by a torsion angle of O2–N3–C2–N2 =29.5°. A reason for this might be the strong hydrogen bonds between the hydroxylammonium cation and the oxygen atoms O3 and O4 of the nitro group. More hydrogen bonds can be observed between the cation and the nitramino moiety. Even though the nitro group is strongly tilted against the plane, the bond distances of the furazan ring (C1–C2 = 1.4293 Å), the nitro group (N3–C2 = 1.451 Å, O3–N3 = 1.217 Å) and nitramino moiety (N4–N5 = 1.3179 Å, O4–N4 = 1.257 Å) are comparable to parent compound 4 and salt 5.
Compound 3,6,7-Triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazolium 3-nitramino-4-nitrofurazan
8 crystallizes in the monoclinic space group
P2
1 with a cell volume of 619.21 Å
3 and two molecular units per unit cell. The calculated density at 173 K is 1.766 g·cm
−3, which is lower than the density of salts
5 and
6. The molecular unit of compound
8 is shown in
Figure 6.
In compound
8, the furazan ring is also planar (N2–O1–N1–C1 = 0.27°), whereas the nitro (O5–N3–C2–C1 = 157.9°) and nitramino (N5–N4–C1–C2 = 50.6°, O2–N5–N4–C1 = 7.4°) moieties are strongly tilted out of the furazan plane. The cation has a planar structure except for the two protons located at N13 as reported previously in the literature [
4]. All the three amino groups in the cation form hydrogen bonds to the nitro and nitramino groups of the anion, resulting in a strong hydrogen-bonding network. This effect was expected because it was already observed for other energetic salts of the 3,6,7-triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazolium cation [
4] and can explain the higher thermal stability of compound
8 compared to that of salts
5 and
6. The bond distances of the furazan ring (C1–C2 = 1.422 Å), the nitro group (C2–N3 = 1.441 Å, O4–N3 = 1.211 Å) and the nitramino group (O3–N5 = 1.227 Å, N4–N5 = 1.300 Å) are virtually the same as in parent compound
4 or salts
5 and
6. The bond distances of the cation fit the values of the literature [
4].
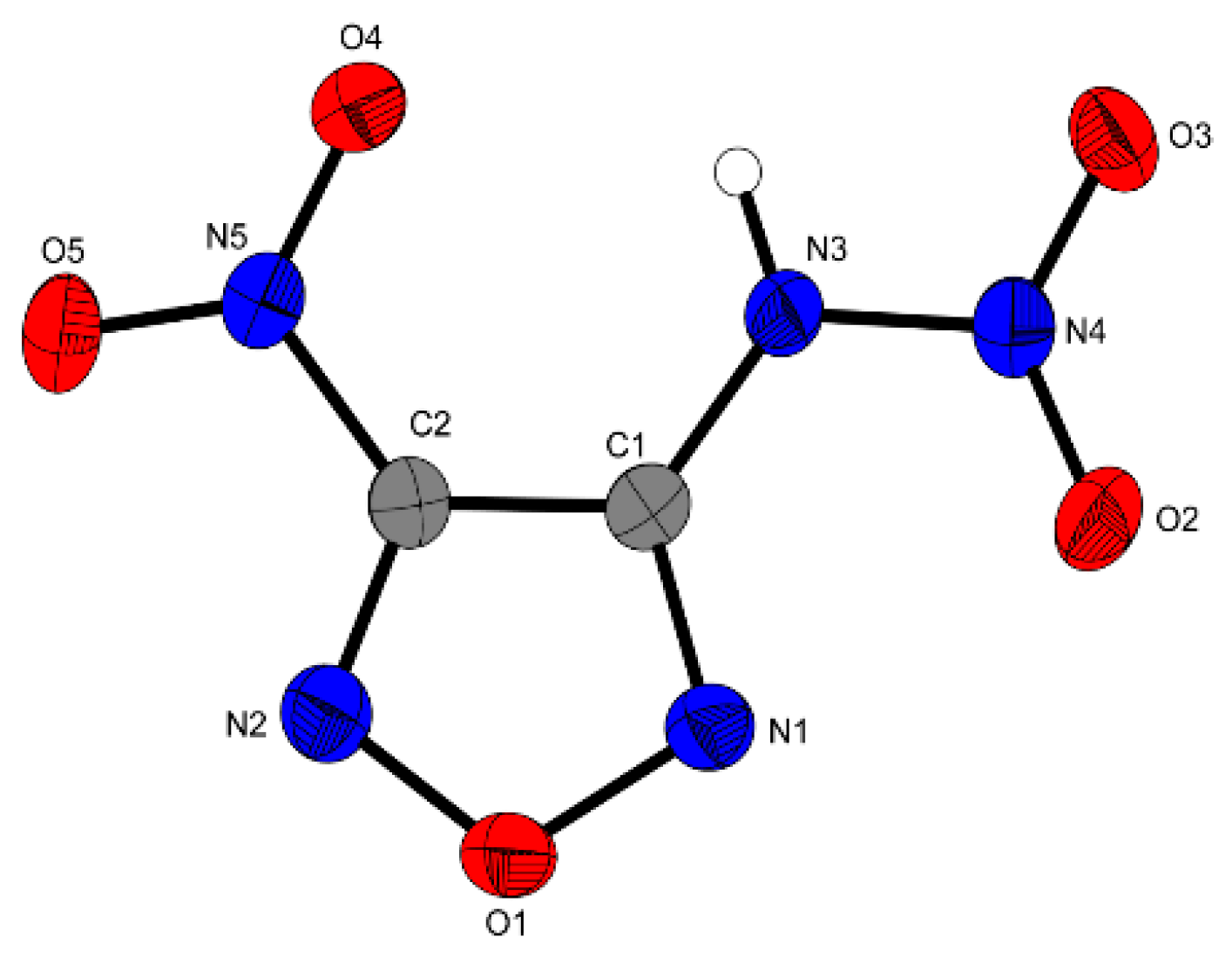
Figure 6.
Molecular unit of 8. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.422(3), N4–C1 1.381(3), N4–N5 1.300(3), O3–N5 1.277(3), C2–N3 1.441(3), O4–N3 1.211(3); selected bond angles (°): N1–O1–N2 111.64(19), N2–C2–N3 138.4(2), O2–N5–N4 123.3(2), N4–C1–C2 134.1(2); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N10−H10…O3: 0.79(3), 2.08(3), 2.869(2), 175(3); N11–H11A…N2 0.87(4), 2.42(4), 3.190(3), 148(3); N12–H12A…O5 0.88(3), 2.58(3), 3.345(3), 147(3); selected torsion angles (°): N2–O1–N1–C1 1.2(3), O2–N5–N4–C1 7.4(4), N5–N4–C1–C2 50.6(4), O5–N3–C2–C1 157.9(2).
Figure 6.
Molecular unit of 8. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.422(3), N4–C1 1.381(3), N4–N5 1.300(3), O3–N5 1.277(3), C2–N3 1.441(3), O4–N3 1.211(3); selected bond angles (°): N1–O1–N2 111.64(19), N2–C2–N3 138.4(2), O2–N5–N4 123.3(2), N4–C1–C2 134.1(2); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N10−H10…O3: 0.79(3), 2.08(3), 2.869(2), 175(3); N11–H11A…N2 0.87(4), 2.42(4), 3.190(3), 148(3); N12–H12A…O5 0.88(3), 2.58(3), 3.345(3), 147(3); selected torsion angles (°): N2–O1–N1–C1 1.2(3), O2–N5–N4–C1 7.4(4), N5–N4–C1–C2 50.6(4), O5–N3–C2–C1 157.9(2).
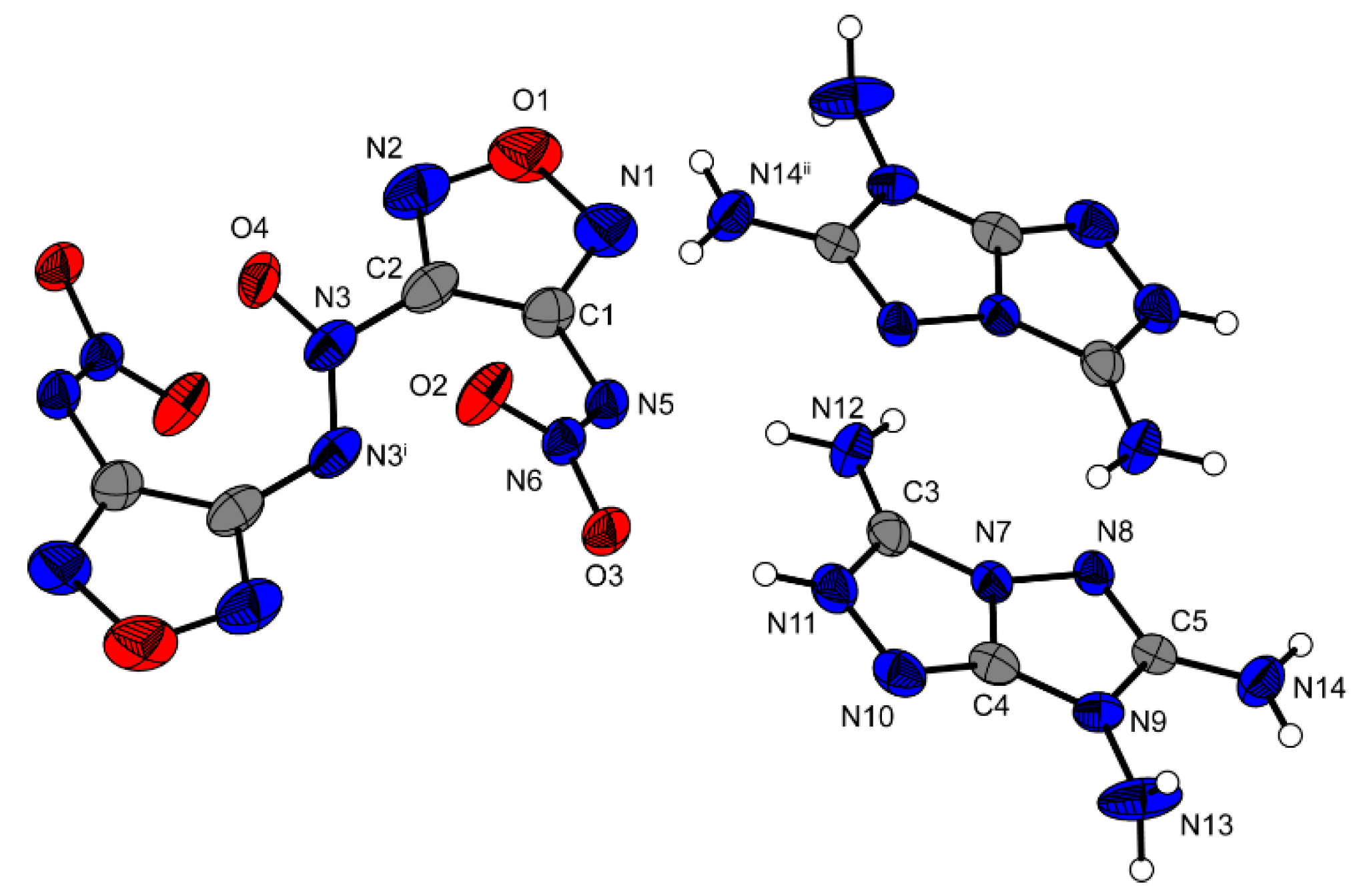
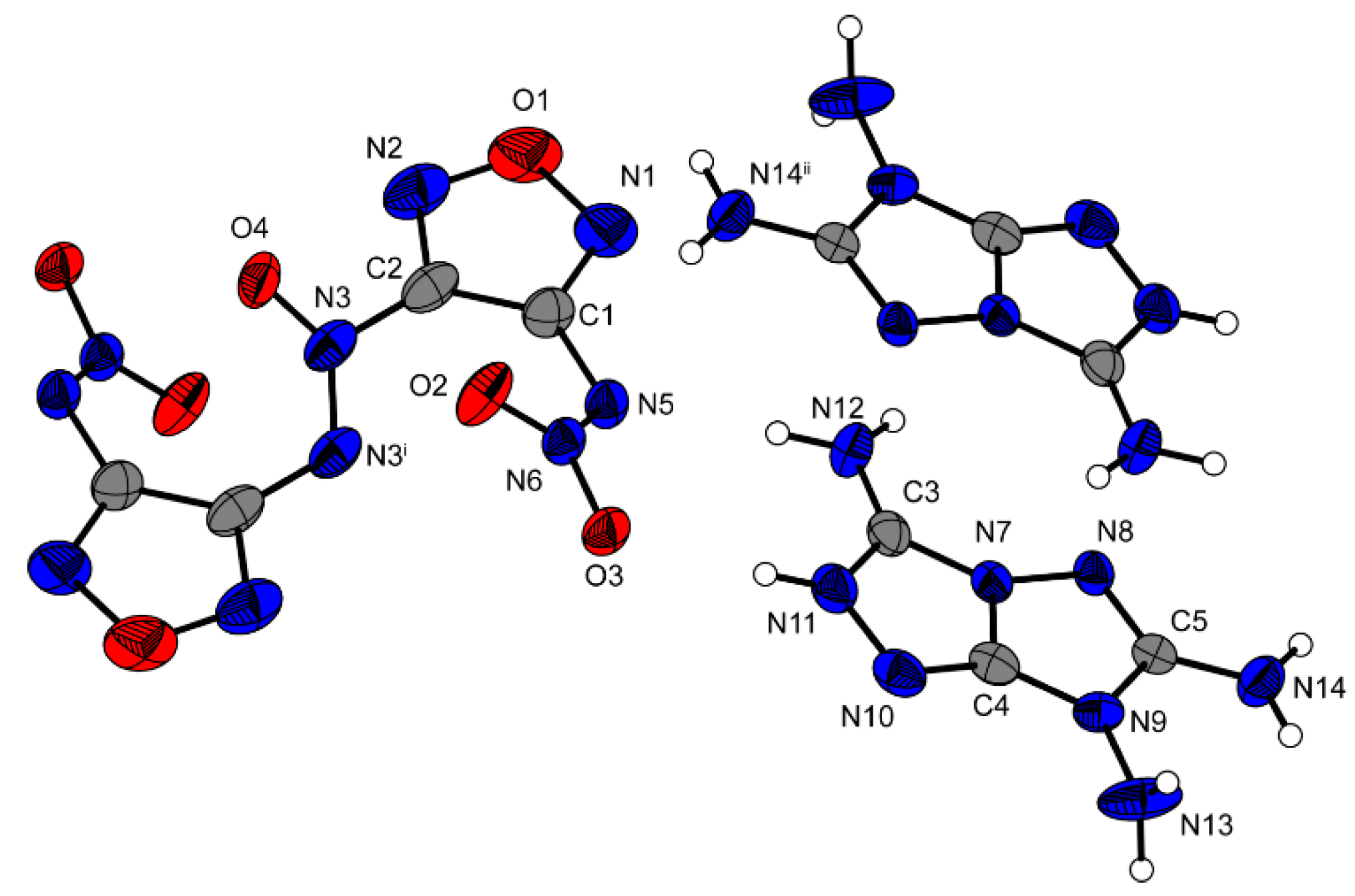
Di-3,6,7-triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazolium dinitraminoazoxyfurazan
12 crystallizes in the triclinic space group
P−1 with a cell volume of 561.13 Å
3 and one molecular units per unit cell. The calculated density at 173 K is 1.807 g·cm
−3, which is somewhat lower than the density of the corresponding hydroxylammonium salt: 1.883 g·cm
−3 (X-ray analysis at 173 K). [
6] The molecular unit of compound
12 is displayed in
Figure 7.
In compound
12, the N=N connected furazan rings are tilted against each other by a torsion angle of 38.2° (N3i–N3–NC2–C1). O4 is in the same plane as the furazan ring which is located further away (O4–N3–C3i–C2i = 1.0°) and twisted against the furazan ring which is closer to O4 (O4–N3–C2–C1 = 37.0°). The two nitramino moieties are also tilted out of the furazan plane by 44.0° (N6–N5–C1–C2). The two cations have a planar structure except for the two protons located at N13 as reported previously in the literature [
4]. As in salt
8, an intensive hydrogen bond network is formed between the three amino groups of the cation and the nitro and nitramino groups of the anion, which makes the salt 12 stable up to 266 °C The bond distances of the anion (C1–C2 = 1.432 Å, N5–C1 = 1.380 Å, N5–N6 = 1.306 Å, O2–N6 = 1.244 Å, C2–N3 = 1.416 Å, N3–O4 = 1.199 Å), are in accordance with other dinitraminoazoxyfurazanes [
6]. The bond distances of the cation are in good agreement with literature values. [
4]
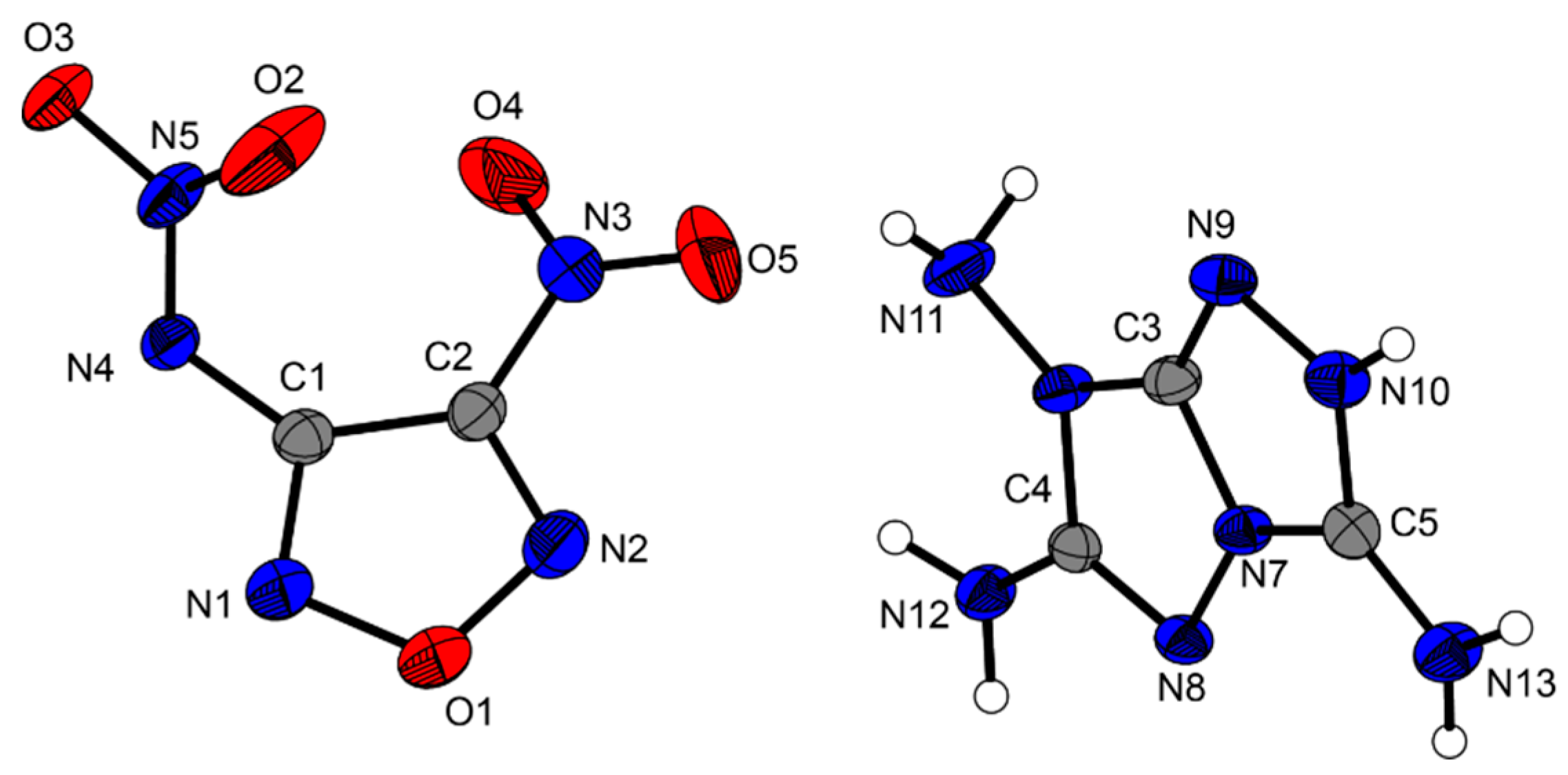
Figure 7.
Molecular unit of 12. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.432(4), N5–C1 1.380(3), N5–N6 1.306(3), O2–N6 1.244(3), C2–N3 1.416(4), N3–O4 1.199(4), N3–N3i 1.268(3); selected bond angles (°): N3i–N3–C2 115.2(2), N4–N3–C2 111.9(3), N6–N5–C1 113.2(2); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N11−H11…O3: 0.89(2), 1.95(3), 2.814(3), 164(3); N12–H12A…N5 0.90(4), 2.11(2), 3.001(3), 171(3); N12–H12B…O2 0.86(4), 2.48(3), 3.163(3), 137(3); selected torsion angles (°): N3i–N3–C2–C1 38.2(4), O4–N3–C3i–C2i 1.0(4), O4–N3–C2–C1 37.0(4), N6–N5–C1–C2 44.0(4), C1–N5–N6–O2 5.7(3).
Figure 7.
Molecular unit of 12. Ellipsoids are drawn at the 50% probability level. Selected bond distances (Å): C1–C2 1.432(4), N5–C1 1.380(3), N5–N6 1.306(3), O2–N6 1.244(3), C2–N3 1.416(4), N3–O4 1.199(4), N3–N3i 1.268(3); selected bond angles (°): N3i–N3–C2 115.2(2), N4–N3–C2 111.9(3), N6–N5–C1 113.2(2); selected hydrogen bond distances [Å] and angles [°] (D−H…A, d(D−H), d(H…A), d(D…A), (D−H…A)): N11−H11…O3: 0.89(2), 1.95(3), 2.814(3), 164(3); N12–H12A…N5 0.90(4), 2.11(2), 3.001(3), 171(3); N12–H12B…O2 0.86(4), 2.48(3), 3.163(3), 137(3); selected torsion angles (°): N3i–N3–C2–C1 38.2(4), O4–N3–C3i–C2i 1.0(4), O4–N3–C2–C1 37.0(4), N6–N5–C1–C2 44.0(4), C1–N5–N6–O2 5.7(3).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}