3.1. Preparation of iBody Conjugates
3.1.1. General Procedures
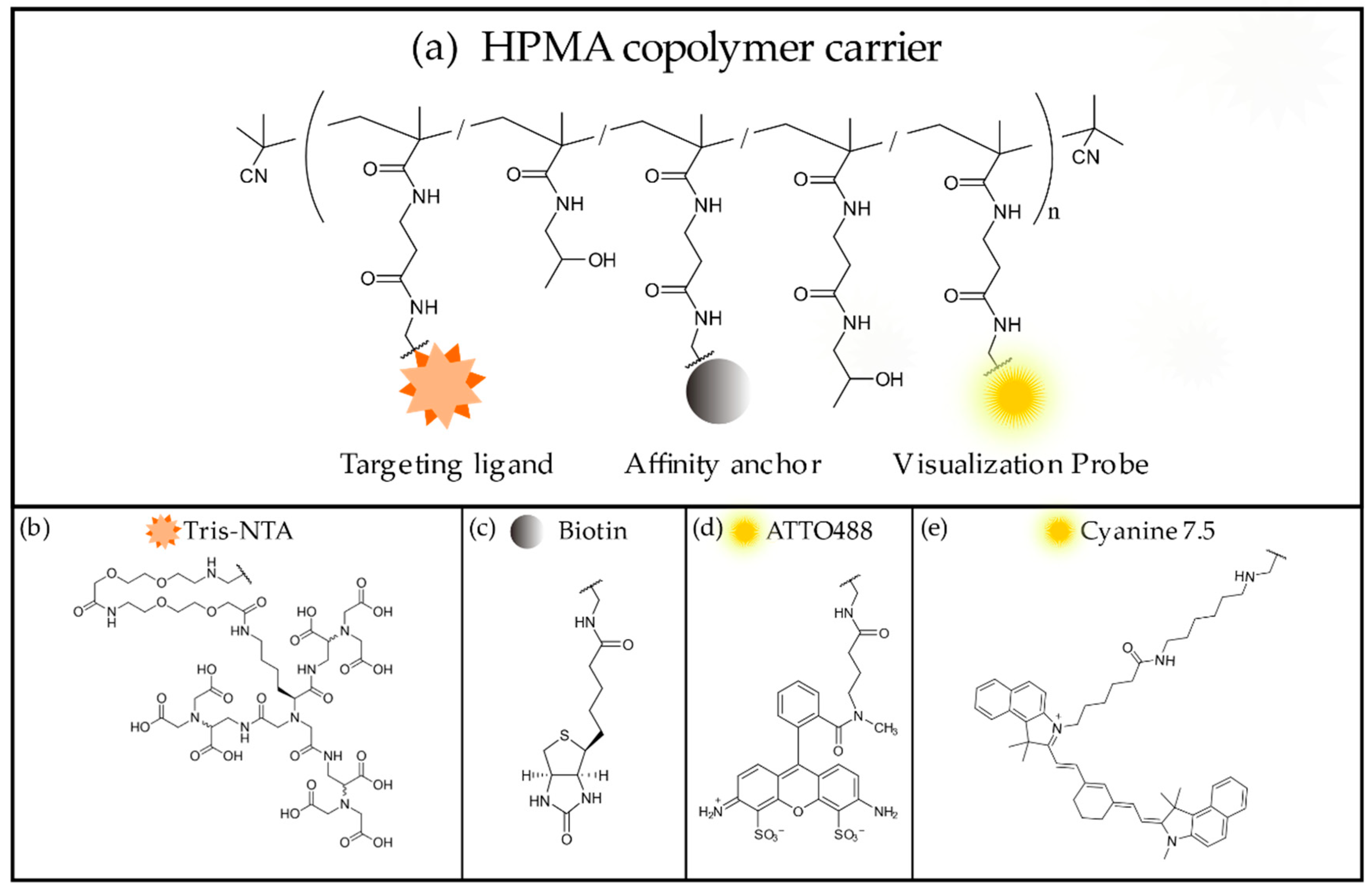
Detailed experimental procedures for the synthesis of the HPMA copolymer precursor and the tris-NTA ligands, as well as the process of ligand conjugation, have been described previously [
1]. Generally, the anti-polyHis polymer conjugates iBody 1 and 2 were prepared by reaction of the copolymer precursor poly(HPMA-co-Ma-ß-Ala-TT) containing thiazolidine-2-thione reactive groups (TT) along the polymer chain with a mixture of fluorophore molecules (fluorophore-NH
2), affinity anchors (biotin-NH
2), and targeting ligands (NH
2-tris-NTA) in a desired ratio.
3.1.2. Synthesis of iBody 1
Copolymer precursor poly(HPMA-co-Ma-ß-Ala-TT) (15 mg, 9.96 × 10−6 mol TT groups; Mw = 64,400 g/mol, Mn = 58,500 g/mol, Ð = 1.10), NH2-tris-NTA (3.0 mg, 2.36 × 10−6 mol), Cy7.5-amine (1.0 mg, 1.22 × 10−6 mol) and N-(2-aminoethyl) biotinamide hydrobromide (biotin-NH2) (1.5 mg, 4.08 × 10−6 mol) were dissolved in 0.3 mL of N,N-dimethylacetamide (DMA). N,N-diisopropylethylamine (DIPEA) (3.5 µL, 1.53 × 10−5 mol) was added, and the reaction was carried out at room temperature (RT) for 4 h. Then, 2 µL of 1-aminopropan-2-ol were pipetted into the mixture to remove residual TT reactive groups, and the reaction was stirred for 10 min. Copolymer conjugate poly(HPMA-co-Ma-ß-Ala-TrisNTA-co-Ma-ß-Ala-Cy7.5-co-Ma-ß-Ala-biotin) (iBody 1) was isolated by precipitation into a mixture of acetone:diethyl ether (3:1), filtered off, washed with acetone and diethyl ether and dried in vacuo. Polymer conjugate was purified on a Sephadex LH-20 chromatography column in methanol. Finally, methanol was evaporated, and the conjugate was dissolved in water, purified on a PD-10 column and lyophilized.
3.1.3. Synthesis of iBody 2
Copolymer precursor poly(HPMA-co-Ma-ß-Ala-TT) (15.1 mg; 1.00 × 10−5 mol TT groups Mw = 64,400 g/mol, Mn = 58,500 g/mol, Ð = 1.10), tris-NTA (3.5 mg, 2.75 × 10−6 mol), ATTO488-NH2 (1.69 mg, 1.97 × 10−6 mol) and N-(2-aminoethyl)biotinamide hydrobromide (biotin-NH2) (1.63 mg, 4.44 × 10−6 mol) were dissolved in 0.3 mL of dimethyl sulfoxide (DMSO). N,N-diisopropylethylamine (DIPEA) (8.0 µL, 4.58 × 10−5 mol) was then added, and the reaction was carried out for 4 h at room temperature. Next, 2 µL of 1-aminopropan-2-ol was added to remove the residual TT reactive groups, and the reaction was stirred for 10 min. Copolymer conjugate poly(HPMA-co-Ma-ß-Ala-TrisNTA-co-Ma-ß-Ala-ATTO488-co-Ma-ß-Ala-biotin) (iBody 2) was isolated by precipitation into a mixture of acetone:diethyl ether (3:1), filtered off, washed with acetone and diethyl ether and dried in vacuo. The polymer conjugate was purified on a Sephadex LH-20 chromatography column in methanol. Finally, methanol was evaporated, and the conjugate was dissolved in water, purified on a PD-10 column and lyophilized.
3.1.4. Synthesis of iBody 3
Copolymer precursor poly(HPMA-co-Ma-ß-Ala-TT) (50 mg; 3.76.00 × 10−5 mol TT groups Mw = 73,000 g/mol, Mn = 60,000 g/mol, Ð = 1.22), ATTO488-NH2 (2.5 mg, 2.91 × 10−6 mol) and N-(2-aminoethyl)biotinamide hydrobromide (biotin-NH2) (6.0 mg, 1.63 × 10−5 mol) were dissolved in 0.5 mL of dimethyl sulfoxide (DMSO). N,N-diisopropylethylamine (DIPEA) (6.8 µL, 3.9×10−5 mol was then added, and the reaction was carried out for 4 h at room temperature. Next, 3 µL of 1-aminopropan-2-ol were added to remove the residual TT reactive groups, and the reaction was stirred for 10 min. Copolymer conjugate poly(HPMA-co-Ma-ß-Ala-ATTO488-co-Ma-ß-Ala-biotin) (iBody 3) was isolated by precipitation into a mixture of acetone:diethyl ether (3:1), filtered off, washed with acetone and diethyl ether and dried in vacuo. The polymer conjugate was purified on a Sephadex LH-20 chromatography column in methanol. Finally, methanol was evaporated, and the conjugate was dissolved in water, purified on a PD-10 column and lyophilized.
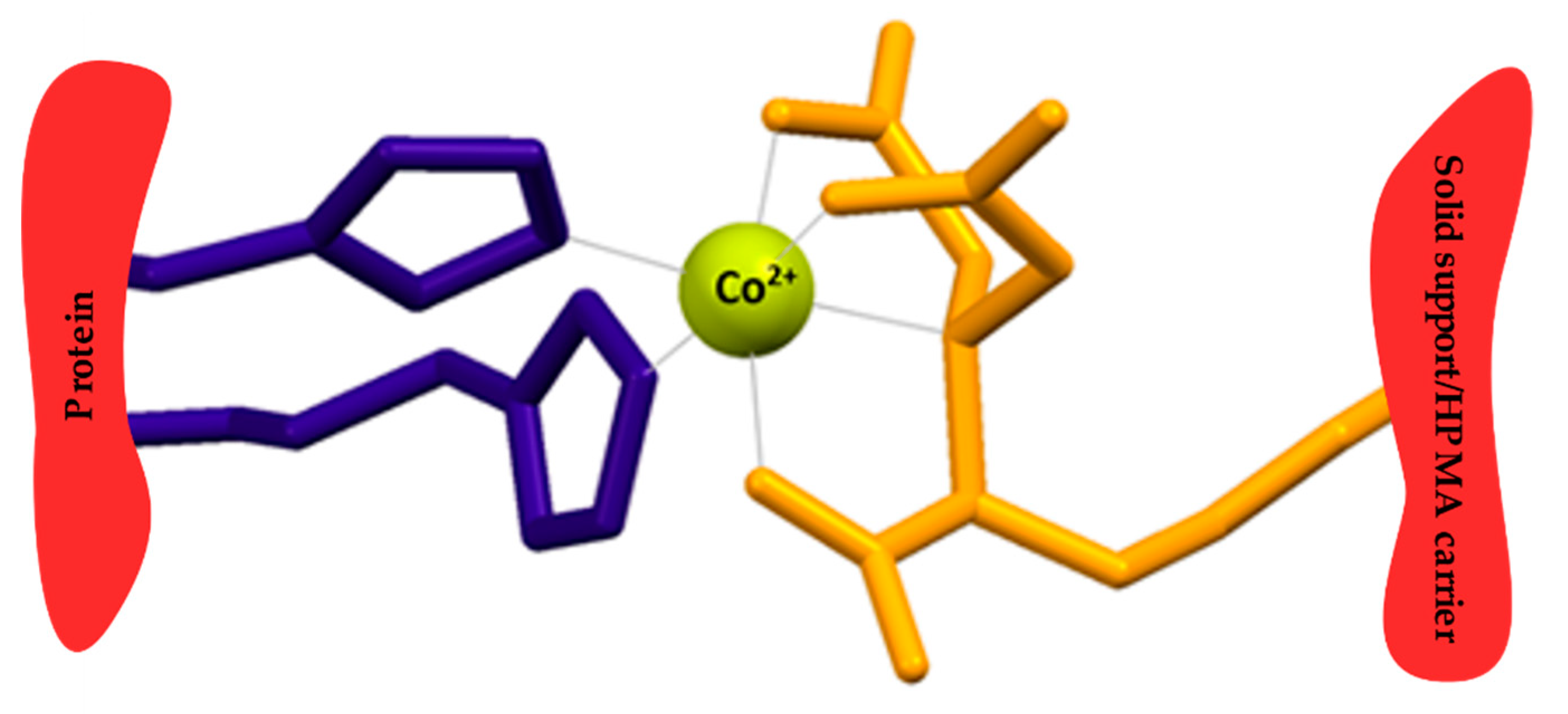
3.1.5. Charging Tris-NTA with Metal Cations
Lyophilized anti-polyHis iBody 1 and iBody 2 were weighed and dissolved in 10 mM CoCl2 or NiCl2. Samples were incubated on a rotator at room temperature for 1 h. The solutions were then transferred into dialysis chambers (10 kDa cutoff) and allowed to equilibrate in 5 L of water at 4 °C for 36 h with multiple (6×) exchanges of water. After dialysis, the concentrations of the charged anti-polyHis iBodies in solution were experimentally determined by amino acid analysis.
3.1.6. Determination of the Composition and Molecular Weight of iBodies
The weight-average molecular weights (Mw), number-average molecular weights (Mn), and dispersity (Ð) of the polymer precursor and conjugates were determined using a HPLC (Shimadzu, Japan) system equipped with a UV-DAD detector, an Optilab®NEO differential refractometer, Viscostar III and DAWN-Heleos II multi-angle light scattering (all from Wyatt Technology, USA) detector and a size-exclusion chromatography on a Superose 6 column. The Mw, Mn and Đ were calculated using Astra 7 software. The refractive index increment dn/dc = 0.167 mL/g was used for calculation. These experiments were conducted with 0.3 M sodium acetate, pH 6.5, buffer at a flow rate of 0.5 mL/min.
The content of TT reactive groups in the polymer precursors and fluorescent dyes in the conjugates were determined spectrophotometrically (TT ε305nm = 10,600 L·mol−1·cm−1, methanol), (ATTO488 ε502nm = 90,000 L·mol−1·cm−1, water) and (Cy7.5 ε788nm = 223,000 L·mol−1·cm−1, water). The biotin content was determined colorimetrically using the HABA/Avidin Reagent Kit (Sigma-Aldrich) the results were corrected for the effect of ATTO488 absorbance at 500 nm.
The content of tris-NTA in iBodies 1 and 2 was determined by amino acid analysis of the hydrolyzed polymer conjugates (6 M HCl, 115 °C, 18 h in a sealed vial). A reversed-phase column (Chromolith®HighResolution RP-18e, 100 × 4.6 mm) (Merck, Germany), following pre-column derivatization with 2,3-naphthalenedicarboxaldehyde (NDA)/NaCN (source of cyanide ions), and a fluorescence detector (excitation at 229 nm, emission at 490 nm) were used for the analysis. Gradient elution with 10%–100% solvent B (300 mL of 0.17 M sodium acetate and 700 mL of methanol) was performed for 35 min at a flow rate of 1.0 mL/min. Solvent A was 0.05 M sodium acetate buffer, pH 6.5.
3.2. Expression and Purification of the His-Tagged SUMO1 Proteins
3.2.1. Plasmid Preparation and Cloning
We selected the pET-15b expression plasmid, which encodes a 6×His-tag, for production of recombinant SUMO1 proteins with 6×His and 10×His fusion tags. We used as a template the pHisTEV30a-SUMO1 plasmid encoding the protein of interest (along with a Tobacco Etch Viru (TEV) protease cleavage site and 10×His cleavage) prepared in-house.
First, the protein coding sequence was amplified from pHisTEV30a-SUMO1 using Taq polymerase and primers 5′-aaacatatgtctgaccaggag-3′ (Fwd, Tm = 57 °C) and 5′-aaactcgagctaaccccc-3′ (Rev, Tm = 59 °C) including NdeI and XhoI restriction sites. To obtain the pET-15b vector coding for 6×His-tagged SUMO1 protein, the amplified PCR product was cleaved with XhoI and NdeI and ligated into pET-15b via corresponding restriction sites. Next, the newly cloned pET-15b-6×HisSUMO1 plasmid served as a template for PCR amplification of the 10×His-tagged SUMO1 protein coding sequence the using primers 5′-taccatgggcagcagccatcatcatcatcatcatcatcatcatcacagca-3′ (Fwd, Tm = 89.8 °C) and 5′-atctcgagctaaccccccgtttgttcctgataaacttcaatcacatcttc-3′ (Rev, Tm = 83.0 °C) including NcoI and XhoI restriction sites. The obtained PCR product was cleaved and ligated into pET-15b via NcoI and XhoI restriction sites, yielding pET-15b-10×HisSUMO1 vector. To verify the sequences of the obtained plasmids, control digestion reactions and Sanger sequencing were performed.
3.2.2. Expression of Recombinant SUMO Proteins
Recombinant SUMO1 proteins with N-terminally fused polyhistidine affinity tags of different lengths were expressed in E. coli. BL21-DE3-RIL competent cells were first transformed with the two prepared plasmids, pET-15b-6×HisSUMO1 and pET-15b-10×HisSUMO1, then plated on LB agar containing ampicillin (100 µg/µL) and allowed to grow at RT for 48 h. A single colony was picked from each plate and separately transferred into liquid media containing ampicillin (8 mL LB, 100 µg/mL ampicillin). After allowing the transformed bacterial cells to grow at 37 °C on a shaker for 12 h, each of the pre-grown cultures was used to inoculate 500 mL of LB media. The cultures were allowed to grow at 16 °C on a shaker. After cultures reached an OD600 of 0.6, protein expression was induced by addition of 0.4 mM Isopropyl ß-D-1-thiogalactopyranoside (IPTG)and allowed to progress overnight. Afterwards, both samples were centrifuged, and the pellets were resuspended in lysis buffer (50 mM Tris, pH 8, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA)). Protease inhibitor cocktail (Sigma Aldrich, Czech Republic) was added, and the samples were homogenized with an Avestin EMULSIFLEX C3 instrument (ATA Scientific, Australia).
3.2.3. Purification of Recombinant His-SUMO Proteins
Cell lysates from expression cultures were collected and used for purification on nickel-NTA resin. Washed resin (0.5 mL) was added to each lysate, and the samples were allowed to incubate for 2 h at RT. The resin with bound proteins was washed three times (50 mM Tris, pH 8.0, 300 mM NaCl, 20 mM imidazole), and the proteins were eluted using buffer with a high concentration of imidazole (50 mM Tris, pH 8.0, 300 mM NaCl, 250 mM imidazole). Elution fractions containing the desired products were transferred into Tris-buffered saline (TBS; 50 mM Tris, pH 7.6, 137 mM NaCl) by dialysis. All fractions (flow-throughs and elutions) were collected during the procedure and used for analysis with silver-stained SDS-PAGE and Western blotting (
Supplementary Materials, Figure S1, Table S1). Amino acid analysis was performed to determine the concentrations of both proteins with high precision.
3.3. Testing the Anti-polyHis iBodies
3.3.1. Determining the Binding Constants of iBodies with ELISA
To measure the binding constants of iBodies to His-tag and evaluate the differences in strength of cobalt- or nickel-mediated interactions with 6×His and 10×His fusion tags, simple sandwich ELISA was performed in a 96-well, flat-bottom, MaxiSorp, nontransparent plate (Thermo Fisher Scientific, Czech Republic). SUMO1 protein (20 ng per well) was immobilized directly onto the plate (1 h, RT) and blocked with 1% BSA in TBS (4 °C, O/N). iBody 1 or iBody 2 were then bound to the protein in a concentration series (60 min, RT) (see details in
Table S2). In the last layer of the sandwich, neutravidin-HRP was bound via biotin and used for detection. Between binding steps of each new species, wash steps were performed, i.e., 4×TBS with 0.1% Tween20 (TBST). Immediately after addition of the HRP substrate, luminescence at 428 nm was measured on a TECAN Infinite M1000 instrument (TECAN, Germany).
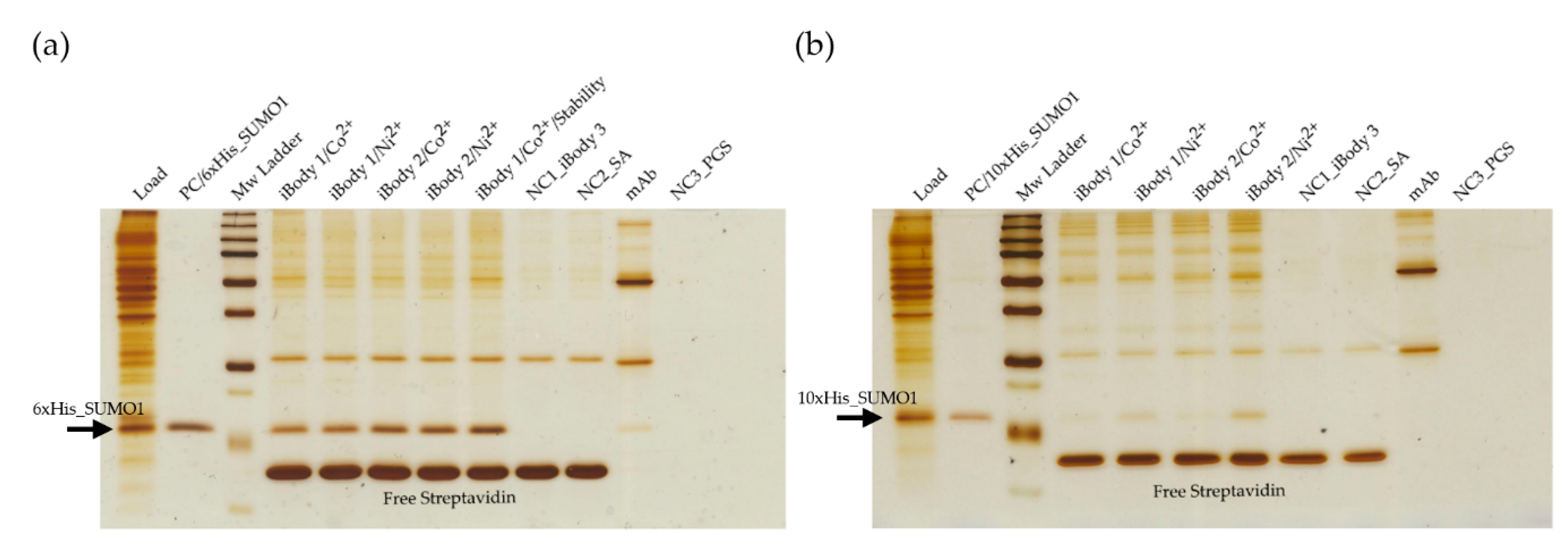
3.3.2. Immobilization on iBodies and Isolation of His-SUMO1 Proteins from Cell Lysate
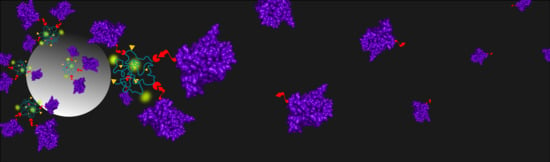
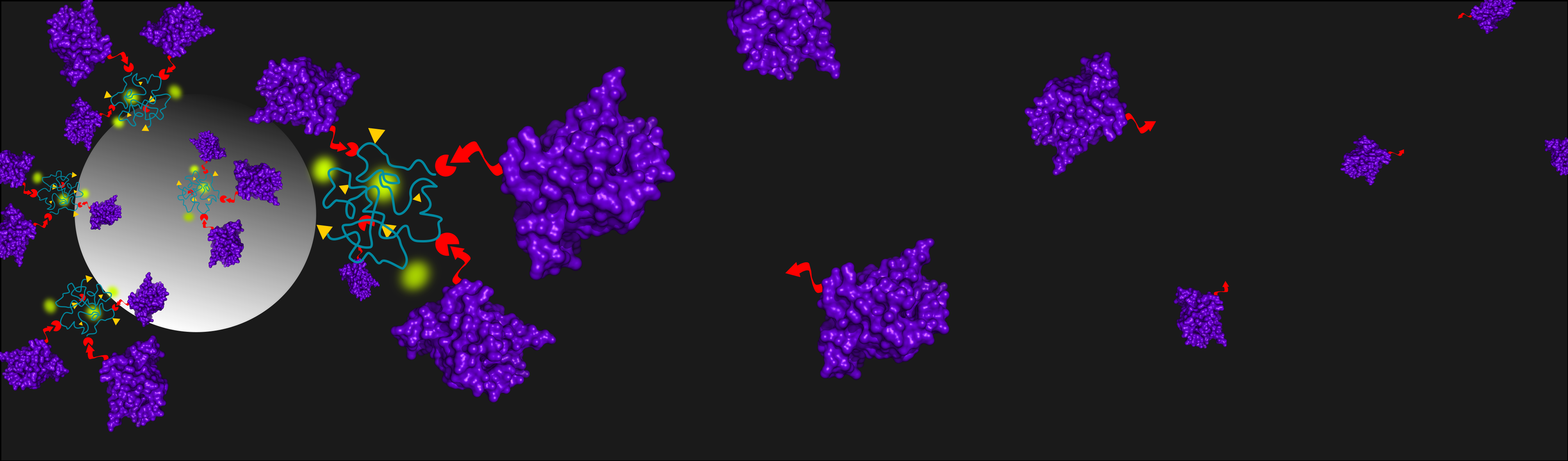
iBody 1 (Ni2+ or Co2+ charged), iBody 1 (stability testing, Co2+ charged), iBody 2 (Ni2+ or Co2+ charged), iBody 3 (negative control), and anti-His-tag mAb (Sigma Aldrich, Czech Republic) were immobilized on specialized beads and used for affinity pull-down of SUMO1 proteins. The iBodies were immobilized on streptavidin agarose Ultra Performance beads (Solulink, USA) via their biotin ligands. The mAb was immobilized on protein-G sepharose beads 4 fast flow (GE Healthcare, USA) via the Fc domain. Aliquots of 300 µL of each 150 nM species to be bound were incubated with 12 µL prewashed beads (TBS, 4 °C, O/N) for 90 min at RT.
Eukaryotic cell lysate (total protein concentration ~ 7 mg/mL) was 4x diluted in TBST and spiked with either 6×His SUMO1 or 10×His SUMO1 (final SUMO1 concentration of 20 ng/µL). Each immobilized species was then mixed with the spiked cell lysate (each sample containing 4 µg of SUMO1) and allowed to incubate (1.5 h, RT). Immunoprecipitated proteins were eluted from resins by incubation in 30 µL SDS buffer (10 min, 98 °C). Each of the samples from pull-down (12 µL) along with negative controls (iBody 3, blank streptavidin agarose beads and blank protein-G sepharose beads) and positive controls (spiked cell lysate and purified SUMO1) were separated on a 16% polyacrylamide gel (SDS-PAGE, 200 V, 70 min, 4 °C) and visualized by silver staining.
3.3.3. Visualization of His-Tagged Proteins on Western Blot
First, 5 µL and 10 µL (corresponding to 50 ng and 100 ng) of 6×His- and 10×His-SUMO1 were loaded onto 16% polyacrylamide gels and resolved by SDS-PAGE (200 V, 70 min, 4 °C). Proteins were then transferred onto a nitrocellulose membrane (BioRad, Czech Republic) using routine WB procedure (100 V, 60 min). After blotting, membranes were blocked with a TBS/0.5% Casein Blocker (CB) solution (60 min, RT). To detect the proteins, membranes were incubated in iBody incubation buffer (TBS, 0.05% CB, 1 mM imidazole, 0.05% SDS, 0.3% Tween) containing 4 nM iBody 1 or 2. For detection with mAb-CF770 (Sigma-Aldrich, Czech Republic), 4 nM antibody in TBST solution was used. After 20 min incubation at RT, blots were washed four times with TBST and allowed to dry. A LI-COR Odyssey CLx instrument (LI-COR Biotechnology, USA) was used for visualization of NIR fluorophore conjugates (iBody 1 and mAb-CF770). A Typhoon FLA 9500 instrument (GE Healthcare, USA) was used to visualize the proteins detected with ATTO488 carrying conjugates (iBody 2).

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}