Synthesis and Crystal Structure of 1-Chloro-2-methyl-4-nitrobenzene

Abstract
:1. Introduction
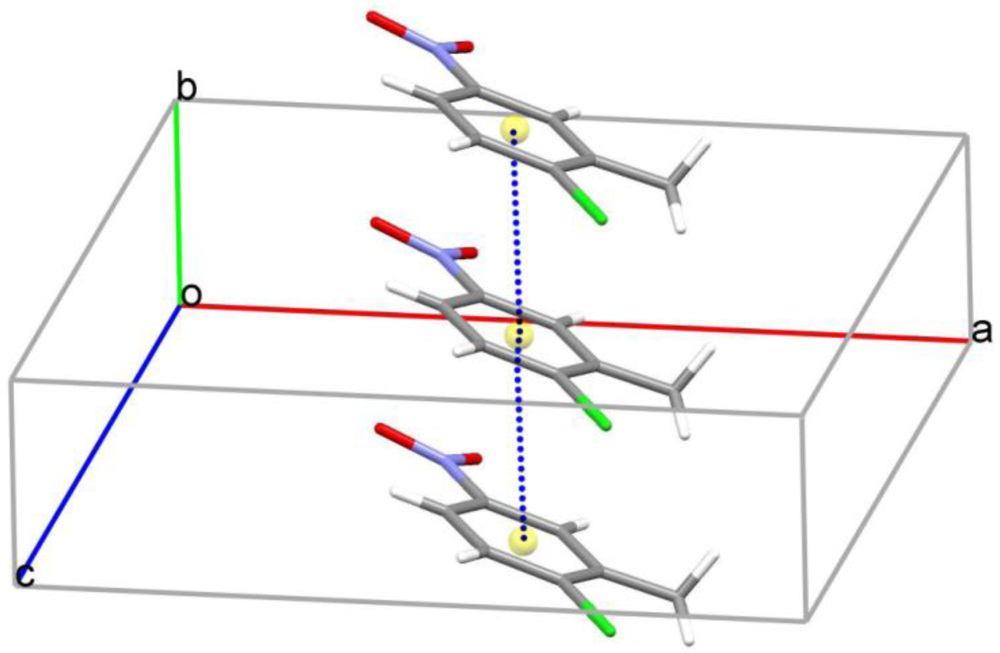
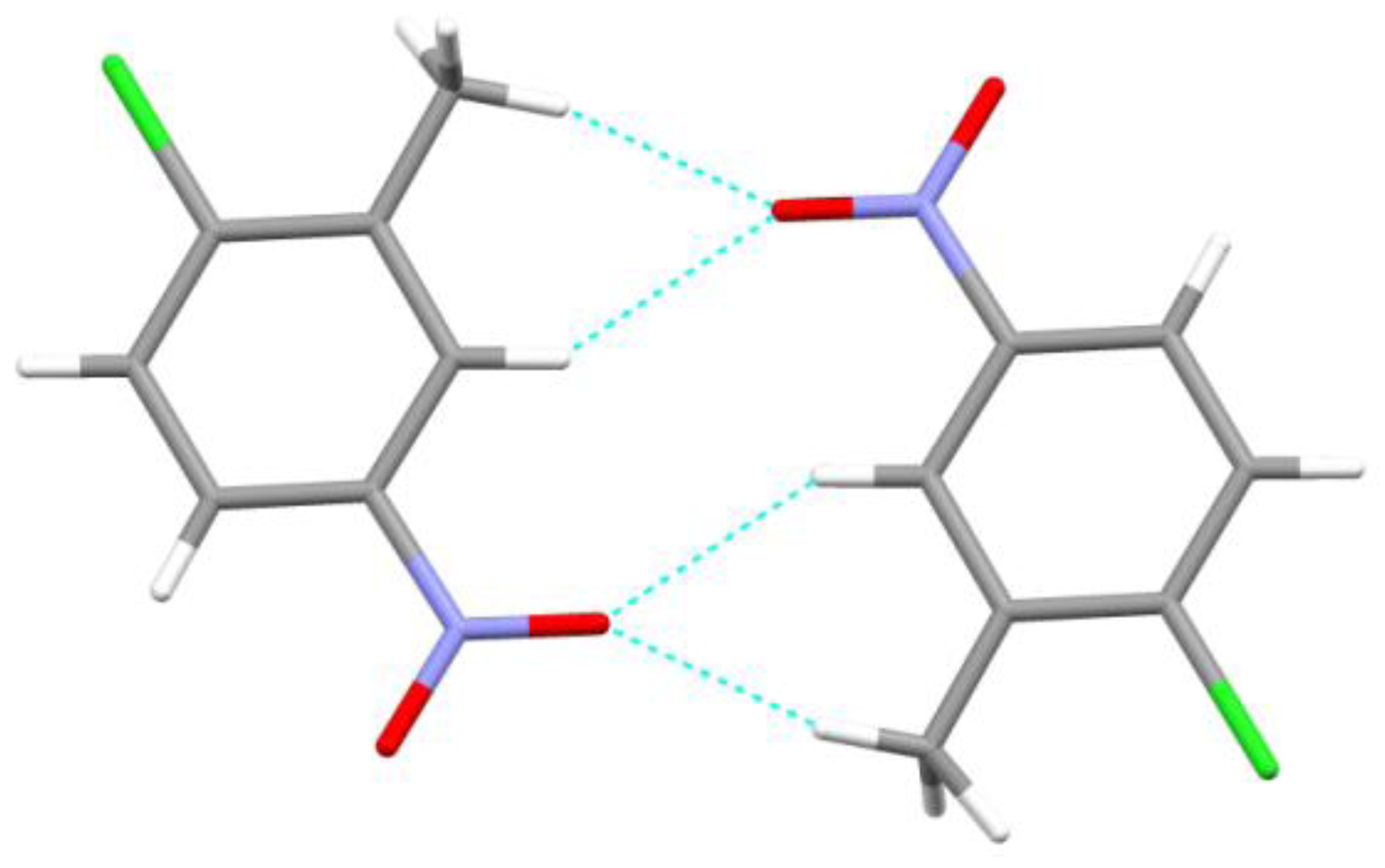
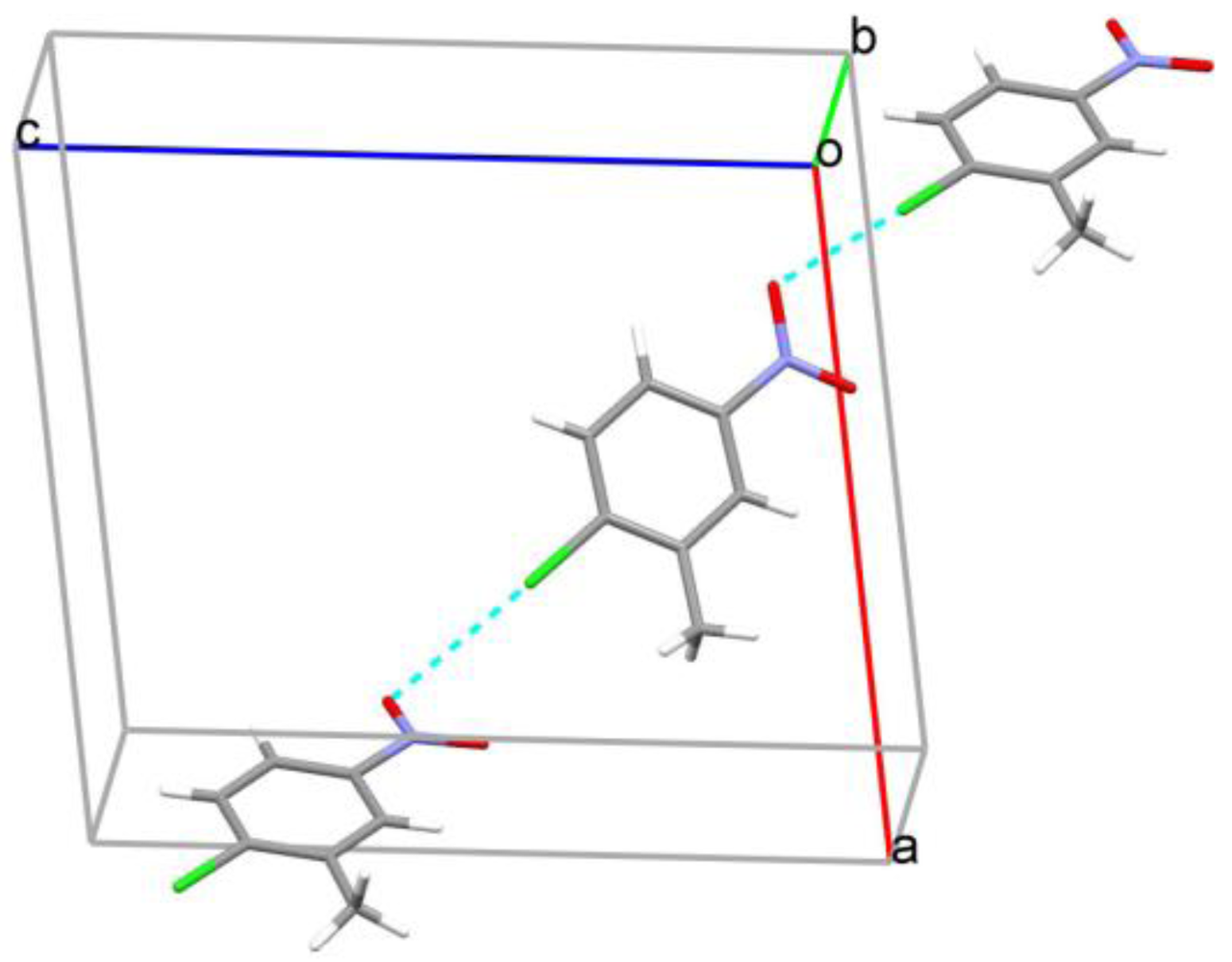
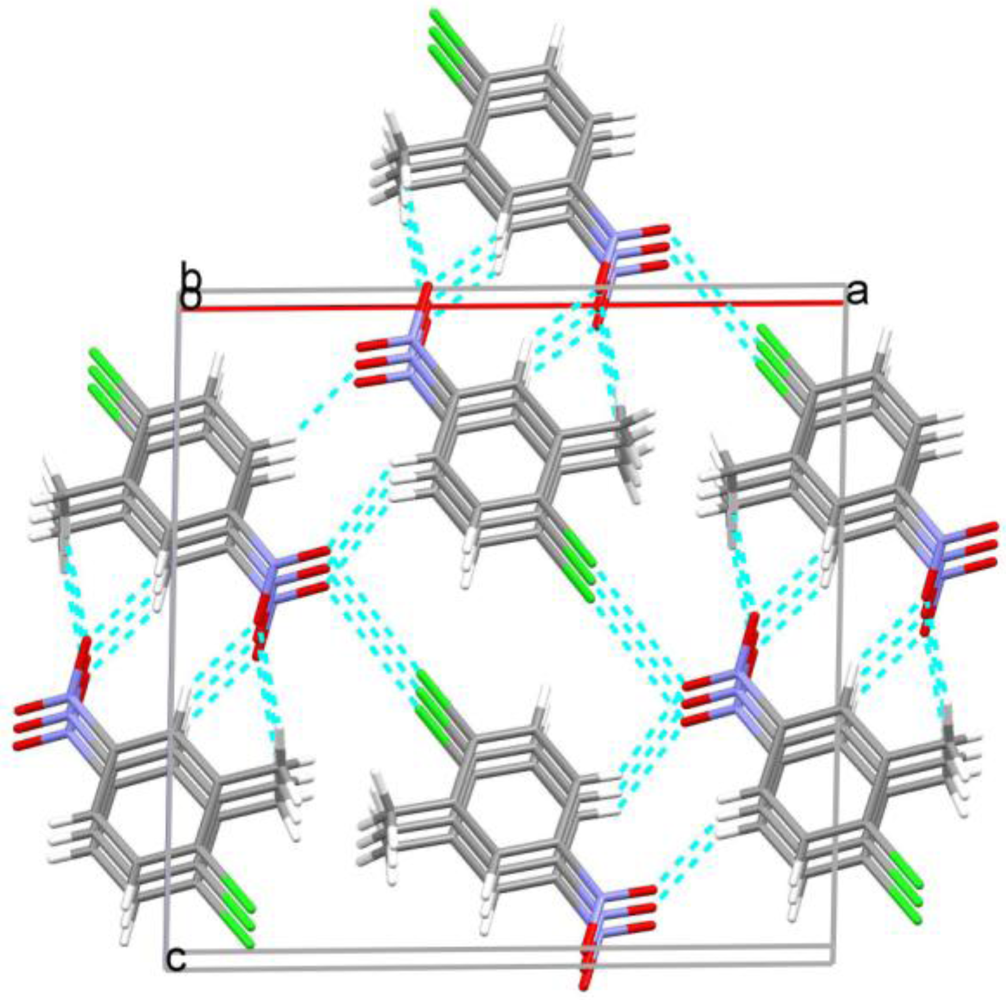
2. Results and Discussion

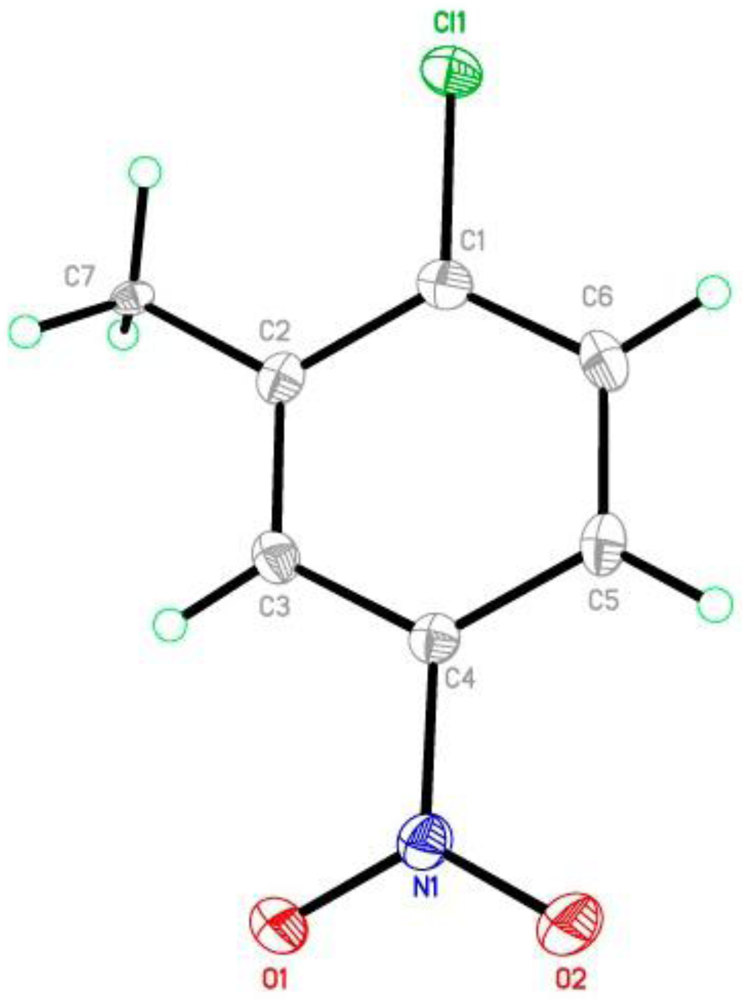




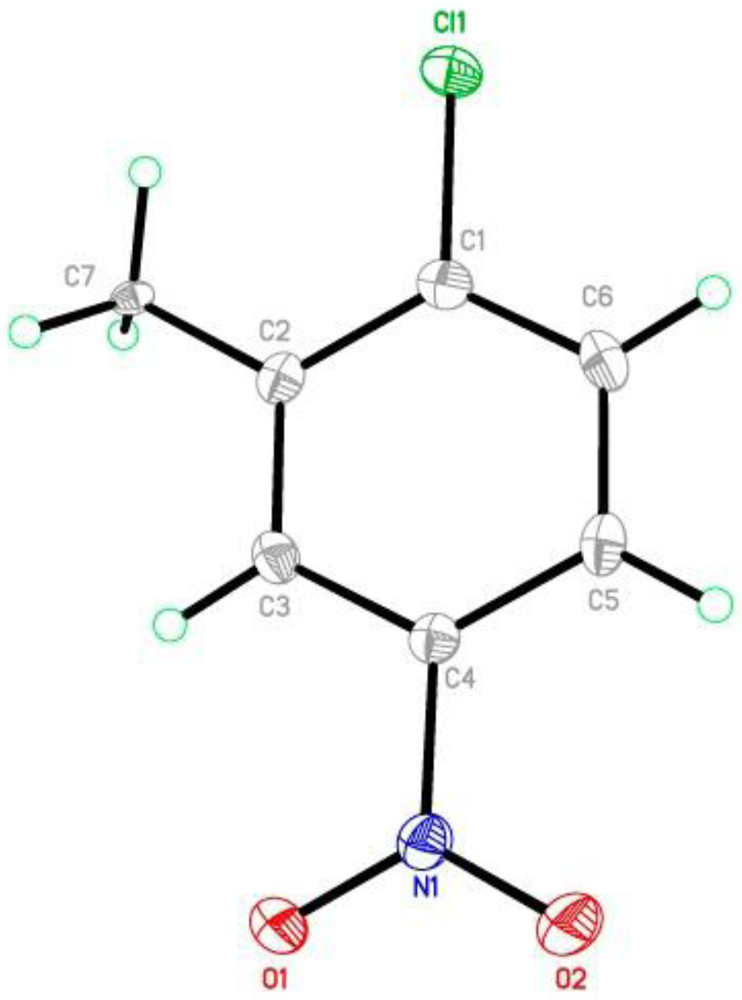
{kind=link}
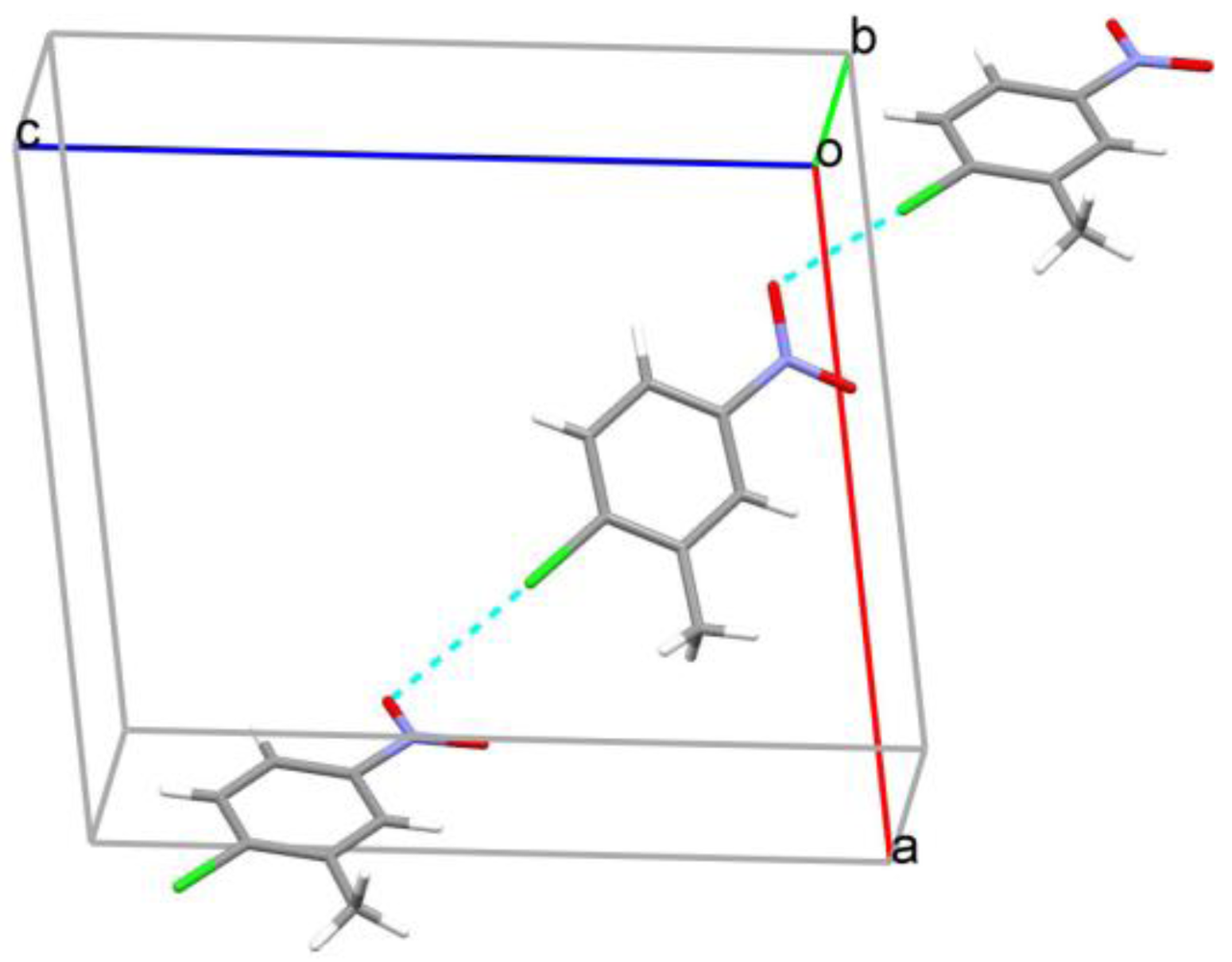
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| C5—H5···O2 i | 0.95 | 2.69 | 3.129(4) | 109 |
| C7—H7D···O1 ii | 0.98 | 2.20 | 3.137(4) | 161 |
| C3—H3···O1 ii | 0.95 | 2.57 | 3.320(4) | 136 |
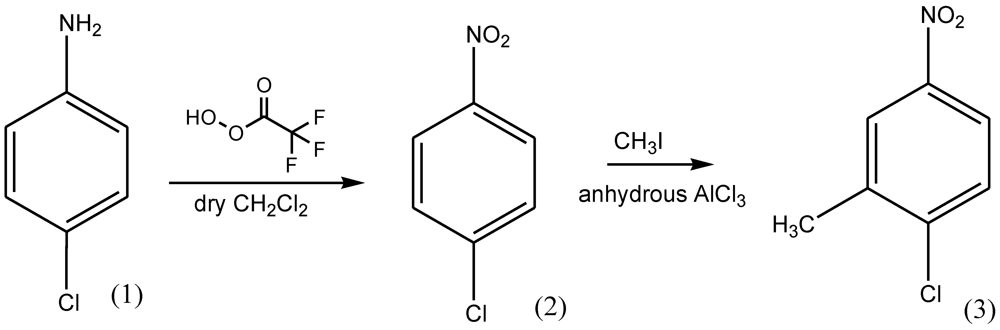
3. Experimental Section
4. Conclusions
Acknowledgments
References and Notes
- Katritzky, A.R.; Oliferenko, P.; Oliferenko, A.; Lomaka, A.; Karelson, M. Nitrobenzene toxicity; QSAR correlations and mechanistic interpretations. J. Phys. Org. Chem. 2003, 16, 811–817. [Google Scholar]
- Ramos, E.U.; Vaal, M.A.; Hermens, J.L.M. Structure-toxicity relationships for benzenes evaluated with Tetrahymena pyriformis. Environ. Toxicol. Pharmacol. 2001, 11, 149–158. [Google Scholar]
- Harttner, D.R. Toxicity of Nitroaromatic Compounds; Hemisphere Publishing Corp.: Washington, D.C., USA, 1985. [Google Scholar]
- Schackmann, A.; Müller, R. Reduction of nitroaromatic compounds by different Pseudomonas species under aerobic conditions. Appl. Microbiol. Biotechnol. 1991, 34, 809–813. [Google Scholar]
- Shimizu, M.; Yasui, T.; Matsumoto, N. Structural specificity of aromatic compounds with special reference to mutagenic activity in Salmonellatyphimurium-a series of chloro- or fluoro-nitrobenzene derivatives. Mutat. Res. 1983, 116, 217–238. [Google Scholar] [CrossRef]
- Spiess, T.F.; Desiere, P.; Fischer, J.C.; Spain, H.-J.; Lenke, H. A new 4-nitrotoluene degradation pathway in a Mycobacterium strain. Appl. Environ. Microbiol. 1998, 64, 446–452. [Google Scholar]
- Susarla, S.; Masunaga, S.; Yonezawa, Y. Transformations of chloronitrobenzenes in anaerobic sediment. Chemosphere 1996, 32, 967–977. [Google Scholar] [CrossRef]
- Takenaka, S.; Murakami, S.; Shinke, R. Metabolism of 2-aminophenol by Pseudomonas sp. AP-3: modified meta-cleavage pathway. Arch. Microbiol. 1998, 170, 132–137. [Google Scholar] [CrossRef]
- Weisburger, E.K.; Russfield, F.; Homburger, J.; Weisburger, J.H.; Boger, E.; Van Dongen, C.G.; Chu, K.C. Testing of twenty-one environmental aromatic amines or derivatives for long-term toxicity or carcinogenicity. Environ. Pathol. Toxicol. 1978, 2, 325–356. [Google Scholar]
- Katsivela, E.; Wray, V.; Pieper, D.H.; Wittich, R. Initial reactions in the biodegradation of 1-chloro-4-nitrobenzene by a newly isolated bacterium, Strain LW1. Appl. Environ. Microbiol. 1999, 65, 1405–1412. [Google Scholar]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths determined by X-Ray and Neutron Diffraction. Part I. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar]
- Henichart, J.-P.; Bernier, J.-L.; Vaccher, C.; Houssin, R.; Warin, V.; Baert, F. Cyclisation de dinitrodiarylamines en nitro-3 carbazoles inhibition sterique de la substitution nucleophile SNAr par effet. Tetrahedron Lett. 1979, 20, 945–948. [Google Scholar]
- Henichart, J.-P.; Bernier, J.-L.; Vaccher, C.; Houssin, R.; Warin, V.; Baert, F. Acces aux dinitro-2,4 diarylamines par substitution nucleophile SNAr-limites de la reaction et application a la synthese de cyanocarbazoles. Tetrahedron 1980, 36, 3535–3541. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. H bond motifs. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- APEX2, SAINT and SADABS. Bruker AXS Inc.: Madison, Wisconsin, USA, 2009.
- Hunter, K.A.; Simpson, J. TITAN2000; University of Otago: Dunedin, New Zealand, 1999. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saeed, A.; Simpson, J. Synthesis and Crystal Structure of 1-Chloro-2-methyl-4-nitrobenzene. Crystals 2012, 2, 137-143. https://doi.org/10.3390/cryst2010137
Saeed A, Simpson J. Synthesis and Crystal Structure of 1-Chloro-2-methyl-4-nitrobenzene. Crystals. 2012; 2(1):137-143. https://doi.org/10.3390/cryst2010137
Chicago/Turabian StyleSaeed, Aamer, and Jim Simpson. 2012. "Synthesis and Crystal Structure of 1-Chloro-2-methyl-4-nitrobenzene" Crystals 2, no. 1: 137-143. https://doi.org/10.3390/cryst2010137
APA StyleSaeed, A., & Simpson, J. (2012). Synthesis and Crystal Structure of 1-Chloro-2-methyl-4-nitrobenzene. Crystals, 2(1), 137-143. https://doi.org/10.3390/cryst2010137